

Молекулярная биология, 2020, T. 54, № 6, стр. 955-967
Гипертрофическая кардиомиопатия как олигогенное заболевание: аргументы транскриптомики
Н. М. Баулина a, *, И. С. Киселёв a, О. С. Чумакова a, О. О. Фаворова a
a Национальный медицинский исследовательский центр кардиологии Министерства здравоохранения
Российской Федерации
121552 Москва, Россия
* E-mail: tasha.baulina@gmail.com
Поступила в редакцию 10.07.2020
После доработки 14.07.2020
Принята к публикации 14.07.2020
Аннотация
Гипертрофическая кардиомиопатия (ГКМП) – самая распространенная генетически детерминированная патология сердца, часто сопровождающаяся фатальными осложнениями. Сегодня на смену традиционному взгляду о моногенном происхождении ГКМП приходит представление об олигогенной природе этого заболевания, клинический фенотип которого определяется не только отдельными мутациями в генах, кодирующих белки саркомера в кардиомиоцитах, но и вкладом других генов (другие саркомерные гены, несаркомерные белоккодирующие гены-модификаторы, гены регуляторных некодирующих РНК). Информативным подходом для выяснения природы ГКМП становится полнотранскриптомный анализ, который позволяет оценить экспрессию всех генов и влияние мутаций в гене на количество кодируемой им РНК, а также выявить механизмы, вовлеченные в регуляцию генной экспрессии. В обзоре представлен анализ опубликованных данных о спектрах генов, дифференциальная экспрессия которых выявляется в миокарде при развитии ГКМП у человека и модельных животных. Отдельное внимание уделено генам некодирующих белок регуляторных РНК: микроРНК и длинных некодирующих РНК, ‒ которые могут быть вовлечены в патогенез заболевания. Проанализированы работы, посвященные изучению уровней микроРНК в крови больных ГКМП для поиска доступных диагностических и прогностических биомаркеров заболевания. Совокупность рассматриваемых данных, несмотря на их относительную немногочисленность, свидетельствует об эффективности транскриптомных исследований при изучении молекулярных механизмов патогенеза ГКМП.
ВВЕДЕНИЕ
Гипертрофическая кардиомиопатия (ГКМП) – самая распространенная в мире генетически детерминированная патология сердца с частотой встречаемости 0.2‒0.5% [1‒3]. Это заболевание миокарда связано с повышенным риском внезапной сердечной смерти (ВСС) и прогрессирующей сердечной недостаточностью, в том числе в молодом возрасте. В России таких больных более 500 000 человек. Спустя 60 лет после начала изучения ГКМП причины и механизмы его развития во многих случаях остаются невыясненными, что затрудняет разработку адекватных методов лечения.
ГКМП в большинстве случаев возникает у людей в возрасте 30‒40 лет, без особых различий по полу или этнической принадлежности [4]. У больных изменяется структура клеток сердечной мышцы – кардиомиоцитов, ‒ вследствие чего происходит ассиметричное утолщение (гипертрофия) стенок левого желудочка сердца (ЛЖ) с одновременным уменьшением его внутреннего объема и в результате нарушается сократительная функция сердца. Болезнь характеризуется плохо предсказуемым клиническим течением. Большая часть больных проживает нормальную или почти нормальную жизнь без инвалидизации и хирургических вмешательств, тогда как у некоторых, по непонятным пока причинам, развиваются фатальные осложнения: ВСС, прогрессирующая сердечная недостаточность и эмболические инсульты, связанные с мерцательной аритмией. Чем раньше происходит дебют заболевания, тем выше вероятность ВСС. Так, ГКМП стала ведущей причиной смерти спортсменов в возрасте до 35 лет [5]. У больных старшего возраста превалируют инсульты и регистрируется повышенная общая смертность [6].
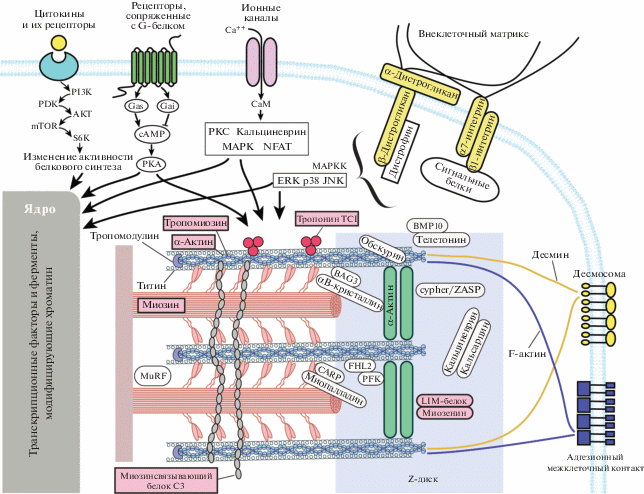
В формировании патологического фенотипа кардиомиоцитов при ГКМП участвуют белки сократительного аппарата, формирующие саркомер сердечной мышцы – базовую сократительную единицу, входящую в состав сократительных волокон, ‒ а также саркомерсвязывающие белки, участвующие в стабилизации трехмерной структуры саркомера, и некоторые несаркомерные белки (рис. 1 ). К числу последних относят белки, участвующие в модуляции сократительной активности миокарда в ответ на внешние стимулы. Быстрое изменение сократимости происходит через активацию Ca2+-зависимого сигналинга, а каскады сигналов, реализующиеся через посттрансляционные модификации белков, в основном заканчиваются в ядре, где вовлечены в модуляцию экспрессии генов [7].
ВЫЯВЛЕНИЕ ПАТОГЕННЫХ ВАРИАНТОВ ГЕНОМА ПРИ ГКМП
Представление о ГКМП как о наследственном заболевании сложилось с 1989 года, когда с помощью анализа сцепления в большой семье обнаружили первый генетический локус (14q1), ассоциированный с развитием этой патологии [8]. В этом локусе у больных ГКМП в гене MYH7, кодирующем тяжелую цепь миозина, идентифицировали миссенс-мутацию, приводящую к замене p.Arg403Glu. Это позволило объяснить менделевский (моногенный) характер наследования ГКМП в изучаемой семье [9]. Одновременно стала ясна генетическая гетерогенность заболевания: при изучении четырех семей косегрегацию вариантов в локусе MYH7 с ГКМП наблюдали только в двух, что указывало на существование альтернативных дефектов, приводящих к ГКМП [10]. После этих основополагающих открытий репертуар генетических локусов, ассоциированных с ГКМП, постепенно расширялся и к концу следующего десятилетия включал в себя еще семь генов, кодирующих саркомерные сократительные белки кардиомиоцитов (MYL2, MYL3, ACTC1) и саркомерсвязывающие белки (MYBPC3, TNNT2, TNNI3, TPM1). Появление высокопроизводительного секвенирования (Next Generation Sequencing, NGS) произвело революцию в возможностях исследования генетической архитектуры менделевских заболеваний и позволило обнаружить при ГКМП мутации в ряде других генов, кодирующих саркомерсвязывающие белки (TNNC1, ACTN2, CSRP3, MYOZ2). Около 80% мутаций, найденных в перечисленных выше генах, оказались сосредоточены в MYH7 и MYBPC3, продукты которых входят в состав толстых миозиновых филаментов, тогда как частота встречаемости мутаций в каждом из 11 остальных генов составляет от 1 до 10% (табл. 1) [11]. Примерно у 5% больных ГКМП находят по несколько мутаций в генах саркомерных белков, что приводит к ранней манифестации заболевания и тяжелому течению [12]. В совокупности у больных ГКМП уже идентифицировано более 450 мутаций в саркомерных генах [13].
