

Молекулярная биология, 2020, T. 54, № 6, стр. 883-921
Ключевые молекулярные механизмы старения, биомаркеры и потенциальные интервенции
Е. Н. Прошкина a, И. А. Соловьёв a, b, М. В. Шапошников a, А. А. Москалев a, b, c, *
a Институт биологии Коми научного центра Уральского отделения Российской академии наук
167982 Сыктывкар, Россия
b Сыктывкарский государственный университет им. Питирима Сорокина
167001 Сыктывкар, Россия
c Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: amoskalev@ib.komisc.ru
Поступила в редакцию 08.05.2020
После доработки 21.05.2020
Принята к публикации 21.05.2020
Аннотация
В обзоре рассмотрены механизмы старения на молекулярном, клеточном, тканевом и системном уровнях. Первичные молекулярные повреждения вызывают клеточный ответ, направленный на компенсацию нарушений, однако постепенно выходят из строя сами механизмы восстановления и поддержания гомеостаза. Когда количество ошибок в регуляторных сетях достигает критического порога, на системном уровне происходит фазовый переход из состояния здоровья в состояние болезни. Рассмотрены подходы к количественной оценке процесса старения (биомаркеры старения), а также интервенции, перспективные с точки зрения возможности замедления процессов старения и уменьшения риска развития возрастзависимых заболеваний.
ВВЕДЕНИЕ
В связи с увеличением среднего возраста населения проблема профилактики преждевременного старения и лечения возрастных заболеваний выходит на первый план в современном здравоохранении [1]. Перспективным подходом к реализации этой цели считается воздействие на ключевые молекулярные механизмы, связанные со старением – основным фактором риска возрастзависимых заболеваний, направленное на подавление патологических процессов и активацию защитных систем клетки и организма в целом [2, 3]. В настоящее время практически отсутствуют клинически проверенные терапевтические вмешательства в ход молекулярных процессов естественного и ускоренного старения, однако имеется большое количество перспективных доклинических исследований [1, 4]. Кроме того, для трансляции в клинику потребуется разработка системы биомаркеров старения, пригодных как для оценки состояния организма, так и для определения эффективности геропротекторной терапии [5].
Основатель геронтологии Бенджамин Гомперц вывел закон смертности, впоследствии названный его именем: “Возможно, что смерть может быть следствием двух обычно сосуществующих причин: одна – случайность, без предшествующей предрасположенности к смерти или изнашиванию; другая – изнашивание или возрастающая неспособность противостоять разрушению” [6]. Тем самым Гомперц не только отделил внешние причины смертности от внутренних (старения), но и предвосхитил разделение механизмов старения на накопление повреждений и ошибок (“изнашивание”) и утрату способности восстанавливать ошибки из-за поломок самих систем репарации.
В обзоре с современных позиций рассмотрена комплексная динамика процесса старения на разных уровнях биологической организации – от первичных повреждений на молекулярном уровне и ответа клетки на них, который отчасти компенсирует нарушения, но может вносить новые ошибки (в том числе, приводящие к нарушению регулирования систем поддержания гомеостаза), до системного уровня, когда количество накопленных ошибок вызывает фазовый переход из состояния здоровья в состояние болезни.
Цель обзора состояла в обобщении современных взглядов на ключевые механизмы старения, а также на биомаркеры, характеризующие биологический возраст организма и связанные со старением патологические процессы, и на геропротекторные интервенции, нацеленные на профилактику преждевременного старения.
МЕХАНИЗМЫ СТАРЕНИЯ
Повреждения и ошибки на молекулярном уровне в процессе старения
В течение жизни организмы сталкиваются с воздействием экзогенных и эндогенных повреждающих факторов. В первую очередь эти факторы вызывают изменения на молекулярном уровне, приводя к нарушению целостности и структурной организации генетического материала, модификациям и агрегированию внутриклеточных и внеклеточных белков, повреждению мембран, митохондриальной дисфункции. В молодом возрасте компенсаторные и гомеостатические механизмы позволяют сохранить баланс в молекулярно-клеточных процессах, предотвращают функциональные и фенотипические нарушения. Однако в результате неспособности этих механизмов устранить все ошибки и возрастное ухудшение их работы накапливается непоправимый ущерб, усиливающий дегенеративные процессы (системное воспаление, хроническая гипоксия, нарушение целостности барьеров) и преодолевающий функциональный порог. Это приводит к старению и развитию возрастзависимых заболеваний [7].
Генетическая нестабильность. В тканях стареющих животных и человека повышается частота повреждений ДНК и соматических мутаций, возникает так называемая нестабильность генома, выражающаяся во всплеске точечных мутаций, разрывов и сшивок цепей ДНК, транспозиций и транслокаций, анеуплоидий [8, 9]. При этом разные соматические клетки накапливают мутации с разной скоростью. В результате в стареющем организме образуются клоны клеток с несколько отличающимся генотипом и формируется соматический мозаицизм [9–11].
Соматический мутагенез как ключевой механизм старения был предложен Лео Сцилардом в 1959 году, однако доказать накопление мутаций с возрастом в тканях оказалось непросто. Основным аргументом долгие годы оставались признаки ускоренного старения у людей с врожденными мутациями генов репарации ДНК (например, синдром Вернера, синдром Кокейна, синдром Блума, пигментная ксеродерма, атаксия-телеангиэктазия и другие) [12–14].
Позже выяснилось, что виды с экстремальным долгожительством, например голые землекопы [15], летучие мыши Брандта [16], киты [17], слепыши [18] и попугаи [19], имеют адаптивные особенности механизмов репарации, повышающие стабильность их ДНК. По-видимому, надежные способы защиты ДНК – одна из причин “бессмертия” клеток зародышевой линии [20].
Однако появление современных методов анализа, в частности, секвенирование генома единичных клеток [21], а также секвенирование транскриптов [22], позволило увидеть соматический мутационный ландшафт тела человека, в том числе в возрастной динамике [23].
Источником повреждений ДНК и мутагенеза являются разнообразные внешние факторы (включая физические и химические агенты, вирусные инфекции) и внутриклеточные причины – спонтанные гидролитические реакции, конверсия метилированного цитозина в тимин, транспозиция мобильных генетических элементов (МГЭ), активные формы кислорода (АФК), ошибки репликации и репарации ДНК [8, 9].
Существует несколько уровней защиты клетки от повреждения ДНК и накопления мутаций: перехват и обезвреживание молекул, повреждающих ДНК, репарация повреждений и элиминация клеток из делящегося пула в ответ на постоянное повреждение ДНК через запуск клеточного старения и апоптоза [24]. Мутации или нарушение этих путей могут привести к ускоренному или преждевременному старению и возрастному снижению функциональной способности жизненно важных органов. При старении нарушается работа компенсаторных механизмов, которые в норме должны обеспечивать адекватный ответ на действие соответствующих источников повреждений и поддерживать стабильность генома.
Кроме того, существенный вклад вносят истощение пула NAD+ [25] и недостаточный синтез ДНК-нуклеотидов [26]. Некоторые механизмы накопления клеток с повреждениями ДНК реализуются на тканевом уровне. Например, жесткость внеклеточного матрикса через дисморфию клеточного ядра может провоцировать поломки хромосом [27]. Переключение клеток с метаболизма глюкозы на бета-окисление жирных кислот способствует перекисному окислению липидов, повышая уровни повреждений ДНК [28]. К тканевым механизмам нужно отнести снижение способности сенесцентных клеток индуцировать апоптоз [29] и ослабление иммунитета, способствующего их элиминации [30].
Немаловажную роль играют и такие факторы, как алкоголь [31], табакокурение [32] и нарушение суточных ритмов [33].
Повышенная частота анеуплоидий связана с хромосомной нестабильностью, вызванной снижением точности сегрегации хромосом из-за недостаточной активности кинезина-13 – фермента, деполимеризующего микротрубочки [34].
С возрастом наблюдается снижение каталитической активности белков репарации ДНК, связанное не только со снижением способности быстро восстанавливать поврежденные участки, но и с повышением частоты ошибок репарации, вносящей новые повреждения [8, 9].
Одна из функций конститутивного гетерохроматина – защита эухроматина от повреждений. В ядре повреждающие агенты поглощаются и блокируются конститутивным гетерохроматином, а поврежденная ДНК удаляется из ядра в цитоплазму через комплексы ядерных пор. Накопление повреждений ДНК при старении, вероятно, связано с возрастным истощением и дерегуляцией гетерохроматина [35–37]. В свою очередь, высвобождаемые в цитоплазму фрагменты ДНК могут распознаваться как ассоциированные с вирусами и провоцировать воспалительные реакции [38]. При старении снижается активность белков, обеспечивающих стабильность гетерохроматина, например деацетилазы гистонов SIRT6 и белка гетерохроматина HP1 [36]. Кроме того, истощение конститутивного гетерохроматина тесно связано с укорочением теломер и активацией МГЭ.
А.М. Оловников еще в начале 70-х годов прошлого века предположил, что концевые участки хромосом, называемые теломерами, играют буферную роль и укорачиваются при каждом клеточном делении. В конечном итоге этот процесс приводит к клеточному старению [39, 40]. В настоящее время хорошо описана роль укорочения теломер в репликативном старении. Однако длина теломер в различных тканях и типах клеток может варьировать, а скорость укорочения теломер изменяется в течение жизни индивида [41]. Вклад в генетическую нестабильность вносят и многочисленные повреждения теломерной ДНК. Помимо того, что теломеры входят в состав конститутивного гетерохроматина и находятся на периферии клеточного ядра [36], их повреждения не распознаются соответствующими сенсорами из-за присутствия шелтеринового комплекса [42, 43].