Таблица 1.
Частота встречаемости при ГКМП мутаций в отдельных генах, кодирующих белки сократительного аппарата кардиомиоцитов или саркомерсвязывающие белки, от общего числа обнаруженных в этих генах мутаций
| № | Ген | Кодируемый белок | Частота встречае-мости мутаций | ||
|---|---|---|---|---|---|
| Название | Хромосомная локализация | Функция | Клеточная локализация | ||
| 1 | MYBPC3 | 11p11.2 | Миозинсвязывающий белок С3 | Саркомер, толстый филамент | ~40% |
| 2 | MYH7 | 14q11.2 | Тяжелая β-цепь миозина 7 | Саркомер, толстый филамент | ~40% |
| 3 | TNNT2 | 1q32.1 | Сердечный тропонин T2 | Саркомер, тонкий филамент | ~10% |
| 4 | TNNI3 | 19q13.42 | Сердечный тропонин I3 | Саркомер, тонкий филамент | <5% |
| 5 | MYH6 | 14q11.2 | Тяжелая β-цепь миозина 6 | Саркомер, толстый филамент | <4% |
| 6 | MYL2 | 12q24.11 | Легкая цепь миозина 2 | Саркомер, толстый филамент | <1% |
| 7 | MYL3 | 3p21.31 | Легкая цепь миозина 3 | Саркомер, толстый филамент | <1% |
| 8 | ACTC1 | 15q14 | Сердечный α-актин 1 | Саркомер, тонкий филамент | <1% |
| 9 | TNNC1 | 3p21.1 | Сердечный тропонин С1 | Саркомер, тонкий филамент | <1% |
| 10 | TPM1 | 15q22.2 | α-Тропомиозин | Саркомер, тонкий филамент | <1% |
| 11 | ACTN2 | 1q43 | α-Актинин 2 | Саркомер, Z-диск | <1% |
| 12 | CSRP3 | 11p15.1 | Сердечный LIM-белок | Саркомер, Z-диск | <1% |
| 13 | MYOZ2 | 4q26 | Миозенин 2 | Саркомер, Z-диск | <1% |
Помимо этих генов, мутации также найдены в генах несаркомерных белков, продукты которых представлены на рис. 1 . Эти мутации изменяют не только структуру и функцию белков саркомера, но и биофизические свойства кардиомиоцитов [14‒19], влияют на удержание кальция в кардиомиоцитах [20] и энергетический баланс клетки [21‒24].
Стремительное накопление огромного массива данных в эру NGS привело к переоценке степени патогенности известных генетических вариантов в сторону ее смягчения [25]. До сих пор остается неясным, что предопределяет высокую клиническую гетерогенность ГКМП даже среди носителей одной мутации и членов одной семьи, включая монозиготных близнецов [26‒28]. Даже при семейных формах ГКМП таргетное секвенирование саркомерных генов позволяет установить молекулярный диагноз примерно у 70% больных [29]. Что касается больных, не несущих мутаций в этих генах, только у пятой части из них (около 5% от всех случаев семейной ГКМП) полногеномный поиск ассоциаций (genome-wide association study, GWAS) выявляет потенциально патогенные варианты в других областях генома [30]. Как следствие этих наблюдений, на сегодняшний день складывается представление, что развитие различных форм ГКМП обусловлено не только мутациями в генах саркомерных и несаркомерных белков, но также эпигенетическими факторами и факторами окружающей среды [31].
В целом, представление о моногенной природе ГКМП сегодня выглядит упрощенным. Совокупность накопленных данных свидетельствует о том, что в развитие заболевания, кроме классических саркомерных генов, вносят вклад и другие гены, которые принято называть генами-модификаторами. У одного больного может быть выявлено более одного гена-модификатора [32]. В отношении мутаций в двух главных саркомерных генах наблюдали модифицирующие эффекты полиморфных вариантов других генов, кодирующих белки саркомера и его окружения [33‒35]. В качестве модификаторов выступают также гены, ассоциированные с развитием вторичной гипертрофии миокарда, которые в основном кодируют белки ренин-ангиотензин-альдостероновой системы (РААС) [36‒38]. Теория олигогенной природы ГКМП, по крайней мере у части больных, для которых не удается найти патогенные мутации в генах саркомерных белков, получает все большее распространение [32, 39]. Тот факт, что при ГКМП одни и те же патогенные мутации участвуют в формировании нескольких фенотипов, хорошо укладывается в представление о совместном действии нескольких генов [40]. Таким образом, итоговый фенотип ГКМП может складываться из сложного комплекса межгенных взаимодействий и механизмов регуляции экспрессии генов на различных уровнях.
ПОЛНОТРАНСКРИПТОМНЫЙ АНАЛИЗ ЭКСПРЕССИИ ГЕНОВ ПРИ ГКМП
Одним из важнейших и неожиданных результатов исследования человеческого генома в XXI веке стал вывод о том, что доля геномной ДНК, кодирующей белки, составляет менее 2% всей ее последовательности [41]; при этом до 90% генома выполняет определенную биологическую функцию, связанную в основном с регуляцией экспрессии генов [41, 42]. Среди выявленных функционально значимых участков генома наиболее представлены гены некодирующих (точнее, нетранслируемых) регуляторных РНК (ncРНК), которые отличаются большим разнообразием структуры и функций [43].
Информативным подходом для выяснения природы таких наследственно обусловленных заболеваний, как ГКМП, может быть полнотранскриптомный анализ экспрессии генов. Действительно, такой свободный от гипотез (hypothesis-free) подход позволяет определить транскрипционную активность всех белоккодирующих и белокнекодирующих генов при различных фенотипах и оценить влияние мутации в гене на количество кодируемого им продукта при формировании кардиомиопатии, а также выявить механизмы, вовлеченные в регуляцию экспрессии генов. Можно надеяться, что на основании оценки активности биологических процессов в кардиомиоцитах будут выявлены неизвестные звенья патогенеза ГКМП и разработаны новые подходы к лечению этого заболевания.
Изменение экспрессии белоккодирующих генов
Полнотранскриптомные исследования ГКМП пока немногочисленны; в большинстве из них проводят поиск в миокарде дифференциально экспрессирующихся белоккодирующих генов на клеточных и животных моделях. Всего в нескольких работах проведен полнотранскриптомный анализ мРНК из биологического материала пациентов, направленный на поиск генов-модификаторов, вовлеченных в формирование фенотипа и влияющих на прогноз течения ГКМП у человека; при этом клиническая гетерогенность заболевания усложняет поиск потенциальных транскриптомных маркеров развития и течения ГКМП.
Биологический материал человека. В исследовании Bos и соавт. [44] проведен анализ мРНК в образцах гипертрофированных участков миокарда больных ГКМП и в образцах нормального миокарда с помощью микрочипа HumanHT-12 v3 Expression BeadChip (“Illumina, Inc.”, США). У всех больных с целью поиска патогенетических вариантов секвенировали гены, кодирующие белки сократительного аппарата кардиомиоцитов и саркомерсвязывающие белки (ACTC1, MYBPC3, MYH6, MYH7, MYL2, MYL3, TNNC1, TNNI3, TNNT2 и TPM1). Наблюдали дифференциальную экспрессию 22% анализируемых генов (8443 из 37 846) при сравнении больных ГКМП с контрольной группой (FDR p-value <0.05, разница в экспрессии не менее чем в 1.5 раза). При сравнении экспрессии генов у больных ГКМП с выявленными мутациями в генах MYBPC3 и MYH7 между собой, а также при сравнении каждой из этих подгрупп с больными ГКМП, негативными по носительству мутаций в обоих генах, показаны различия в экспрессии примерно 5% анализируемых генов [44]. В работе, проведенной Malgija и др. [45], у больных ГКМП выявлены изменения в экспрессии 758 генов (310 генов с повышенной экспрессий и 448 с пониженной экспрессией) в сравнении с контрольной группой. Проведенный анализ белок-белковых взаимодействий продуктов этих генов установил вовлеченность Smad-зависимого TGF-β-сигнального пути в развитие ГКМП. Ранее Ren и соавт. [46] выявили у больных ГКМП 62 гена с пониженной и 195 с повышенной экспрессией, из которых 91 ген характеризуется дифференциальной экспрессией, специфической для миокарда. Наиболее выраженные при ГКМП изменения в экспрессии (более чем в 4 раза по сравнению с контрольной группой) обнаружены для генов CHRDL2, FGF12, DHRS7C, SEZ6L, ESM1, COL10A1, SFRP4 и CA3. Примечательно, что снижение экспрессии гена FGF12, кодирующего фактор роста фибробластов 12, наблюдали позднее Bos и соавт [44]. При сравнении транскрипционных профилей миокарда трех больных ГКМП и трех условно здоровых добровольцев обнаружена группа генов, исходя из данных о дифференциальной экспрессии которых, можно с точностью до 90% классифицировать одно из распространенных осложнений ГКМП ‒ хроническую сердечную недостаточность [47].
Рис. 1.
Белки, участвующие в сократительной активности, росте и формировании фенотипа кардиомиоцитов. На рисунке изображена часть саркомера от M-линии до Z-диска – точек прикрепления миозиновых и актиновых филаментов соответственно. Представлены также белки основных сигнальных каскадов, участвующих в модуляции сократительной активности миокарда в ответ на внешние стимулы. Саркомерные белки, гены которых наиболее часто содержат мутации, ведущие к развитию ГКМП, представлены в рамках на розовом фоне. AKT (AKT Serine/Threonine Kinase) – AKT серин/треониновая протеинкиназа; BAG3 (Bcl-2-associated athanogene-3) – Bcl-2-ассоциированный атаноген-3; BMP10 (bone morphogenetic protein-10) – костный морфогенетический белок-10; CaM (calmodulin) – кальмодулин; ERK (extracellular signal-regulated kinase) – киназа, регулируемая внеклеточными сигналами; FHL2 (four and a half LIM-domain protein-2) – белок с четырьмя с половиной LIM-доменами 2; JNK (Jun N-terminal kinase) – Jun-N-терминальная киназа; MAPK (mitogen-associated protein kinase) – митогенассоциированная протеинкиназа; mTOR (mechanistic target of rapamycin) – мишень рапамицина млекопитающих; MuRF (muscle specific ring finger protein) – Е3-убиквитинлигаза TRIM63; NFAT (nuclear factor associated with T cell activation) – ядерный фактор, ассоциированный с активацией Т-клеток; PDK (phosphoinoside-dependent kinase) – фосфоинозидзависимая киназа; PFK (phosphofructokinase) – фосфофруктокиназа; PI3K (phosphoinositide 3-kinase) – фосфоинозитид-3-киназа; PKA (protein kinase A) – протеинкиназа А; PKC (proteinkinase C) – протеинкиназа С; S6K (ribosomal S6 protein kinase) – протеинкиназа рибосомного белка S6.
В результате секвенирования РНК изогенных индуцированных плюрипотентных стволовых клеток (ИПСК), полученных от больных ГКМП с мутациями в гене MYBPC3, Seeger и др. [48] выявили 125 генов с повышенной и 173 гена с пониженной экспрессией по сравнению с контрольными ИПСК. Среди наиболее дисрегулированных генов, которые можно рассматривать в качестве транскриптомных сигнатур ГКМП, идентифицированы гены, вовлеченные в удержание кальция в кардиомиоцитах (ATP2A2, ATP2B2 и CASQ1), ассоциированные с гипертрофией миокарда (GP130, JAK2, RRAS, MEK1, TWEAKR и NPPB), а также гены, экспрессия которых меняется в ответ на стресс (HSPB1, HSPB6, HSPB7, IGF1 и IGF2) и продукты которых участвуют в структурной организации саркомера и механосенсорных комплексов (CSRP3 и TCAP). Анализ обогащения по функциональной принадлежности (enrichment analysis) показал перепредставленность выявленных дифференциально экспрессирующихся генов в сигнальных путях, вовлеченных в белковый транспорт, трансляцию, метаболизм и в контроль качества мРНК посредством ее нонсенс-опосредованного распада. Кроме того, для фенотипа ГКМП с мутациями в гене MYBPC3 Seeger и др. [48] выявили характерную активацию нонсенс-опосредованного распада мРНК, а также нарушение экспрессии генов, продукты которых участвуют в сократительной активности миокарда. Ранее Han и соавт. [49] провели анализ транскриптома ИПСК, полученных от пациентов с ГКМП, и обнаружили ряд сигнальных путей, вовлеченных в развитие заболевания, в том числе сигнальный путь Notch.