В результате нарушения структуры конститутивного гетерохроматина происходит активация МГЭ [44, 45], что усиливает генетическую нестабильность, нарушение или изменение экспрессии генов [46–48].
Кроме того, на стабильность генома влияет организация ядерной ламины. Снижение количества ламина B1, накопление токсических уровней преламина А и экспрессия прогерина (патогенная форма ламина А) приводят к дефектам структуры ядра и связаны со старением клеток и организма [9, 49, 50]. Мутации в генах ядерной ламины вызывают синдромы преждевременного старения, называемые ламинопатиями (в частности, синдром Хатчинсона–Гилфорда) [51, 52]. Состояние ламины в контексте старения влияет на скорость укорочения теломер, активность генов и сигнальных путей, организацию хроматина и паттерны метилирования ДНК [9, 50].
Состояние генетической нестабильности сопровождается нарушением транскрипции жизненно важных генов, клеточного метаболизма, вызывает клеточное старение. Это способствует накоплению дисфункциональных клеток и генетической гетерогенности, нарушению регенеративного потенциала и физиологических функций тканей, снижению адаптационной способности и толерантности организма к стрессу, повышенному риску возрастных заболеваний и старению [53].
Повреждения ДНК провоцируют активацию сигнальных путей, которые могут управлять судьбой клеток, включая их старение [54] и апоптоз [55], а также дисфункцию митохондрий [56], повышенную выработку АФК [57], гиперреактивность врожденного иммунитета и воспаление [58].
Последствия накопления повреждений ДНК нередко тканеспецифичны. Повреждение ДНК в макрофагах приводит к развитию воспаления [59], в нейронах – к когнитивным расстройствам [60], в остеопрогениторных клетках – способствует снижению костной массы [61]. Особенно стоит выделить накопление повреждений ДНК и мутаций в стволовых клетках, поскольку оно влияет на их регенеративный потенциал и создает риск возникновения опухолевых стволовых клеток. Чем меньше делятся прогениторные клетки, тем меньше мутаций они накапливают [62].
Эпигенетические изменения. Остановку клеточного роста, вызванную повреждением ДНК, поддерживают особые ядерные структуры – ДНК-шрамы [63]. Как предполагает Дэвид Синклер, эти изменения служат причиной перераспределения факторов реорганизации хроматина и, в конечном итоге, способствуют появлению “эпигенетического шума”, который нарушает паттерны экспрессии генов, необходимых для оптимального функционирования клеток и восстановления после повреждения [64].
Эпигенетические процессы регулируют экспрессию генов, влияя на организацию хроматина и доступность ДНК для факторов транскрипции. С одной стороны, наблюдаемые при старении эпигенетические изменения носят компенсаторный характер, препятствуя непоправимому повреждению ДНК и позволяя клетке жить в условиях нарастающего стресса, а с другой, они вызывают побочные процессы, негативно влияющие на клетки и организм, усиливая старение и развитие возрастзависимых заболеваний [65].
В 70-х годах прошлого столетия Б.Ф. Ванюшин с коллегами показали, что при старении в различных органах и тканях грызунов снижается уровень метилирования ядерной ДНК [66, 67]. Гипометилирование наблюдается преимущественно в повторяющихся геномных областях и в МГЭ из-за снижения активности ДНК-метилтрансфераз. При этом происходит дифференциальное гиперметилирование специфических геномных локусов, в том числе, соответствующих генам репарации и репликации ДНК, биосинтеза белков, генам-супрессорам опухолевого роста и апоптоза, генам-мишеням Polycomb, генам, регулирующим дифференцировку. Наконец, наблюдается увеличение межиндивидуальных различий между паттернами метилирования ДНК (эпигенетический дрейф) и скорости эпимутаций [68–73].
Для эпигенома стареющих клеток характерно снижение общего количества гистонов и изменение соотношения их модификаций. Уменьшение количества гистонов приводит к снижению числа нуклеосом и открытию хроматина [69, 74]. Деконденсации хроматина способствует уменьшение количества репрессивных меток, таких как триметилирование гистона H3 в Lys9 и Lys27. С другой стороны, наблюдается увеличение активирующих меток, в частности, триметилирования H3K4 и ацетилирования H4K16. Однако некоторые модификации гистонов варьируют при старении в разных клетках и у разных индивидов [65, 69, 75]. Такие изменения касаются также теломер и центромер [69]. Помимо метилирования и ацетилирования существуют и другие модификации гистонов, но они слабо изучены в контексте старения. Стоит отметить, что при старении происходит снижение ферментативных модификаций гистонов (в частности, ацетилирования), и накопление неферментативных посттрансляционных модификаций [76]. Кроме того, на возрастное изменение структуры хроматина влияет включение вариантов гистонов, например, H3.3, H2A.J, H2A.Z [65, 77, 78].
Старение сопровождается изменениями количества и активности белков, модифицирующих гистоны и архитектуру хроматина [69]. В контексте старения наиболее изучены гистондеацетилазы и гистон-ацетилтрансферазы. В частности, NAD+-зависимые гистондеацетилазы SIRT1 и SIRT6 являются известными детерминантами продолжительности жизни и старения, активность которых снижается с возрастом вместе с уменьшением уровня NAD+ [69, 75, 79].
На начальных этапах старения снижение доли гетерохроматина может выступать в качестве компенсаторного механизма, стимулируя активность генов ответа на стресс и ингибируя mTOR. По мере того как старение прогрессирует, и хроматин открывается дальше, эти пути утрачивают способность компенсировать нарушение транскрипции и нестабильность генома, усиливая дегенеративные процессы в клетке [69, 75]. В то же время в некоторых стареющих клетках хроматин, напротив, подвергается аномальной конденсации с образованием очагов гетерохроматина, ассоциированных со старением [69, 75]. Стоит отметить, что долгожительство обеспечивается скоординированной эпигенетической регуляцией [80].
Возрастные эпигенетические изменения касаются также паттернов экспрессии некодирующих РНК, включая длинные некодирующие РНК (днРНК), микроРНК, малые интерферирующие РНК, piwi РНК [69, 81–84]. При этом, если активность одних некодирующих РНК снижается (например, днРНК H19 – негативного регулятора фактора роста IGF-1), то других увеличивается (например, микроРНК lin-4, miR-34, miR-290, miR-335). На процесс старения они действуют с помощью подавления экспрессии их мишеней, относящихся к различным связанным со старением сигнальным путям, репрессии МГЭ, регуляции структуры хроматина, поддержанию теломер [69, 81–84]. Кроме того, существуют кольцевые РНК, которые накапливаются во время старения и действуют в качестве “губок” микроРНК или связываются с РНК-ассоциированными белками [85, 86]. Некоторые некодирующие РНК, главным образом микроРНК, могут секретироваться из клеток в составе экзосом и влиять на старение через межклеточные коммуникации [87, 88].
Утрата протеостаза. Старение сопровождается ухудшением способности клеток поддерживать протеостаз. Модифицированные и неправильно свернутые белки и белковые агрегаты находят почти во всех тканях пожилого организма, особенно в слабо пролиферирующих [89–92]. Неферментативные посттрансляционные модификации накапливаются с возрастом во всех компартментах клетки и приводят к нарушению протеостаза [76]. Наиболее изученная в контексте старения модификация – неферментативное гликозилирование (гликирование) – приводит к образованию сшивок белковых молекул, формированию конечных продуктов гликирования (КПГ), токсичных промежуточных продуктов (продукты Амадори, глиоксаль), а также АФК [93]. Гликирование белков происходит в результате взаимодействии аминокислотных остатков (лизина, аргинина) с восстанавливающими формами некоторых сахаров (галактоза, фруктоза, глюкоза) или метилглиоксалем (продуктом гликолиза). Установлено, что при старении повышается количество окисленных, карбонилированных [94, 95] и карбамилированных белков [95]. Структурные и функциональные характеристики белков, содержащих химические модификации, изменяются. С одной стороны, это приводит к нарушению их работы в результате конформационных изменений, которые могут находиться вблизи активного сайта фермента. С другой стороны, они перестают распознаваться ферментами, субстратами которых были раньше, что приводит к накоплению этих белков и перегрузке систем деградации белков. Химически модифицированные белки зачастую образуют межмолекулярные связи с формированием белковых олигомеров и агрегатов, вызывая активацию реакции на стресс, воспаление, нарушение клеточных функций (включая синтез белка и работу механизмов клеточной защиты), клеточные повреждения и апоптоз [91, 93, 96, 97]. Накопление белковых агрегатов в различных органах и тканях связано с развитием специфических болезней, называемых амилоидозами, а также с рядом возрастзависимых заболеваний [89, 90, 98]. Кроме того, продукты гликирования активируют рецепторы конечных продуктов гликирования (RAGE), вызывая усиленную выработку АФК, активацию сигнальных путей p21, митоген-активируемых протеинкиназ (MAPK), JAK/STAT, воспалительные реакции [93, 94]. Стоит отметить, что наибольший вклад в процесс старения вносит повреждение долгоживущих белков, включая белки комплекса ядерных пор и внеклеточного матрикса. Они накапливаются в течение всей жизни клетки или даже организма, вызывая глубокие структурные и функциональные изменения [97, 99]. Тем не менее, существуют механизмы оборота белков. Например, белки ядерных пор могут замещаться в ответ на голодание или при генетических вмешательствах с помощью вакуолярных протеаз и аутофагии [100].