Животные модели. Транскрипционные профили достаточно хорошо описаны для моделей ГКМП, полученных с использованием различных видов лабораторных животных [7]. Недавно в транскриптомном исследовании, проведенном на мышиной модели ГКМП в сравнении с мышами дикого типа, обнаружены изменения в некоторых сигнальных путях [50]. Так, при ГКМП повышена экспрессия генов, продукты которых участвуют 1) в проведении сигналов от эукариотического фактора инициации 2 и интегрина, 2) в нуклеации актина с помощью актинсвязанного белкового комплекса семейства синдрома Вискотта–Олдрича, 3) в Rho-зависимой регуляции подвижности актиновых филаментов и 4) в активации рецептора витамина D/рецептора ретиноида X. Напротив, экспрессия генов, вовлеченных в процессы деградации аминокислот валина и метионина, цикл трикарбоновых кислот, в инозитолфосфатную систему передачи сигналов, заметно подавлена у мышей с ГКМП. Последнее может быть результатом изменения энергетического баланса гипертрофированных кардиомиоцитов и способствовать сохранению энергии с учетом повышенной сократительной активности сердца. При отдельном сравнении самцов и самок мышей с ГКМП с мышами дикого типа наблюдали сходные паттерны большинства дифференциально экспрессирующихся генов, продукты которых вовлечены в канонические сигнальные пути. Исключение составили 7 генов: Abca1, Tspyl4, Tet1, Sacs, Ier5, Eif5b и Jun, ‒ изменения в экспрессии которых оказались разнонаправленными у самцов и самок. В другом исследовании проведен сравнительный анализ транскриптомов мышей с мутациями R92W-TnT и R403Q-αMyHC в генах, кодирующих сердечный тропонин T2 и тяжелую β-цепь миозина-6 соответственно. Оказалось, что транскрипционные профили кардиомиоцитов молодых мышей с мутациями в этих генах отличались от таковых в контрольной группе. При анализе сигнальных путей выявлена активация профибротического сигнального пути фактора роста тромбоцитов-β у мышей с мутацией в гене Tnnt2, которая определяет тяжелое течение ГКМП у молодых особей. Можно предположить, что блокаторы рецепторов ангиотензина, не оказывающие значимого позитивного эффекта в общем пуле больных с ГКМП [51, 52], могут быть эффективными для лечения пациентов с мутацией в гене TNNT2 на ранней стадии заболевания [53]. В результате транскрипционного профилирования у мышей с ГКМП, обусловленной мутациями в генах Myh6 или Tpm1, у трансгенных мышей, экспрессирующих фосфоламбан человека, у мышей, нокаутных по гену Fxn1, кодирующему фратаксин, и у мышей с поперечным сужением аорты Sasagawa и др. [54] обнаружили изменения в экспрессии генов Gstk1, Myh7, Ctgf, Postn и Rtn4, характерных для всех 5 моделей, при сравнении с соответствующими контролями. Авторы считают, что подавление экспрессии гена GSTK1, кодирующего глутатион-S-трансферазу-κ1, может быть общим механизмом, лежащим в основе ГКМП различной этиологии, возможно, за счет усиления окислительного стресса и экспрессии генов саркомера.
Таким образом, в транскриптомных исследованиях, проведенных к настоящему моменту на миокарде и ИПСК больных ГКМП, а также на модельных животных, выявлены отдельные гены и сигнальные пути, включающие их белковые продукты, которые вовлечены в развитие патологической гипертрофии миокарда. Однако полученные в разных работах результаты трудно сопоставить, что в первую очередь связано с различиями в их дизайне и небольшим объемом накопленных данных. В связи с этим рано ожидать появления новых способов лечения этого заболевания, основанных на знании транскриптома миокарда.
Изменение экспрессии генов некодирующих РНК
В транскриптоме, наряду с мРНК, транскрибированной с белоккодирующих генов, присутствуют также регуляторные ncРНК: микроРНК и длинные ncРНК (lncРНК), ‒ кодируемые соответствующими генами.
Экспрессия генов микроРНК. МикроРНК ‒ малые ncРНК, формирующие координированную регуляторную систему, которая на посттранскрипционном уровне контролирует экспрессию множества генов, участвующих в фундаментальных биологических процессах, таких как дифференцировка клеток, пролиферация, апоптоз, реакция на стресс и др. [55]. Сиквенсспецифическая регуляция мРНК-мишеней с помощью микроРНК ‒ одно из важнейших звеньев при реализации наследственной информации, которое может вносить существенный вклад в пенетрантность мутаций, связанных с развитием ГКМП, и определять общую гетерогенность фенотипов заболевания.
Профили экспрессии микроРНК при ГКМП на полнотранскриптомном уровне изучали как у человека, так и на животных моделях в гипертрофированной ткани сердца при сравнении со здоровой тканью или с миокардом при дилатационной кардиомиопатии (табл. 2 ). Профилирование микроРНК в ткани сердца пациентов с ГКМП, несущих мутацию MYBPC3, выявило существенные отличия уровней 13 микроРНК (как снижение, так и повышение) по сравнению с контрольными образцами [56]. Song и соавт. [57] обнаружили дифференциальную экспрессию 13 микроРНК в ткани сердца больных ГКМП по сравнению с контрольной группой, причем наибольшее снижение выявили для miR-451. В этой работе показано также, что микроРНК регулирует гипертрофию миокарда и аутофагию, влияя на трансляцию белка гамартина (TSC1). На различных этапах развития ГКМП у мышей обнаружено подавление экспрессии mir-1 и усиление экспрессии mir-21 [58]. Аналогичные результаты получены на тканях сердца человека [57, 59]. Участие miR-133 в развитии ГКМП выявлено как у мышей, так и у человека [58, 59]. Carè и др. [60] не только наблюдали снижение экспрессии mir-133 и mir-1 в тканях сердца модельных животных, но и показали, что их повышенная экспрессия подавляет развитие гипертрофии миокарда, тогда как снижение уровня mir-133, напротив, стимулирует этот процесс.
Таблица 2.