У млекопитающих количество функциональных белковых молекул может регулироваться за счет изменения их растворимости, так при превышении физиологической концентрации белки начинают агрегировать [92]. Поэтому в качестве компенсаторной реакции на накопление поврежденных и неправильно свернутых белков уменьшается количество рибосом и снижается синтез белков. В связи с уменьшением трансляции энергетические затраты могут перераспределяться и направляться на поддержание жизнеспособности клеток [92, 101].
При старении во многих тканях нарушается работа механизмов восстановления окисленных белков (система метионин-сульфоксид-редуктазы), контроля качества белков (ответ на неправильно свернутые белки) и их деградации (убиквитин-протеасомная и аутофагийно-лизосомная системы) [92, 102–104]. Например, для стареющих клеток характерны изменения в ответных реакциях на денатурированные белки как в цитоплазме, так и в специфических компартментах клетки (эндоплазматической сети (ЭПС), митохондриях). С возрастом снижается экспрессия АТP-зависимых шаперонов (HSP70 и HSP60), но повышаются уровни малых белков теплового шока и их накопление во фракции агрегатов [92, 105]. Такие изменения отражают сдвиг в защитных стратегиях клетки – от предотвращения агрегации к изоляции опасных видов белков. Предполагается, что если денатурированные белки не могут подвергаться повторному сворачиванию или деградации, то образующиеся белковые молекулы более токсичны для клетки, чем нерастворимые агрегаты [92]. В результате накопления несвернутых белков повышаются также уровни маркеров стресса ЭПС. При этом способность активировать ответ на несвернутые белки в ЭПС подавляется. Активность шаперонов ЭПС снижается при старении, в то время как уровни проапоптотических медиаторов возрастают [103, 104].
Эффективность протеолиза и аутофагии, количество и экспрессия структурных компонентов протеасомы и ферментов, осуществляющих деградацию, снижаются с возрастом [94, 106, 107]. Эти две системы не только предотвращают накопление нефункциональных белков и белковых агрегатов, но и обеспечивают оборот макромолекул и органелл в клетке. Они также необходимы для регуляции обмена веществ и энергии (в зависимости от содержания АМP/АТP, аминокислот, свободных жирных кислот в клетке), поддержания стабильности генома, иммунитета, подавления воспаления и канцерогенеза [98, 102, 106, 108]. Однако при дерегуляции протеостаза избыточное накопление аутофагосом с последующим запуском гибели клеток может вызывать патогенные процессы, например, утрату нейронов при нейродегенеративных заболеваниях [102].
Дисфункция митохондрий. У человека и многих видов животных при старении снижается активность электрон-транспортной цепи (ЭТЦ) и интенсивность окислительного фосфорилирования (в том числе за счет повреждения комплексов ферментов ЭТЦ или накопления мутаций в мтДНК), в результате чего нарушается выработка NAD+ [109–111]. Дисбаланс в работе ЭТЦ приводит к увеличению продукции АФК. С возрастом АФК, которые выполняют роль сигнальных молекул, индуцируют запуск компенсаторных реакций. Однако по мере повышения уровня АФК перестают выполнять первоначальную функцию и в сочетании со снижением антиоксидантной защиты усугубляют возрастные нарушения [112]. Эти изменения сопровождаются дисбалансом окислительно-восстановительных реакций в митохондриях, нарушением их целостности и морфологическими аномалиями, потерей мембранного потенциала, нарушением буферизации кальция и индукцией апоптоза [109, 111]. Нарушается работа регуляторных митохондриальных белков. Например, у пожилых людей снижается активность SIRT3, который деацетилирует многие ферменты, участвующие в энергетическом метаболизме, антиоксидантной защите, ответе на повреждение ДНК, аутофагии [113, 114].
При старении повышаются уровни повреждений основных макромолекул, содержащихся в митохондриях, включая мтДНК и белки; в результате перекисного окисления липидов изменяется состав митохондриальных мембран [96, 111, 115–117]. В различных тканях организма увеличивается частота клеток с точечными мутациями мтДНК. Однако основными причинами этих мутаций являются не окислительные повреждения, а ошибки репликации и спонтанное дезаминирование оснований. Возникая в раннем возрасте, мутации либо теряются в результате сегрегации мтДНК при клеточном делении, либо накапливаются в процессе клональной экспансии. В результате в тканях стареющего организма увеличивается доля клеток с митохондриальной дисфункцией [116, 118]. Повышению частоты повреждений и мутаций мтДНК способствует снижение активности белков, обеспечивающих ее целостность. К таким белкам относятся ферменты эксцизионной репарации оснований и теломерный белок TERT, который обнаруживается в митохондриях пролиферирующих и постмитотических клеток [38, 110, 117, 119, 120]. Поддержанию стабильности митохондриального генома способствует избирательная деградация поврежденных копий мтДНК, поэтому при старении наблюдается снижение их количества [118]. Стоит отметить, что многие белки, необходимые для функционирования митохондрий, кодируются ядерными генами. Соответственно, нестабильность ядерного генома, нарастающая при старении, играет критическую роль в функционировании митохондрий [116]. Кроме того, фрагменты мтДНК, высвобождаемые при повреждении или деградации митохондрий, могут перемещаться в ядро и встраиваться в геном как некодирующие последовательности, называемые ядерно-митохондриальными псевдогенами (NUMT-псевдогены). Их внедрению способствуют двухцепочечные разрывы ДНК, количество которых увеличивается при воздействии генотоксических агентов и старении. В свою очередь, NUMT-псевдогены изменяют структуру генома и влияют на экспрессию генов, внося вклад в развитие различных заболеваний и в процесс старения [121].
У стареющих животных и пожилых людей наблюдается увеличение количества модифицированных белков (главным образом, возникающих в результате окислительных повреждений) и образование белковых агрегатов. В первую очередь повреждению подвергаются белки ЭТЦ [96, 122]. Так как большинство митохондриальных белков кодируется ядерным геномом, транслируется и дозревает в цитоплазме, после чего импортируется в митохондрии, нарушение клеточного протеостаза вносит существенный вклад в дисбаланс протеома митохондрий при старении [122]. С другой стороны, митохондрии характеризуются высокой скоростью оборота белков и имеют собственные механизмы ответа на несвернутые белки: митохондриальные протеазы и шапероны. С возрастом происходит снижение экспрессии протеаз и индуцибельности шаперонов. Дефекты в этих защитных белках связаны с различными заболеваниями (например, с синдромом Перро), включая расстройства, связанные со старением [122]. Дополнительная цель митохондриального ответа на несвернутые белки состоит в том, чтобы облегчить работу поврежденных митохондрий путем смещения метаболизма от зависимого от митохондрий окислительного фосфорилирования к цитоплазматическому гликолизу [122].
Кроме того, в митохондриях действуют специфические компенсаторные механизмы: поддержание баланса между слиянием и делением митохондрий, а также между биогенезом и деградацией (митофагией, специфичной для органелл формой аутофагии). В результате их работы митохондриальная дисфункция при старении нарастает нелинейно, наблюдается пороговая реакция на нарушение работы митохондрий [123]. Предполагается, что именно оборот митохондрий играет ключевую роль в поддержании стабильности мтДНК и функционирования митохондрий [118]. В здоровых клетках митохондрии слиты в большую сеть, в результате чего ослабляется эффект небольших количеств повреждений. Благодаря делению митохондрий поврежденный материал удаляется из митохондриальной сети. Но деление может предшествовать митофагии или апоптозу [122, 124]. Долголетию организма способствует пластичность митохондриальной сети, в то время как старение характеризуется дисбалансом этих процессов [124]. Также с возрастом изменяется соотношение между биогенезом митохондрий и митофагией [102, 109]. Снижается общее число митохондрий, нарушается их биосинтез (например, в результате дерегуляции белков семейства PGC-1, нестабильности генома и дисфункции теломер) [102, 110]. Митофагия, в свою очередь, обеспечивает рециркуляцию митохондрий и индуцируется при чрезмерной активации митохондриального ответа на несвернутые белки и других событиях, сопровождающих митохондриальную дисфункцию [102, 122, 125, 126].
Нарушение работы митохондрий и повышенная выработка АФК могут приводить к повреждению макромолекул в других компартментах клетки [110, 111]. Снижение выработки АТP нарушает работу энергозависимых процессов, включая обеспечивающие клеточную защиту [90]. Кроме того, митохондрии выступают в качестве сигнальной платформы. В частности, специфические митохондриальные липиды (кардиолипин и фосфатидилэтаноламин) могут модулировать события передачи сигналов, регулирующих метаболизм клетки. Митохондрии связаны с механизмами врожденного иммунитета, особенно с цитозольным РНК-распознающим путем, который опосредуется митохондриальным противовирусным сигнальным белком (MAVS) [127]. Эти органеллы могут передавать в ядро ретроградные сигналы (АФК, катионы кальция, измененное клеточное соотношение АМP/АТP, NAD+, продукты цикла Кребса, специфические транскрипционные факторы и другие белки) для активации компенсаторных и защитных механизмов. Такая сигнализация вызывает изменения в паттернах генной экспрессии и влияет на пути, связанные со старением, долголетием и стрессоустойчивостью [38, 111, 116, 127, 128]. Состояние мтДНК и ее гетероплазмия могут влиять на экспрессию и уровень мутаций ядерной ДНК, эпигеномные и метаболические процессы. Эпигеномные изменения наблюдаются и при запуске митохондриального ответа на несвернутые белки [116, 129]. Наконец, окисленные фрагменты мтДНК и формиловые пептиды, высвобождаемые в цитоплазму и во внеклеточное пространство при повреждении митохондрий, воспринимаются соответствующими рецепторами как сигналы, ассоциированные с бактериальным заражением. Их количество увеличивается с возрастом, что влечет за собой активацию воспалительных и иммунных реакций [38, 111, 116].
Молекулярно-клеточные механизмы ответа на связанные со старением повреждения
Процесс старения взаимосвязан с дерегуляцией механизмов восстановления и поддержания гомеостаза организма на всех уровнях организации – от молекулярного до системного, включая экспрессию генов, межклеточную сигнализацию, сигнальные пути нутриентов, регенерацию тканей, воспаление, нейроэндокринную регуляцию.
Дерегуляция экспрессии генов. Изменение экспрессии генов при старении – одна из характеристик инволюции регуляторных механизмов, которая проявляется как локально (сайленсингом или активацией отдельных генов и сигнальных путей в определенных тканях), так и в целом, в нарушении транскрипции, процессинга РНК и трансляции, повышении уровня гетерогенности клеток и транскрипционной активности последовательностей некодирующей ДНК и повторяющихся элементов генома [130–133].
Для многих генов, активность которых изменяется в течение жизни, характерна прямая или обратная U-образная зависимость уровня экспрессии от возраста [133–135]. Так, экспрессия генов, высокая в зрелом возрасте, становится в старости низкой и наоборот. Это явление названо транскрипционным дрейфом [136]. Предполагается, что геропротекторное действие должно сопровождаться снижением транскрипционного дрейфа, что позволяет сохранить в старости более молодой транскриптом [137]. Подобные эффекты удается наблюдать при действии геропротекторов на модели Caenorhabditis elegans [136].
Старение связано также с повсеместной индукцией генов, вовлеченных в процессы воспаления, врожденного иммунного ответа, деградации в лизосомах, и генов, связанных с апоптозом, задержкой клеточного цикла и клеточным старением. Недостаточная экспрессия характерна для генов биосинтеза коллагена и генов, связанных с энергетическим обменом, в частности, митохондриальных [138]. Спектр функциональных классов генов, уровень экспрессии которых снижается с возрастом, часто является тканеспецифичным [131].
Один из характерных признаков старения некоторых тканей млекопитающих – накопление соматических мутаций и эпимутаций и, как следствие, увеличение вариабельности уровней экспрессии генов в отдельных клетках и тканях – транскрипционный шум [139].
Для транскриптома характерно возрастзависимое нарушение баланса между короткими и длинными транскриптами, выражающееся в снижении уровня длинных транскриптов [140] вследствие низкой активности гена фактора сплайсинга Sfpq [140]. Старение также сопровождается появлением дефектов сплайсинга из-за нонсенс-опосредованного распада транскриптов с сохраненными интронами, что ведет к изменениям в соотношении изоформ белков [131].
Уровень некодирующих РНК, участвующих в контроле экспрессии генов, меняется с возрастом в зависимости от ткани и вида организма [130]. Масштабное сокращение конститутивного гетерохроматина проявляется в активации МГЭ и дестабилизации генома. Накопление кДНК ретротранспозонов в цитоплазме способствует формированию стерильного воспаления [48].
На уровне трансляции старение сопровождается снижением активности транскриптов, содержащих 5'-концевой олигопиримидиновый тракт (5'TOP), характерный для мРНК факторов элонгации, рибосомных белков и транскриптов, регулируемых mTOR [133]. Доступность рибосом для многих транскриптов с 5'TOP-мотивом с возрастом снижается в 3 раза и более. Более того, покрытие рибосомами постепенно уменьшается вблизи стартовых кодонов и увеличивается вблизи стоп-кодонов [133].
Нарушение регуляции экспрессии генов приводит не только к изменению уровня активности автономных сигнальных путей, но и путей межклеточной сигнализации.
Понятие “межклеточной сигнализации” настолько общее, что охватывает практически любой известный физиологический механизм [141]. Мы рассмотрим два частных случая дерегуляции межклеточной сигнализации, которые относятся к ключевым признакам старения [142]: дерегуляцию сигнальных путей нутриентов (в этом разделе) и воспаление – в разделе “Молекулярные механизмы иммуностарения и воспаления”.
Нутриенты регулируют не только процессы роста, но и стрессоустойчивость, возникновение различных возрастных заболеваний и продолжительность жизни. Избыток или дисбаланс питательных веществ приводит к ожирению и неблагоприятным метаболическим последствиям, в то время как ограничение питания задерживает начало возрастных заболеваний и увеличивает продолжительность жизни [143]. Стареющий организм страдает одновременного от избытка калорий и нехватки необходимых питательных веществ [144]. При этом основное влияние на долголетие оказывает качественный состав пищи, а не количество калорий [145]. Уровень поступления в организм питательных веществ детектируется с помощью сенсоров и связанных с ними сигнальных путей [146].
Сигнальный путь инсулин/IGF-1 активируется в ответ на поступление углеводов [147], уровень аминокислот определяется с помощью компонентов пути mTOR [148], а липидов – с участием рецепторов, активируемых пролифераторами пероксисом (PPAR) [149]. Энергетический статус клетки оценивается с помощью AMPK (соотношение АТP/АDP) и сиртуинов (соотношение NADH/NAD+) [150].
В зависимости от уровня организации компоненты сигнальных путей нутриентов можно подразделить на циркулирующие системные факторы, представляющие соматотрофную ось (гипофизарный гормон роста, гипоталамический соматолиберин, вырабатываемый в печени IGF-1, и инсулин, образующийся в β-клетках поджелудочной железы), рецепторы клеточной поверхности (IGF-1R, IR, ERBB), внутриклеточные сигнальные компоненты (PI3K/AKT/mTOR, MAPK/ERK) и молекулярные эффекторы (FOXO, AMPK, S6K, GSK3, PPAR) [150].
Сигнальные пути нутриентов тесно взаимосвязаны [150, 151]. Активация сигнального пути инсулин/IGF-1 посредством связывания инсулина, IGF-1 или факторов роста с рецепторами IR, IGF-1R или ERBB приводит к активации путей PI3K/AKT и MAPK/ERK соответственно. Активация AKT вызывает ингибирование киназы 3 гликоген-синтазы (GSK-3) и фактора транскрипции FOXO, а также индукцию mTOR. mTOR запускает процесс трансляции мРНК через rpS6K/rpS6 и белок 4E-BP1, а также подавляет аутофагию путем ингибирования ULK1. Путь ERK/MAPK активирует митогенные факторы, такие как протоонкоген c-MYC, и способствует трансляции, активируя rpS6 посредством фосфорилирования в rpS6K-независимых сайтах [150]. AMPK ингибирует mTOR посредством фосфорилирования TSC2 и Raptor [152, 153]. AMPK активирует FOXO и сиртуины (SIRT1-7) [153]. SIRT1, в свою очередь, активирует AMPK путем деацетилирования и активации LKB1 [153].
AMPK является регулятором метаболизма, энергетического обмена, аутофагии, биогенеза митохондрий, антиоксидантной защиты [152, 153]. Сиртуины участвуют в эпигенетической регуляции транскрипции посредством деацетилирования различных субстратных белков, регулируют многие биологические процессы, включая транскрипцию, репарацию ДНК и стабильность генома, метаболизм и внутриклеточную сигнализацию [154].
Интенсивный метаболизм в период роста и развития организма косвенно способствует нарушению протеостаза и энергетического гомеостаза через подавление аутофагии и стресс эндоплазматической сети, в результате чего усиливается воспаление, дисфункция митохондрий и окислительный стресс – ключевые признаки старения [142, 151, 153]. Кроме того, патологические признаки старения не только способствуют нарушению регуляции механизмов восприятия нутриентов, но и усугубляются данными нарушениями [155].
Старение, особенно в сочетании с ожирением, способствует формированию резистентности к инсулину, что приводит к снижению скорости поглощения глюкозы в тканях-мишенях при нормальном уровне инсулина [156]. Для достижения нормального уровня глюкозы в крови β-клетки поджелудочной железы секретируют избыточные количества инсулина, что приводит к гиперинсулинемии [157]. При дальнейшем усилении инсулинорезистентности функция β-клеток нарушается и становится недостаточной для поддержания нормального уровня глюкозы в крови, что вызывает развитие гипергликемии. Высокий уровень глюкозы в плазме также обусловлен нарушением ингибирующего действия инсулина на выработку глюкозы в печени, снижением синтеза гликогена в гепатоцитах и неспособностью скелетных мышц и адипоцитов поглощать глюкозу [155]. Инсулинорезистентность лежит в основе многих метаболических нарушений, включая сахарный диабет второго типа и метаболический синдром.
Таким образом, сигнальные пути нутриентов в значительной степени модулируют здоровье и продолжительность жизни организма [150]. Имеющиеся данные свидетельствуют о том, что активация анаболических сигнальных путей ускоряет старение, а катаболических – способствует увеличению продолжительности жизни [158]. Известно несколько интервенций, которые позволяют противостоять неблагоприятным последствиям дерегуляции механизмов распознавания нутриентов.
Нарушение нейроэндокринной регуляции. При старении не только возникают изменения на уровне межклеточной сигнализации, но и нарушается регуляция нейрогормональной передачи сигналов [142]. Гипоталамус играет решающую роль в нейроэндокринном взаимодействии между центральной нервной системой и периферией. Например, в норме активация mTORC1 в гипоталамусе снижает экспрессию орексигенных пептидов (NPY, AgRP) посредством S6K1-опосредованного механизма. Старение, ожирение или питание с высоким содержанием жиров подавляют способность лептина и инсулина стимулировать активность mTORC1 и снижать потребление пищи [159, 160]. Старение связано с активацией в нейронах гипоталамуса пути IKK-β/NF-κB, который ингибирует выработку гонадотропин-рилизинг-гормона и вызывает множественные связанные со старением физиологические изменения. Введение гонадотропин-рилизинг-гормона замедляет старение [161]. Замедление старения и увеличение продолжительности жизни у мышей достигаются также при подавлении возрастзависимой активации IKK-β и NF-κB в гипоталамусе или мозге.