МикроРНК, уровни которых, по данным транскриптомного профилирования, изменяются в ткани сердца при развитии ГКМП
| МикроРНКa | Направление изменения экспрессии при ГКМП | Объект исследования | Биологи-ческий материал | Ген, в котором найдена мутация | Метод скрининго-вого анализа экспрессии | Дифферен-циально экспресси-рованные микроРНК, прошедшие валидацию | Ссыл-ка |
|---|---|---|---|---|---|---|---|
| miR-34b* | Повышена | Человек | Ткань сердца | MYBPC3 | TaqMan MiRNA array (“Thermo Fisher Scientific”, США) | miR-34b* miR-184 miR-204 miR-222* miR-497 |
[56] |
| miR-96 | |||||||
| miR-181-a2* | |||||||
| miR-184 | |||||||
| miR-204 | |||||||
| miR-222* | |||||||
| miR-371-3p | |||||||
| miR-383 | |||||||
| miR-497 | |||||||
| miR-708 | |||||||
| miR-10a* miR-10b miR-10b* |
Понижена | miR-10b miR-10b* |
|||||
| miR-21 miR-130b miR-132 |
Повышена | Человек | Миокард левого желудочка | MYH7 или не найдена | SurePrint Human miRNA Microarray (“Agilent”, США) | [57] | |
| miR-139-3p | Понижена | miR-451 | |||||
| miR-139-5p | |||||||
| miR-144 | |||||||
| miR-144* | |||||||
| miR-150 | |||||||
| miR-363 | |||||||
| miR-451 | |||||||
| miR-486-3p | |||||||
| miR-1246 | |||||||
| miR-3141 | |||||||
| miR-92a miR-125a-3p miR-199a-3p miR-208a miR-223 miR-451 miR-483-5p miR-590–5p |
Повышена | Человек | Миокард левого желудочка | MYH7 | TaqMan MiRNA array (“Thermo Fisher Scientific”) | miR-92a miR-590–5p |
[59] |
| miR-1 miR-30b miR-93 miR-133a miR-133b miR-191 miR-208b miR-218 miR-374 miR-454 miR-495 |
Понижена | miR-1 miR-30b miR-133b miR-191 miR-208b miR-218 miR-374 miR-454 miR-495 |
|||||
| miR-3671 | Повышена | Человек | Миокард желудочка | MYBPC3 или MYH7 | HiSeq X Ten (“Illumina”, США) | Не проводили | [61] |
| miR-30d miR-154 miR-487a miR-654 miR-3193 |
Понижена | Не проводили | |||||
| mir-10b mir-21 mir-31 mir-34b-3p mir-132 mir-142-3p mir-146b mir-199a-3p mir-210 mir-214 mir-221 mir-222 mir-674 |
Повышена | Мышь | Миокард желудочка | Tnni3 и Myh6 | TaqMan Low Density Array (“Thermo Fisher Scientific”) | mir-1 mir-21 mir-31 mir-34b-3p mir-132 mir-142 mir-214 mir-222 mir-331 |
[58] |
| mir-1 mir-30b mir-30c mir-30e mir-133a mir-133b mir-150 mir-200c mir-450a-5p mir-486-3p mir-499 mir-542-3p |
Понижена | mir-30-5p mir-30b-5p mir-30c mir-30e mir-133a mir-133b mir-150 mir-486-5p mir-499-5p |
Важно отметить, что повышенные уровни miR-133 и miR-21 наблюдали не только в миокарде, но также и в плазме пациентов с ГКМП [62]. Это означает, что уровни микроРНК в плазме в той или иной степени могут отражать патологические процессы, происходящие в миокарде. Следовательно, их анализ в плазме, как легкодоступном биологическом материале, может быть использован для определения фенотипа заболевания. Помимо miR-133 и miR-21 в плазме пациентов с ГКМП повышено содержание еще некоторых микроРНК, среди которых miR-199a-5p, miR-27a и miR-29a. Их уровни коррелировали со степенью гипертрофии, а уровень miR-29a еще и со степенью фиброза, что позволило Roncarati и др. [62] рассматривать последнюю в качестве потенциального биомаркера для оценки ремоделирования миокарда при ГКМП. Повышение уровня miR-29a наблюдали также в сыворотке больных ГКМП при сравнении с контрольной группой [63]. Fang и др. [64] сравнили экспрессию 84 микроРНК у больных ГКМП и здоровых добровольцев и обнаружили различия в уровнях 16 циркулирующих микроРНК, в том числе и описанных выше miR-21, miR-29a, miR-133 и miR-200a, причем уровень большинства из них коррелировал с выраженностью фиброза.
Таким образом, различия между уровнями микроРНК, выявленные в зависимости от стадии заболевания, клинических особенностей течения ГКМП и обнаруженных в геноме пациента мутаций, позволяют рассматривать микроРНК в качестве потенциальных биомаркеров ГКМП. Следует отметить, что, несмотря на противоречивость результатов, которые получены в исследованиях, проведенных с использованием разных технологий, на модельных животных и с участием больных, для ряда микроРНК наблюдали сходную картину. Как видно из данных, представленных в табл. 2 , для 9 микроРНК однонаправленное изменение экспрессии (ее повышение или понижение) при ГКМП наблюдали в двух разных исследованиях.
Экспрессия генов lncРНК. Другими посттранскрипционными регуляторами экспрессии генов служат lncРНК, которые представляют собой большую гетерогенную группу РНК, нуклеотидная последовательность которых, как правило, не содержит открытой рамки считывания. Хотя функции большинства lncРНК неизвестны, для уже охарактеризованных молекул этого класса показана их роль в регуляции разнообразных биологических процессов, таких как пролиферация, апоптоз, дифференцировка и метаболизм, посредством влияния на экспрессию генов [65].
Дифференциальная экспрессия ряда генов lncРНК при ГКМП выявлена в нескольких исследованиях [66‒69]. При сравнении больных ГКМП и здоровых добровольцев наблюдали различие в уровнях митохондриальных lncРНК uc004cov.4 и uc022bqu.1 в цельной крови и сыворотке [66]. Последующий анализ их диагностического потенциала показал, что циркулирующие lncРНК обеспечивают среднее качество классификации при попытке дискриминировать больных от здоровых (AUC > 0.6). Дисрегулированные мРНК, коэкспрессирующиеся с lncРНК, вовлечены преимущественно в регуляцию процессов окислительного фосфорилирования и передачи сигналов РААС [67]. Кроме того, Sonnenschein и соавт. [70] обнаружили снижение уровней еще одного типа РААС РНК ‒ кольцевых РНК circDNAJC6, circTMEM56 и circMBOAT2 ‒ в сыворотке больных ГКМП, которое обеспечивает хорошее качество дискриминации больных от здоровых (AUC от 0.722 до 0.949). Уровни circTMEM56 и circDNAJC6 коррелировали с тяжестью заболевания.
Системный подход при анализе полнотранскриптомных профилей
Системный подход, основанный на одновременном изучении полнотранскриптомных профилей экспрессии белоккодирующих генов и генов их возможных регуляторов − ncРНК − у одних и тех же индивидов, обладает очевидным преимуществом за счет существенного повышения информативности анализа. Примерами таких исследований могут служить работы нескольких групп исследователей [61, 71‒73], в которых определяли характеристики сетей, образуемых мРНК, lncРНК или микроРНК. Знание структуры таких сетей позволяет предсказывать не только роль отдельных молекул, но и целых сигнальных путей в патогенезе заболевания. Идентифицировано новое патогенетическое звено ГКМП ‒ петля прямой связи между miR-17-5p, FASN и STAT3, ‒ которая занимает центральное место в патогенезе заболевания [72]. Авторами этой работы также предложена диагностическая панель, включающая три транскрипционных фактора и четыре микроРНК, использование которой обеспечивает очень хорошее качество классификации больных ГКМП и здоровых индивидов (AUC > 0.8). С помощью одновременного анализа экспрессии белоккодирующих генов и генов микроРНК установлено участие miR-20 в развитии гипертрофии миокарда путем снижения уровня мРНК гена MFN2 [73]. В еще одном системном анализе обнаружили вовлеченность ряда генных подсетей: подсети, образованной генами митохондриальной ДНК, десмоплакин-образующей подсети, а также MYH7- и MYBPC3-образующей подсети – в прогрессирование ГКМП [[61]].
Одновременный анализ уровней мРНК и ncРНК успешно применен для исследования функции митохондрий при ГКМП и поиска сигнальных путей, которые могли бы объяснить различия в пенетрантности тех или иных мутаций в саркомерных генах [53]. В этой работе показано, что мутации в генах саркомерных белков приводят у лабораторных мышей к окислительному стрессу и нарушениям в работе митохондрий, что в свою очередь способствует активации Tgfb-зависимого сигналинга, в том числе при участии mir-29, и стимулирует фиброз и осложненное течение заболевания. В хорошем соответствии с этими результатами установлено активное участие TGF-β в формировании профибротического фенотипа при участии двух сигнальных путей: SMAD-зависимого и через индукцию экспрессии фактора роста соединительной ткани, CTGF [45]. Полученные с использованием такого комплексного подхода данные представляют большую ценность для создания диагностикумов и лекарственных препаратов направленного действия.
ЗАКЛЮЧЕНИЕ
Совокупность представленных в обзоре данных свидетельствует о том, что в основе патогенетических механизмов, вовлеченных в формирование ГКМП, лежит разбалансировка различных сигнальных путей в кардиомиоцитах. Она может быть обусловлена не только мутациями в генах саркомерных белков, но и изменениями в экспрессии отдельных генов, определяющих олигогенный характер наследования ГКМП. Разнообразие вовлеченных в формирование ГКМП сетей ген-генных взаимодействий, построенных на основании анализа экспрессии белоккодирующих генов и генов ncРНК, свидетельствует о возможности развития ГКМП по разным патогенетическим механизмам, что, скорее всего, и определяет высокую клиническую гетерогенность заболевания, морфологию гипертрофии и выраженность фиброза. В то же время вполне вероятно, что увеличение объема экспериментальных данных и их последующее сопоставление позволит выявить универсальные регуляторные звенья, координирующие процесс формирования ГКМП.
В отличие от других сердечно-сосудистых заболеваний, исследования ГКМП в области трансляционной медицины только начинаются. Поиск биомаркеров этого достаточно распространенного гетерогенного заболевания будет продолжен, а их идентификация позволит понять механизмы патогенеза ГКМП и применить эти знания для диагностики и прогнозирования его течения, а также для создания новых лекарственных препаратов.
Исследование выполнено при финансовой поддержке Российского научного фонда (грант № 20-15-00353).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Elliott P.M., Anastasakis A., Borger M.A., Borggrefe M., Cecchi F., Charron P., Hagege A.A., Lafont A., Limongelli G., Mahrholdt H., McKenna W.J, Mogensen J., Nihoyannopoulos P., Nistri S., Pieper P.G., Pieske B., Rapezzi C., Rutten F.H., Tillmanns C., Watkins H. (2014) 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 35, 2733–2779.
Maron B.J., Gardin J.M., Flack J.M., Gidding S.S., Kurosaki T.T., Bild D.E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4 111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 92, 785–789.
Semsarian C., Ingles J., Maron M.S., Maron B.J. (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254.
Charron P., Elliott P.M., Gimeno J.R., Caforio A.L.P., Kaski J.P., Tavazzi L., Tendera M., Maupain C., Laroche C., Rubis P., Jurcut R., Calo L., Helio T.M., Sinagra G., Zdravkovic M., Kavoliuniene A., Felix S.B., Grzybowski J., Losi M.A., Asselbergs F.W., Garcia-Pinilla J.M., Salazar-Mendiguchia J., Mizia-Stec K., Maggioni A.P., Investigators E.C.R. (2018) The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur. Heart J. 39, 1784–1793.
Maron B.J., Doerer J.J., Haas T.S., Tierney D.M., Mueller F.O. (2009) Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980‒2006. Circulation. 119, 1085–1092.
Lorenzini M., Anastasiou Z., O’Mahony C., Guttman O.P., Gimeno J.R., Monserrat L., Anastasakis A., Rapezzi C., Biagini E., Garcia-Pavia P., Limongelli G., Pavlou M., Elliott P.M., Hypertrophic Cardiomyopathy Outcomes investigators (2019) Mortality among referral patients with hypertrophic cardiomyopathy vs the general European population. JAMA Cardiol. e194534.
Raghow R. (2016) An “Omics” perspective on cardiomyopathies and heart failure. Trends Mol. Med. 22, 813‒827.
Jarcho J.A., McKenna W., Pare J.A., Solomon S.D., Holcombe R.F., Dickie S., Levi T., Donis-Keller H., Seidman J.G., Seidman C.E. (1989) Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 321, 1372–1378.
Geisterfer-Lowrance A.A., Kass S., Tanigawa G., Vosberg H.P., McKenna W., Seidman C.E., Seidman J.G. (1990) A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 62, 999–1006.
Solomon S.D., Jarcho J.A., McKenna W., Geisterfer-Lowrance A., Germain R., Salerni R., Seidman J.G., Seidman C.E. (1990) Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease. J. Clin. Invest. 86, 993–999.
Seidman C.E., Seidman J.G. (2011) Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res. 108, 743–750.
Lopes L.R., Rahman M.S., Elliott P.M. (2013) A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 99, 1800–1811.
Wolf C.M. (2019) Hypertrophic cardiomyopathy: genetics and clinical perspectives. Cardiovasc. Diagn. Ther. 9, S388–S415.
Palmer B.M., Wang Y., Teekakirikul P., Hinson J.T., Fatkin D., Strouse S., Vanburen P., Seidman C.E., Seidman J.G., Maughan D.W. (2008) Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 294, H1939–H1947.
Green E.M., Wakimoto H., Anderson R.L., Evan-chik M.J., Gorham J.M., Harrison B.C., Henze M., Kawas R., Oslob J.D., Rodriguez H.M., Song Y., Wan W., Leinwand L.A., Spudich J.A., McDowell R.S., Seidman J.G., Seidman C.E. (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 351, 617–621.
Tyska M.J., Hayes E., Giewat M., Seidman C.E., Seidman J.G., Warshaw D.M. (2000) Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 86, 737–744.
Tardiff J.C. (2005) Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail. Rev. 10, 237–248.
Sequeira V., Wijnker P.J., Nijenkamp L.L., Kuster D.W., Najafi A., Witjas-Paalberends E.R., Regan J.A., Boontje N., Ten Cate F.J., Germans T., Carrier L., Sadayappan S., van Slegtenhorst M.A., Zaremba R., Foster D.B., Murphy A.M., Poggesi C., Dos Remedios C., Stienen G.J., Ho C.Y., Michels M., van der Velden J. (2013) Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 112, 1491–1505.
Linke W.A. (2008) Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 77, 637–648.
Fatkin D., McConnell B.K., Mudd J.O., Semsarian C., Moskowitz I.G., Schoen F.J., Giewat M., Seidman C.E., Seidman J.G. (2000) An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J. Clin. Invest. 106, 1351–1359.