Повышение активности гипоталамо-гипофизарно-надпочечниковой оси в пожилом возрасте способствует формированию воспалительных стрессовых реакций, ведущих к дерегуляции иммунной системы [162] и дисбиозу [163].
Стоит отметить, что Владимир Дильман еще в 1958 г. предположил, что старение вызвано постепенной утратой чувствительности гипоталамуса к ингибирующему действию периферических гормонов [164].
Эпифиз (шишковидная железа), расположенный в головном мозге, синтезирует гормон мелатонин, регулятор фотопериодичности физиологических функций и мощный нейронный антиоксидант. По-видимому, у млекопитающих эпифиз участвует в регуляции процессов старения, поскольку введение мелатонина способно увеличивать продолжительность жизни животных [165]. Известно, что у млекопитающих в результате постепенной кальцификации эпифиза при старении снижается биосинтез мелатонина [166].
При старении происходит системное снижение образования половых гормонов – тестостерона у мужчин и эстрадиола у женщин. С возрастом уменьшается способность клеток Лейдига вырабатывать тестостерон в результате снижения синтеза сАМР в ответ на лютеинизирующий гормон и снижение митохондриального транспорта холестерина, вызванное окислительным стрессом и сенесценцией [167]. Причина гормональных изменений, нерегулярности и возрастного прекращения менструальных циклов понятна пока лишь в самых общих чертах [168]. В норме гипоталамус выделяет гонадотропин-рилизинг-гормон, который стимулирует выработку гипофизом лютеинизирующего и фолликулостимулирующего гормонов (ФСГ). Эти гормоны обеспечивают созревание фолликулов в яичниках и их гормональную активность. Фолликулы яичников, в свою очередь, начинают вырабатывать гормоны эстрадиол и ингибин B. Ингибин B подавляет образование ФСГ в гипофизе, и цикл замыкается. Однако количество фолликулов с возрастом снижается, так как их запас ограничен, и секреция ингибина B становится все меньше, а выброс ФСГ все больше. Некоторые фолликулы становятся нечувствительными к избытку ФСГ, что, видимо, способствует нерегулярности циклов и снижению выработки эстрадиола и ингибина B. Таким образом замыкается порочный круг.
Нарушения молекулярно-клеточных механизмов регенерации тканей. Истощение пула стволовых клеток. Одно из негативных последствий старения – снижение регенеративного потенциала тканей [169], обусловленное истощением пула стволовых клеток [170, 171] и связанное с системными факторами старения, механизмами старения самих стволовых клеток, межклеточного матрикса и клеток микроокружения (ниши стволовых клеток) [172, 173].
Системные факторы включают повышение в тканях уровня трансформирующего фактора роста β (TGF-β) [174] и медиаторов воспаления [175], активацию NF-κB-зависимого сигнального пути [138], аккумуляцию в тканях сенесцентных клеток, секретирующих воспалительные факторы, регуляторы роста и металлопротеиназы [176]. Напротив, для поддержания функциональной активности стволовых клеток необходим постоянный уровень циркулирующего окситоцина [177] и дифференцировочного фактора 11 (GDF11) [178, 179], однако он снижается с возрастом. В экспериментах по гетерохронной трансплантации и парабиозу GDF11 способствует омоложению мышечных [178] и нейрональных стволовых клеток [179].
К внутренним причинам старения стволовых клеток относятся накопление токсических метаболитов (АФК), увеличение уровня повреждений ДНК и репликативного стресса, эпигенетические изменения, потеря протеостаза, митохондриальная дисфункция [170, 172].
Изменения клеточного состава стволовых ниш. Гомеостаз стволовых клеток в значительной степени регулируется сигналами, исходящими от ниши стволовых клеток, состоящей из разных типов клеток (например, фиброадипогенные предшественники и миофибриллы для мышечных стволовых клеток) [180], секретируемых ими факторов (WNT/β-катенин, KITL/KIT, EDNs/EDNRB, TGF-β/TGF-βR, α-MSH/MC1R и Notch) и внеклеточного матрикса [181].
При старении изменяется клеточный состав ниш [182], нарушаются сигнальные взаимодействия стволовых клеток с нишей посредством морфогенов и факторов роста [183], ослабляется адгезия между стволовыми клетками и их микроокружением [184], изменяются компоненты внеклеточного матрикса [185], увеличивается уровень провоспалительных маркеров [186]. В результате стволовые клетки утрачивают способность к самообновлению, становятся неустойчивыми к неблагоприятным воздействиям и подверженными аберрантной дифференцировке [187–189].
Например, воздействие генотоксического стресса на стволовые клетки меланоцитов не индуцирует апоптоз или клеточное старение, а запускает их дифференцировку в зрелые меланоциты, что ведет к истощению их пула и проявляется в необратимом поседении волос [190]. Аберрантная дифференцировка мышечных стволовых клеток в фиброгенные способствует формированию фиброза мышечной ткани, ассоциированного со старением [191].
Клеточное старение. Клеточное старение – необратимая остановка клеточного цикла, ограничивающая пролиферативный потенциал клеток [192]. Сенесцентные клетки вовлечены в подавление опухолей, эмбриональное развитие и заживление ран [193]. Накопление сенесцентных клеток в различных тканях является одним из механизмов старения организма [194] и формирования возрастных патологий [195]. Клеточное старение способствует старению организма путем снижения регенеративного потенциала тканей (вследствие истощения пула стволовых клеток) и индукции хронического воспаления (вследствие формирования секреторного фенотипа, ассоциированного со старением (SASP – Senescence Associated Secretory Phenotype)) [196].
Клеточное старение может индуцироваться в ответ на внешние или внутренние стимулы [197]. К внешним стимулам относятся сигналы от других стареющих клеток [198] и воспалительные факторы [199], индукторы деления клеток (гормон роста и другие) [200], метаболические сигналы (высокий уровень глюкозы) [201], факторы стресса (ионизирующая радиация) [202]. Внутренние факторы включают укорочение [203] и повреждение теломер [204], повреждение ДНК [205], хромосомную нестабильность [206], дисфункцию митохондрий [195] и ядерной оболочки [199], повышение уровней АФК [207], индукцию онкогенов [208] и некоторые другие факторы [197, 209].
Молекулярные механизмы индукции клеточного старения и воспаления пересекаются через активацию сигнальных путей TLR/NF-κB, cGAS-STING, AGE-RAGE и сборку инфламмасомы NLRP3. Более подробно они рассмотрены в разделе “Молекулярные механизмы иммуностарения и воспаления”.
Клеточное старение характеризуется нарушением уровня экспрессии генов [210], устойчивостью к апоптозу [211], конститутивным ответом на повреждение ДНК [205], повышенной активностью β-галактозидазы, ассоциированной со старением [212], высоким уровнем экспрессии ингибиторов циклинзависимой киназы (CDK) p16INK4A (CDKN2A) [213] и p21CIP1 (CDKN1A) [214], низким уровнем белка ядерной ламины ламина B1 [199], накоплением липофусцина [215], p53-зависимым высвобождением ядерного белка алармина (HMGB1) [216], очагами повреждения ДНК возле теломер [42], очагами гетерохроматина, ассоциированного со старением [217], дисфункцией митохондрий [207], повышенным уровнем секреции провоспалительных факторов SASP [176, 218]. Более подробно SASP рассмотрен в разделе “Секреторный фенотип сенесцентных клеток”.
Дерегуляция апоптоза. Сенесцентные клетки характеризуются устойчивостью к гибели от вырабатываемых ими проапоптотических факторов [211], несмотря на активацию апоптотических сигнальных путей [219]. Устойчивость сенесцентных клеток к апоптозу (на фоне снижения иммунного клиренса) позволяет им сохраняться в тканях продолжительное время, ведет к нарушению функций тканей и лежит в основе специфических возрастных дегенеративных заболеваний, таких как остеоартрит, фиброз легких, атеросклероз, сахарный диабет и болезнь Альцгеймера [220]. В число факторов, обеспечивающих устойчивость сенесцентных клеток к апоптозу, входят эфрины (EFNB1 или 3), PI3Kδ, p21, BCL-xL или активированный плазминогеном ингибитор-2 [219, 220]. Еще один фактор устойчивости сенесцентных клеток к гибели в ответ на повреждение ДНК – комплекс FOXO4 с p53 в их ядрах [221].
Молекулярные механизмы иммуностарения и воспаления. Для старения человека характерны дерегуляция иммунной системы (иммуностарение) и вялотекущее хроническое воспаление, которое получило название “инфламейджинг” (inflammation – воспаление и aging – старение) [222].
Инволюция тимуса. Физиологический процесс атрофии тимуса считается основным фактором, способствующим возрастной дерегуляции иммунной системы [223]. Инволюция тимуса приводит к снижению эффективности тимопоэза, нарушению миграции наивных Т-клеток на периферию, сокращению разнообразия антигенного репертуара Т-клеток и снижению адаптивного иммунитета в пожилом возрасте. Одной из основных причин возрастной инволюции тимуса может быть нарушение межклеточной коммуникации, выражающееся в снижении уровня секреции трофических цитокинов, таких как IL-7, стареющими клетками микроокружения и повышении уровня тимосупрессорных цитокинов, таких как лейкоз-ингибирующий фактор (LIF), онкостатин М, фактор стволовых клеток (SCF) [224]. В качестве терапевтических средств, направленных против инволюции тимуса, рассматриваются прием цинка, введение IL-7, IL-22, фактора роста кератиноцитов (KGF), гормональная терапия (лептин и гормон роста), ингибирование половых стероидов [225, 226].
В качестве биомаркеров старения клиническое значение имеют уменьшение доли наивных T-клеток в результате инволюции тимуса и накопление маркеров “усталости” иммунитета, CD28null CD8 T-клеток вследствие перенесенных инфекций [227, 228].
Хроническое воспаление. Для системного воспаления характерны высокие уровни IL-1 и IL-6, TNF-α и С-реактивного белка [229]. Хроническое воспаление считается фактором риска старческой хрупкости (frailty) и широкого спектра заболеваний, включая гипертензию, сахарный диабет второго типа, атеросклероз, болезнь Альцгеймера и онкологические заболевания [229, 230]. Потенциальные причины воспаления, ассоциированного со старением, включают генетическую предрасположенность, клеточное старение, окислительный стресс, ожирение, дерегуляцию иммунной системы, нарушение целостности гастроинтестинального барьера, изменение состава микробиоты и хронические инфекции [223, 231, 232]. Сигналами активации воспалительного ответа являются молекулярные структуры, ассоциированные с микроорганизмами (MAMP – Microbe-Associated Molecular Patterns) или с повреждениями клеток и тканей (DAMP – Damage-Associated Molecular Patterns) [230, 233]. К MAMP относятся липополисахариды, липопептиды, бактериальные белки и нуклеиновые кислоты бактериального и вирусного происхождения [230]. В состав DAMP входят эндогенные белки (HMGB1, HSP, S100, сывороточный амилоид А и гистоны), небелковые молекулы (АТP, мочевая кислота), свободно циркулирующие ДНК (геномная и митохондриальная) и РНК [233]. Эти воспалительные сигналы распознаются рецепторами врожденного иммунитета, включая Toll-подобные рецепторы (TLR), NOD-подобные рецепторы (NLR), сенсоры ДНК (cGAS и AIM2) и РНК (RIG-I-подобные рецепторы), RAGE и некоторые другие [234].
TLR могут быть активированы нуклеиновыми кислотами в эндосомах, эндогенными белками и компонентами внеклеточного матрикса [234]. TLR передают сигналы через адаптерные белки MyD88 и TRIF, которые рекрутируют и активируют MAPK и киназу IκB (IKK), что приводит к последующей выработке воспалительных цитокинов TNF, IL-6, IL-1 и IL-12 посредством активации факторов транскрипции AP-1 и NF-κB соответственно [230]. Кроме того, TRIF рекрутирует и активирует киназу TBK1, что приводит к продукции интерферона типа I (IFN-I) с помощью фактора транскрипции IRF3 [234].
Активация NLR и сборка инфламмасомы NLRP3 происходит в ответ на различные эндогенные стимулы, такие как кристаллы урата натрия, кристаллы холестерина, β-амилоид (Aβ) и АТP, которые могут индуцировать внутриклеточные дефекты, например, высвобождение содержимого лизосом, повреждение митохондрий, фрагментацию комплекса Гольджи [235]. Сборка инфламмасомы NLRP3 приводит к активации воспалительных каспаз, что запускает созревание и секрецию воспалительных цитокинов IL-1β и IL-18. NLRP3 также может инициировать воспалительную форму гибели клеток (пироптоз) [230].
Активация cGAS происходит не только в ответ на вирусную и бактериальную ДНК, но и, что важно с точки зрения старения, при возрастзависимой активации ретротранспозонов LINE-1, а также при повреждении и утечке в цитоплазму собственной ДНК из фагосом, митохондрий и ядра [236–238]. Активация cGAS индуцирует образование вторичного посредника, cGAMP (циклического GMP-AMP), который активирует STING – стимулятор экспрессии генов IFN (Stimulator of Interferon Genes). Активированный STING, в свою очередь, активирует TBK1 и IKK, что приводит к продукции IFN и воспалительных цитокинов через факторы транскрипции IRF3 и NF-κB [239]. В отличие от cGAS активация AIM2 в ответ на повреждение ДНК или присутствие ДНК в цитоплазме инициирует сборку инфламмасомы [240].
Эндогенными лигандами RAGE являются не только КПГ, но и другие DAMP [234], а предполагаемым адаптерным белком RAGE считается mDia-1 (diaphanous 1) [241]. Пути передачи сигналов RAGE зависят как от лиганда, так и от типа клеток, но, в целом, активация RAGE может инициировать различные сигнальные пути, включая PI3K-AKT, MAPK и JAK-STAT. Эти сигнальные пути индуцируют различные факторы транскрипции, такие как NF-κB, AP-1 и STAT3, которые способствуют экспрессии провоспалительных генов, а также миграции клеток, пролиферации и апоптозу [242].
Активация миелоидных клеток. Один из механизмов, ответственных за хроническое воспаление при старении, связан с повышенным количеством миелоидных клеток в костном мозге, крови и вторичных лимфоидных органах [243]. Избыточное формирование миелоидных клеток обусловлено функциональными изменениями в нишах гемопоэтических стволовых клеток (ГСК), такими как старение микроокружения, включая межклеточный матрикс, несбалансированная дифференцировка мезенхимальных стромальных клеток костного мозга, ремоделирование сосудов, нарушение передачи сигналов с участием β-адренергических рецепторов и секреция провоспалительных факторов [182, 244]. Увеличение уровня нестабильности генома может приводить к злокачественной трансформации миелоидной линии ГСК и развитию миелопролиферативных нарушений [223].
Секреторный фенотип сенесцентных клеток. Сенесцентные клетки имеют определенный секреторный фенотип, именуемый SASP [176, 218], или SMS (Senescence-Messaging Secretome – секретом сенесцентных сообщений) [245]. SASP представляет комбинацию растворимых сигнальных факторов (воспалительные цитокины, хемокины, факторы роста), секретируемых протеаз и нерастворимых секретируемых белков/компонентов внеклеточного матрикса [176]. Недавнее исследование, проведенное под руководством Бёрджит Шиллинг, позволило создать протеомный атлас секретома SASP, который насчитывает свыше полутора тысяч растворимых белков (sSASP) и сопоставимое количество факторов, упакованных в экзосомы (eSASP) [218]. При составлении атласа секретома SASP выявлено несколько потенциальных биомаркеров клеточного старения, которые перекрываются с маркерами старения в плазме человека, включая фактор роста/дифференцировки 15 (GDF15), станниокальцин 1 (STC1) и серпины (SERPIN) [218]. По-видимому, существует многоуровневый контроль активности секретома SASP, который включает сигнальные пути опухолевых супрессоров, NF-κB-зависимый путь и ответ на повреждения ДНК [246, 247]. Сенесцентные клетки влияют на микроокружение паракринно и посредством “эффекта свидетеля”, индуцируя ключевые патологические признаки старения, включая сенесцентность, хроническое воспаление, онкогенез и нарушение самообновления стволовых клеток [205].
Секреторный фенотип адипоцитов. Белая жировая ткань рассматривается в настоящее время как эндокринный орган, состоящий из зрелых и развивающихся адипоцитов, а также фибробластов, эндотелиальных клеток и широкого спектра иммунных клеток, включая макрофаги жировой ткани, нейтрофилы, эозинофилы, тучные клетки, Т- и В-клетки [248]. В норме межклеточная коммуникация между адипоцитами и клетками иммунной системы способствует поддержанию тканевого гомеостаза. В частности, эозинофилы и регуляторные T-клетки секретируют цитокины (IL-10 и IL-4), которые направляют дифференцировку макрофагов жировой ткани к противовоспалительному фенотипу M2, а секретируемый жировыми клетками адипонектин повышает чувствительность к инсулину [248]. При ожирении адипоциты увеличиваются в размерах (гипертрофия адипоцитов), что приводит к их гипоксии с последующей продукцией хемоаттрактантов (CCL2, MIF), привлекающих макрофаги, Т- и В-клетки [249]. Т-клетки активируются, фенотип макрофагов изменяется с M2 на M1, продуцирующих воспалительные цитокины (IL-6 и TNF-α), способствующие формированию инсулинорезистентности [248, 249]. Жировые клетки увеличивают секрецию воспалительных факторов (лептин, липокалин-2 и програнулин) и снижают выработку противовоспалительного адипонектина [250–252]. Наравне с другими механизмами индукции локального и системного воспаления ожирение является одним из ключевых факторов развития таких метаболических заболеваний, как сахарный диабет второго типа, псориаз, атеросклероз, гипертензия и гиперлипидемия [253].