Crilley J.G., Boehm E.A., Blair E., Rajagopalan B., Blamire A.M., Styles P., McKenna W.J., Ostman-Smith I., Clarke K., Watkins H. (2003) Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 41, 1776–1782.
Ashrafian H., McKenna W.J., Watkins H. (2011) Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 109, 86–96.
Ferrantini C., Belus A., Piroddi N., Scellini B., Tesi C., Poggesi C. (2009) Mechanical and energetic consequences of HCM-causing mutations. J. Cardiovasc. Transl. Res. 2, 441–451.
Marston S.B. (2011) How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 4, 245–255.
Walsh R., Thomson K.L., Ware J.S., Funke B.H., Woodley J., McGuire K.J., Mazzarotto F., Blair E., Seller A., Taylor J.C., Minikel E.V., Exome Aggregation C., MacArthur D.G., Farrall M., Cook S.A., Watkins H. (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203.
Van Driest S.L., Ackerman M.J., Ommen S.R., Shakur R., Will M.L., Nishimura R.A., Tajik A.J., Gersh B.J. (2002) Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 106, 3085–3090.
Oliva-Sandoval M.J., Ruiz-Espejo F., Monserrat L., Hermida-Prieto M., Sabater M., Garcia-Molina E., Ortiz M., Rodriguez-Garcia M.I., Nunez L., Gimeno J.R., Castro-Beiras A., Valdes M. (2010) Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart. 96, 1980–1984.
Jansweijer J.A., van Spaendonck-Zwarts K.Y., Tanck M.W.T., van Tintelen J.P., Christiaans I., van der Smagt J., Vermeer A., Bos J.M., Moss A.J., Swan H., Priori S.G., Rydberg A., Tfelt-Hansen J., Ackerman M.J., Olivotto I., Charron P., Gimeno J.R., van den Berg M., Wilde A.A.M., Pinto Y.M. (2019) Heritability in gene-tic heart disease: the role of genetic background. Open Heart. 6, e000929.
Ingles J., Burns C., Bagnall R.D., Lam L., Yeates L., Sarina T., Puranik R., Briffa T., Atherton J.J., Dris-coll T., Semsarian C. (2017) Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ. Cardiovasc. Genet. 10(2), e001620.https://doi.org/10.1161/CIRCGENETICS.116.001620
Bagnall R.D., Ingles J., Dinger M.E., Cowley M.J., Ross S.B., Minoche A.E., Lal S., Turner C., Colley A., Rajagopalan S., Berman Y., Ronan A., Fatkin D., Semsarian C. (2018) Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 72, 419–429.
Maron B.J., Maron M.S., Maron B.A., Loscalzo J. (2019) Moving beyond the sarcomere to explain heterogeneity in hypertrophic cardiomyopathy: JACC review topic of the week. J. Am. Coll. Cardiol. 73, 1978–1986.
Pasipoularides A. (2018) Challenges and controversies in hypertrophic cardiomyopathy: clinical, genomic and basic science perspectives. Rev. Esp. Cardiol. (Engl. Ed.). 71, 132–138.
Burns C., Bagnall R.D., Lam L., Semsarian C., Ingles J. (2017) multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ. Cardiovasc. Genet. 10, S307.
Mouton J.M., van der Merwe L., Goosen A., Revera M., Brink P.A., Moolman-Smook J.C., Kinnear C. (2016) MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients. Hum. Genet. 135, 477–483.
Wooten E.C., Hebl V.B., Wolf M.J., Greytak S.R., Orr N.M., Draper I., Calvino J.E., Kapur N.K., Maron M.S., Kullo I.J., Ommen S.R., Bos J.M., Ackerman M.J., Huggins G.S. (2013) Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 6, 10–18.
Perkins M.J., Van Driest S.L., Ellsworth E.G., Will M.L., Gersh B.J., Ommen S.R., Ackerman M.J. (2005) Gene-specific modifying effects of pro-LVH polymorphisms involving the renin-angiotensin-aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. Eur. Heart J. 26, 2457–2462.
Ortlepp J.R., Vosberg H.P., Reith S., Ohme F., Mahon N.G., Schroder D., Klues H.G., Hanrath P., McKenna W.J. (2002) Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein C gene. Heart. 87, 270–275.
Kolder I.C., Michels M., Christiaans I., Ten Cate F.J., Majoor-Krakauer D., Danser A.H., Lekanne Deprez R.H., Tanck M., Wilde A.A., Bezzina C.R., Dooijes D. (2012) The role of renin-angiotensin-aldosterone system polymorphisms in phenotypic expression of MYBPC3-related hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 20, 1071–1077.
Helms A.S., Day S.M. (2016) Hypertrophic cardiomyopathy: single gene disease or complex trait? Eur. Heart J. 37, 1823–1825.
Cerrone M., Remme C.A., Tadros R., Bezzina C.R., Delmar M. (2019) Beyond the one gene–one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 140, 595–610.
Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature. 489, 57–74.
Costa F.F. (2010) Non-coding RNAs: meet thy masters. Bioessays. 32, 599‒608.
Brosnan C.A., Voinnet O. (2009) The long and the short of noncoding RNAs. Curr. Opin. Cell Biol. 21, 416–425.
Bos J.M., Hebl V.B., Oberg A.L., Sun Z., Herman D.S., Teekakirikul P., Seidman J.G., Seidman C.E., Dos Remedios C.G., Maleszewski J.J., Schaff H.V., Dearani J.A., Noseworthy P.A., Friedman P.A., Ommen S.R., Brozovich F.V., Ackerman M.J. (2020) Marked up-regulation of ACE2 in hearts of patients with obstructive hypertrophic cardiomyopathy: implications for SARS-CoV-2-mediated COVID-19. Mayo Clin. Proc. 95, 1354–1368.
Malgija B., Kumar N.S., Piramanayagam S. (2018) Collective transcriptomic deregulation of hypertrophic and dilated cardiomyopathy – importance of fibrotic mechanism in heart failure. Comput. Biol. Chem. 73, 85–94.
Ren C.W., Liu J.J., Li J.H., Li J.W., Dai J., Lai Y.Q. (2016) RNAseq profiling of mRNA associated with hypertrophic cardiomyopathy. Mol. Med. Rep. 14, 5573–5586.
Liu Y., Morley M., Brandimarto J., Hannenhalli S., Hu Y., Ashley E.A., Tang W.H., Moravec C.S., Margulies K.B., Cappola T.P., Li M., MAGNet consortium (2015) RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 105, 83–89.
Seeger T., Shrestha R., Lam C.K., Chen C., McKeithan W.L., Lau E., Wnorowski A., McMullen G., Greenhaw M., Lee J., Oikonomopoulos A., Lee S., Yang H., Mercola M., Wheeler M., Ashley E.A., Yang F., Karakikes I., Wu J.C. (2019) A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation. 139, 799–811.