Дисбаланс микробиоты. Согласно современным представлениям, микробиом желудочно-кишечного тракта (ЖКТ) может играть ключевую роль в возрастном воспалении [254, 255]. У здорового взрослого человека поддерживается гомеостаз комменсальной микрофлоры ЖКТ (эубиоз), представленной бактероидами, бифидобактериями и лактобациллами [255]. Микрофлора активирует механизмы, способствующие поддержанию физической и функциональной целостности гастроинтестинального эпителиального барьера [256]. Продуцируемый лимфоидными клетками кишечника IL-22 [257] активирует сигнальный путь JAK-STAT в эпителиальных клетках кишечника, индуцируя таким образом экспрессию генов, регулирующих иммунный ответ (TLR) [258], пролиферацию, заживление ран и апоптоз, поддерживает плотные контакты между эпителиальными клетками кишечника (клаудин-1) [256], активирует выработку антимикробных пептидов (дефензины, Reg3β, Reg3γ и S100) [259] и гликопротеинов слизи (муцины Muc1, 3, 10 и 13) [260]. Продуцируемая эпителиальными клетками кишечника щелочная фосфатаза необходима также для поддержания гомеостаза микробиома, сохранения барьерной функции кишечника и уменьшения воспаления [261]. Дендритные клетки индуцируют активацию и дифференцировку наивных В-клеток с образованием плазматических клеток, продуцирующих иммуноглобулин A (IgA) [262]. IgA секретируется в просвет кишечника, где связывается с комменсальными микроорганизмами, ограничивая их прилипание к эпителию и проникновение через гастроинтестинальный барьер [263]. Кроме того, микрофлора ЖКТ продуцирует короткоцепочечные жирные кислоты (SCFA), такие как масляная, пропионовая и уксусная, а также их производные (бутираты, пропионаты и ацетаты соответственно), которые оказывают иммуномодулирующее и противовоспалительное действие на эпителий кишечника и выполняют роль энергетического субстрата [255, 264].
С возрастом нарушается целостность гастроинтестинального барьера и изменяется состав микроорганизмов ЖКТ: снижается разнообразие видов основных таксонов, уменьшается доля комменсалов, увеличивается доля условно-патогенной флоры (стрептококки, стафилококки, энтерококки и энтеробактерии) [254, 265]. Эти изменения способствуют попаданию кишечных бактерий и их токсинов в систему кровообращения, что увеличивает антигенную нагрузку и ведет к активации врожденного иммунитета через сигнальный путь cGAS–STING, хроническому воспалению, иммуностарению, инсулинорезистентности и развитию метаболических заболеваний [266, 267].
Профилактические и терапевтические вмешательства, направленные на поддержание гомеостаза микробиоты кишечника, сосредоточены на использовании пребиотиков и метабиотиков (ингредиенты пищи, которые стимулируют рост и активность микробиоты и продукцию SCFA) [268] и гетерохронной трансплантации микробиомов фекалий [269]. Предполагается, что положительные эффекты таких потенциальных геропротекторов, как метформин [270], рапамицин [271] и ресвератрол [272] могут быть обусловлены их влиянием на состав кишечной микрофлоры.
Виром. Кишечник человека содержит не только бактерии, но и на порядок большее количество ДНК- и РНК-содержащих вирусов, совокупность которых составляет кишечный виром [273]. Виром включает вирусы эукариот, эндогенные ретровирусы, вирусы бактерий (бактериофаги) и вирусы архей [274]. Бактериофаги могут лизировать как комменсальные, так и патогенные бактерии и в значительной степени модулировать состав микробиома кишечника человека [273]. Возрастзависимые параллельные изменения компонентов вирома и бактерий ЖКТ указывают на существование дополнительных уровней регуляции гомеостаза микробиоты [274].
Виром принимает участие в старении, не только определяя ландшафт микрофлоры кишечника, но и повышая риск иммуностарения, развития атеросклероза (цитомегаловирус), онкологических (папилломавирусы) и нейродегенеративных (герпесвирус) заболеваний [228, 275–277].
Системные проявления старения
Возрастные нарушения биологических ритмов. В процессе эволюции у живых организмов сформировались различные молекулярные хронометры, связанные с рецепторными структурами, что позволяет синхронизировать процессы жизнедеятельности различных уровней со световым режимом. В центральном осцилляторе эукариотической клетки главную роль играет комплекс транскрипционно-трансляционных петель обратной связи, в котором сигнал входит через рецепторную молекулу, включенную в состав репрессорного димера или тримера. Этот димер или тример, в свою очередь, блокирует комплексный фактор транскрипции, индуцирующий экспрессию генов различных метаболических путей [278]. Центральный осциллятор контролирует 12–40% кодирующих последовательностей ДНК в геноме разных видов животных [279]. Например, репрессор клеточных часов CRY1 подавляет глюконеогенез в печени, PER2 контролирует липидный обмен, рецепторы ядерных гормонов REV-ERB ограничивают скорость метаболизма жирных кислот и холестерина [278]. Нарушение уровней CLOCK и BMAL1 связывают с ожирением, гиперинсулинемией и диабетом, циркадный посттранскрипционный регулятор Nocturnin также контролирует метаболизм липидов и холестерина [280]. Напротив, мутации per0 и tim0 продлевают жизнь у самцов дрозофилы посредством индукции ассоциированного с UCP4C механизма разобщения мембранного потенциала и окислительного фосфорилирования митохондрий, препятствующего гиперпролиферации клеток кишечника [281], что открывает новые молекулярные мишени для разработки геропротекторов. Функции центральных и периферических часов ослабляются в процессе старения [282, 283]. Нарушение циркадной ритмичности может способствовать риску возрастзависимых заболеваний и развитию синдрома старческой хрупкости [284–286].
Десинхроноз можно регулировать, строго соблюдая режим приема пищи и правила гигиены сна. Недостаточный или фрагментированный сон увеличивает риск множественных патологических состояний, включая сердечно-сосудистые заболевания [287]. Крепкий сон способствует гемопоэзу и оказывает антиатеросклеротическое действие [287]. Сон позволяет сохранять повышенную активность иммунных Т-клеток [288], в то время как недосыпание способствует повышению активности воспалительных Th2-лимфоцитов [289]. Сон способствует синтезу проколлагена в соединительной ткани [290], репарации ДНК в нейронах [291, 292], питанию и очищению тканей мозга за счет омывания спинномозговой жидкостью через лимфатическую систему [293], снижению накопления тау-белка и Aβ, вызывающих болезнь Альцгеймера [294].
Напротив, острая потеря сна приводит к повышению уровня тау-белка в крови [295] и ночных уровней Aβ в спинномозговой жидкости по сравнению с “выспавшимся” контролем [296]. Более короткий и нерегулярный сон связаны с ускоренным эпигенетическим старением [297].
Нарушения циркадных ритмов при старении, известные как возрастной десинхроноз, корректируются в некоторых случаях приемом гормона мелатонина, который при правильном употреблении восстанавливает амплитуду циркадного ритма и синхронизирует ее с режимом освещения. Мелатонин достаточно давно известен в качестве потенциального геропротектора [298].
Возрастные нарушения метаболизма и хронический стресс-ответ. В качестве основных факторов возрастной дерегуляции метаболизма традиционно рассматривают элементы сигнальных путей mTOR, AMPK, SIRT и инсулин/IGF-1 [299]. Наибольший интерес вызывает путь mTOR [300], участвующий в распознавании нутриентов, регуляции клеточного роста и аутофагии [301].
Cенсоры нутриентов, такие как mTOR, AMPK и сиртуины, взаимодействуют с рецептором гамма коактиватора 1α, активируемого пролифератором пероксисом (PGC1α). Данный белок является регулятором транскрипции, который участвует в стимуляции биогенеза митохондрий [302], окислительного фосфорилирования, в контроле деления и слияния митохондрий, а также количества копий митохондриального генома [303, 304].
PGC1α, в свою очередь, контролирует экспрессию ядерных дыхательных факторов 1 и 2 (NRF1 и 2), рецептора-γ, активируемого пролифератором пероксисом (PPARγ) или фактора транскрипции митохондрий A (TFAM) [305]. Когда клетки находятся в условиях ограниченного поступления питательных веществ AMPK и SIRT1 прямо активируют PGC1α посредством фосфорилирования и деацетилирования соответственно. AMPK и mTORC1 разнонаправленно контролируют удаление поврежденных митохондрий посредством митофагии [305, 306].
В удалении дефектных митохондрий важную роль играют факторы транскрипции семейства FOXO, участвующие в регуляции роста, пролиферации, дифференцировки и стрессоустойчивости клеток, а также состояния покоя стволовых клеток [307]. Регуляция белков FOXO, связанная с такими путями, как mTOR, AMPK и сиртуины, посредством снижения калорийности питания [308], приема полифенолов или регулярного выполнения физических упражнений, способствует предотвращению дисфункции митохондрий.
Декомпартментализация клеток и утрата барьерной функции в процессе старения. Нарушения структуры и функции ядерных пор. Комплекс ядерных пор состоит из нуклеопоринов [309]. Нуклеопорины периферических компартментов имеют короткий период жизни, тогда как нуклеопорины каркаса ядерной поры – долгоживущие белки с ограниченной способностью к обновлению и репарации, особенно в постмитотических клетках [310]. Активный ядерный транспорт макромолекул внутрь и за пределы ядра через комплекс ядерных пор обеспечивается GTP-связывающим ядерным белком Ran, который взаимодействует с нуклеопоринами в ядерной оболочке. Комплекс ядерных пор взаимодействует с хроматином в процессе регуляции глобальной организации хроматина и экспрессии генов, специфичной для типа клетки [309]. Поскольку белки ядерных пор накапливают повреждения при старении [311], вероятно, что их неферментативные модификации приводят к связанным со старением изменениям в эпигенетическом ландшафте [312].
Эпителиальные барьеры. С возрастом нарушается целостность эпителиальных барьеров нескольких органов, включая кожу, легкие, ЖКТ и почки [313], что связывают со снижением экспрессии генов белков, влияющих на формирование межклеточных контактов – ZO-1, окклюдина, JAM-A, клаудинов-1/7, кадгеринов E и N, катенинов a и b [313].
Роговой слой кожи обычно имеет “кислотную мантию” (кислые значения рН) [314], обеспечивающую барьерный гомеостаз проницаемости, противомикробную защиту и активацию первичных цитокинов. Повышение pH неизменно увеличивает проницаемость кожного барьера. В состарившихся участках кожи снижается образование IL-1α, необходимого для барьерной функции клеток эпидермиса [313]. Снижение выработки цитокинов и ответных реакций фибробластов и кератиноцитов на цитокины может быть обусловлено клеточным старением, в том числе при повреждении ДНК ультрафиолетом. Ультрафиолетовое излучение, высокие уровни восстанавливающих сахаров (глюкозы, фруктозы, галактозы) в крови вызывают неферментативные изменения матрикса, способствуя снижению эластичности кожи и увеличению ее морщинистости [315].
Почечный барьер. Возрастные нарушения целостности почечного барьера обусловлены изменением уровня экспрессии генов, кодирующих белки межклеточной адгезии (кадгерин/катенин) и компоненты плотных контактов (клаудины) [313, 316–319]. Снижение числа подоцитов, наблюдаемое у стареющих крыс [320], может влиять на гломерулярный фильтрационный барьер, а снижение калорийности питания приводит к отмене этих изменений.
Эндотелиальный барьер в сосудах. В процессе старения нарушается целостность не только эпителиальных, но и эндотелиальных барьеров [321], причиной этому служит дерегуляция экспрессии ряда белков. Например, PPARγ необходим для предотвращения эндотелиальной дисфункции [322]. Гликопротеин CD9 активируется в стареющих эндотелиальных клетках, в гиперплазированной неоинтиме и атеросклеротических бляшках. Так, в артериях людей и крыс и в атеросклеротических бляшках людей и мышей наблюдается возрастное повышение экспрессия CD9. Активация CD9 в клетках вызывала проявление сенесцентного состояния посредством сигнального пути PI3K-AKT-mTOR-p53. Антитела к CD9 мыши заметно снижали образование атеросклеротических поражений у мышей ApoE–/– и Ldlr–/– [323].
Кроме того, выявлены изменения в экспрессии и уровнях важных плазменных компонентов эндотелия, связанные с процессами гемостаза. К ним относятся изменения в метаболизме оксида азота и простаноидов, эндотелина-1, тромбомодулина и фактора Виллебранда. Эти изменения усиливают прокоагулянтный статус, развивающийся при старении, подчеркивая роль эндотелия в развитии возрастных тромбозов [324].
Гематоэнцефалический барьер. Повышенная проницаемость гематоэнцефалического барьера (ГЭБ), занимает, вероятно, центральное место в возрастном повреждении паренхимы головного мозга. Проницаемость ГЭБ для молекул различного размера (флуоресцеин натрия, инулин и других) у мышей с возрастом увеличивается, что сопровождается изменениями в организации плотных контактов, снижении экспрессии клаудина-5 [325]. Это может быть связано с многократным уменьшением уровней экспрессии SIRT1. Сверхэкспрессия или повышение активности SIRT1 с помощью агониста SRT1720 защищают от индуцированной старением сверхпроницаемости эндотелиального барьера головного мозга [325], таким образом SRT1720 способен продлевать жизнь [326].
Дисфункция ГЭБ, в свою очередь, вызывает гиперактивацию TGF-β в астроцитах [327], что может приводить к дисфункции нервной системы и возрастной патологии у грызунов, в том числе к когнитивным нарушениям [328].
Не последнюю роль в нарушении ГЭБ играет дерегуляция транспортных механизмов. К основополагающим механизмам стоит отнести транспорт глюкозы, инсулина и Aβ [329]. При помощи ПЭТ с 18F-флуородеоксиглюкозой показано возрастное снижение транспорта глюкозы во фронтальном и темпоральном отделах коры, что ассоциировано со снижением экспрессии GLUT1 и стойкой когнитивной дисфункцией [330].
Накопление и отложение белка Aβ в мозге рассматривается как основной патологический признак болезни Альцгеймера, способствующий развитию нейродегенерации. ГЭБ экспрессирует системы транспорта Aβ, которые обеспечивают как его поступление, так и выход из ЦНС – связанный с рецептором липопротеинов белок 1 (LRP-1) и P-гликопротеин. Показано, что в микрососудах мозга экспрессия LRP-1 снижается с возрастом и при болезни Альцгеймера [329, 331]. Функция P-гликопротеина снижается у пожилых людей, а также у старых мышей. В совокупности эти изменения в ГЭБ могут способствовать накоплению Aβ в мозге. Также известно, что системное воспаление у молодых мышей может способствовать нарушению оттока Aβ [332].
В мозге инсулин действует как трофический фактор, он контролирует пищевое поведение, обучаемость и память. В мозге инсулин не синтезируется, он поступает из крови с помощью транспортных систем ГЭБ. Концентрация инсулина в тканях мозга в целом снижается с возрастом [333]. Повышение концентрации инсулина в сосудистом пространстве наблюдали в теменной коре, мозжечке и таламусе старых мышей SAMP8, что указывает на возможность избыточного связывания инсулина с эндотелиальными клетками сосудов в этих участках мозга при старении [334].
Общепризнан вклад сосудистых изменений в деменцию и болезнь Альцгеймера. Недавние исследования указывают на разрушение ГЭБ как на биомаркер ранней когнитивной дисфункции человека, включая ранние клинические стадии болезни Альцгеймера. Ген APOE4 кодирует вариант Е4 аполипопротеина Е – важнейший фактор риска болезни Альцгеймера, который приводит к ускоренному распаду ГЭБ и дегенерации перицитов капилляров мозга, поддерживающих целостность ГЭБ [335].
Гастроинтестинальный барьер. Кишечный эпителиальный барьер защищает слизистую оболочку ЖКТ и играет ключевую роль в поддержании гомеостаза хозяина. Этот барьер включает несколько элементов: кишечный эпителий и биохимические и иммунологические продукты, такие как слой слизи, антимикробные пептиды и секреторный иммуноглобулин A (sIgA) [336]. Эти компоненты взаимодействуют с микробным сообществом, населяющим кишечник, вместе они формируют очень сложную систему, которая играет важную роль в поддержании здоровья человека как локально, так и на уровне организма. В течение жизни изменяется регенерация стволовых клеток кишечника, регуляция гомеостаза крипт кишечника, целостность барьера, выработка регуляторных цитокинов и состояние врожденного иммунитета эпителия к патогенным антигенам [337]. Данные о возрастзависимом изменении экспрессии ключевых барьерных генов противоречивы [338].
БИОМАРКЕРЫ СТАРЕНИЯ
Молекулярные биомаркеры повреждений и ошибок, возникающих при старении
Современные методы молекулярной биологии позволяют достаточно точно оценить уровень специфических повреждений ДНК и соматических мутаций в клетках человека (в том числе в клетках, циркулирующих в кровотоке). С этой целью применяют секвенирование одиночных клеток, анализ репарации ДНК и маркеров ответа на повреждение ДНК (например, фосфорилированного гистона γ-H2AX), модифицированных нуклеотидов ДНК (8-гидрокси-2'-дезоксигуанина). Тем не менее, требуется валидация и стандартизация этих методов, прежде чем они смогут использоваться в исследованиях на людях и в клинической практике в качестве биомаркеров старения [21, 141, 339]. Укороченные теломеры считаются одним из ключевых маркеров клеточного старения. Однако на уровне организма длина теломер рассматривается как слабый биомаркер, так как направление и величина изменений средней длины теломер различаются в разных клетках и тканях и крайне неоднородны в популяции даже одного возраста [141, 340]. С другой стороны, скорость укорочения теломер можно использовать для оценки риска развития заболеваний сердечно-сосудистой системы и связанной с ними смертности пожилых людей [341]. Информативным биомаркером старения может быть снижение активности теломеразы на уровне мРНК и фермента [342]. Кроме того, идентифицированы дополнительные маркерные белки, связанные с теломерной дисфункцией (например, CRAMP, статмин, EF-1α, хитиназа). Экспрессия этих белков увеличивается в плазме крови пожилых людей и пациентов с заболеваниями, связанными со старением [343]. В качестве биомаркеров старения можно рассматривать повышенную выработку прогерина и преламина А, снижение уровня ламина В1 [50, 339].
Достаточно точно оценить биологический возраст и состояние здоровья человека позволяют эпигенетические биомаркеры [141, 344]. Метилирование ДНК в GpG-участках считается одним из наиболее надежных маркеров определения хронологического и биологического возраста [69, 73, 141, 345]. Метилирование, легко оцениваемое в циркулирующих клетках (с использованием микрочипов и секвенирования), относительно стабильно в популяции. В настоящее время описаны достаточно точно настроенные эпигенетические подписи (signatures), которые можно использовать для отслеживания старения как отдельных клеток, так и организма (например, PhenoAge и GrimAge) [141, 344–348]. Метилирование ДНК может отражать ускоренное старение человека и предсказывать смертность от всех причин [73, 349–351], риск развития возрастных заболеваний [73, 141, 352, 353], изменение темпов старения, вызванное коррекцией образа жизни [354]. Кроме того, в тканях стареющих мышей и людей повышено содержание 5-гидроксиметилцитозина, образующегося при активном деметилировании ДНК [355, 356].
Маркерами старения могут служить репрессивные (H3K9me3, H3K27me3) и активирующие метки гистонов (H3K4me3, H4K16ac), варианты гистонов (например, H3.3), а также экспрессия хроматин-модифицирующих факторов (в частности, SIRT1, JMJD3) [69, 357, 358]. Выявлены специфические изменения ряда микроРНК