Han L., Li Y., Tchao J., Kaplan A.D., Lin B., Li Y., Mich-Basso J., Lis A., Hassan N., London B., Bett G.C., Tobita K., Rasmusson R.L., Yang L. (2014) Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 104, 258‒269.
Dieseldorff Jones K.M., Vied C., Valera I.C., Chase P.B., Parvatiyar M.S., Pinto J.R. (2020) Sexual dimorphism in cardiac transcriptome associated with a troponin C murine model of hypertrophic cardiomyopathy. Physiol. Rep. 8, e14396.
Axelsson A., Iversen K., Vejlstrup N., Ho C., Norsk J., Langhoff L., Ahtarovski K., Corell P., Havndrup O., Jensen M., Bundgaard H. (2015) Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 3, 123–131.
Maron M.S., Chan R.H., Kapur N.K., Jaffe I.Z., McGraw A.P., Kerur R., Maron B.J., Udelson J.E. (2018) Effect of spironolactone on myocardial fibrosis and other clinical variables in patients with hypertrophic cardiomyopathy. Am. J. Med. 131, 837–841.
Vakrou S., Fukunaga R., Foster D.B., Sorensen L., Liu Y., Guan Y., Woldemichael K., Pineda-Reyes R., Liu T., Tardiff J.C., Leinwand L.A., Tocchetti C.G., Abraham T.P., O’Rourke B., Aon M.A., Abraham M.R. (2018) Allele-specific differences in transcriptome, miRNome, and mitochondrial function in two hypertrophic cardiomyopathy mouse models. JCI Insight. 3, e94493
Sasagawa S., Nishimura Y., Okabe S., Murakami S., Ashikawa Y., Yuge M., Kawaguchi K., Kawase R., Okamoto R., Ito M., Tanaka T. (2016) Downregulation of gstk1 is a common mechanism underlying hypertrophic cardiomyopathy. Front. Pharmacol. 7, 162.
Zhao Y., Wang Z., Zhang W., Zhang L. (2019) MicroRNAs play an essential role in autophagy regulation in various disease phenotypes. Biofactors. 45, 844–856.
Kuster D.W., Mulders J., Ten Cate F.J., Michels M., Dos Remedios C.G., da Costa Martins P.A., van der Velden J., Oudejans C.B. (2013) MicroRNA transcriptome profiling in cardiac tissue of hypertrophic cardiomyopathy patients with MYBPC3 mutations. J. Mol. Cell. Cardiol. 65, 59–66.
Song L., Su M., Wang S., Zou Y., Wang X., Wang Y., Cui H., Zhao P., Hui R., Wang J. (2014) MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 18, 2266–2274.
Bagnall R.D., Tsoutsman T., Shephard R.E., Ritchie W., Semsarian C. (2012) Global microRNA profiling of the mouse ventricles during development of severe hypertrophic cardiomyopathy and heart failure. PLoS One. 7, e44744.
Palacin M., Reguero J.R., Martin M., Diaz Molina B., Moris C., Alvarez V., Coto E. (2011) Profile of microRNAs differentially produced in hearts from patients with hypertrophic cardiomyopathy and sarcomeric mutations. Clin. Chem. 57, 1614–1616.
Care A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N.D., Elia L., Latronico M.V., Hoydal M., Autore C., Russo M.A., Dorn G.W., 2nd, Ellingsen O., Ruiz-Lozano P., Peterson K.L., Croce C.M., Peschle C., Condorelli G. (2007) MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 13, 613–618.
Gao J., Collyer J., Wang M., Sun F., Xu F. (2020) Genetic dissection of hypertrophic cardiomyopathy with myocardial RNA-Seq. Int. J. Mol. Sci. 21, e3040.
Roncarati R., Viviani Anselmi C., Losi M.A., Papa L., Cavarretta E., Da Costa Martins P., Contaldi C., Saccani Jotti G., Franzone A., Galastri L., Latronico M.V., Imbriaco M., Esposito G., De Windt L., Betocchi S., Condorelli G. (2014) Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 63, 920–927.
Derda A.A., Thum S., Lorenzen J.M., Bavendiek U., Heineke J., Keyser B., Stuhrmann M., Givens R.C., Kennel P.J., Schulze P.C., Widder J.D., Bauersachs J., Thum T. (2015) Blood-based microRNA signatures differentiate various forms of cardiac hypertrophy. Int. J. Cardiol. 196, 115–122.
Fang L., Ellims A.H., Moore X.L., White D.A., Taylor A.J., Chin-Dusting J., Dart A.M. (2015) Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Transl. Med. 13, 314.
Yao R.W., Wang Y., Chen L.L. (2019) Cellular functions of long noncoding RNAs. Nat. Cell Biol. 21, 542–551.
Kitow J., Derda A.A., Beermann J., Kumarswarmy R., Pfanne A., Fendrich J., Lorenzen J.M., Xiao K., Baven-diek U., Bauersachs J., Thum T. (2016) Mitochondrial long noncoding RNAs as blood based biomarkers for cardiac remodeling in patients with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 311, H707–H712.
Yang W., Li Y., He F., et al. (2015) Microarray profiling of long non-coding RNA (lncRNA) associated with hypertrophic cardiomyopathy. BMC Cardiovasc. Disord. 15, 62.
Liu X., Ma Y., Yin K., Li W., Chen W., Zhang Y., Zhu C., Li T., Han B., Liu X., Wang S., Zhou Z. (2019) Long non-coding and coding RNA profiling using strand-specific RNA-seq in human hypertrophic cardiomyopathy. Sci. Data. 6, 90.
Hu X., Shen G., Lu X., Ding G., Shen L. (2019) Identification of key proteins and lncRNAs in hypertrophic cardiomyopathy by integrated network analysis. Arch. Med. Sci. 15, 484–497.
Sonnenschein K., Wilczek A.L., de Gonzalo-Calvo D., Pfanne A., Derda A.A., Zwadlo C., Bavendiek U., Bauersachs J., Fiedler J., Thum T. (2019) Serum circular RNAs act as blood-based biomarkers for hypertrophic obstructive cardiomyopathy. Sci. Rep. 9, 20350.
Li J., Wu Z., Zheng D., Sun Y., Wang S., Yan Y. (2019) Bioinformatics analysis of the regulatory lncRNA-miRNA-mRNA network and drug prediction in patients with hypertrophic cardiomyopathy. Mol. Med. Rep. 20, 549–558.
Shi H., Li J., Song Q., Cheng L., Sun H., Fan W., Li J., Wang Z., Zhang G. (2019) Systematic identification and analysis of dysregulated miRNA and transcription factor feed-forward loops in hypertrophic cardiomyopathy. J. Cell. Mol. Med. 23, 306–316.
Sun D., Li C., Liu J., Wang Z., Liu Y., Luo C., Chen Y., Wen S. (2019) Expression profile of microRNAs in hypertrophic cardiomyopathy and effects of microRNA-20 in inducing cardiomyocyte hypertrophy through regulating gene MFN2. DNA Cell Biol. 38, 796–807.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология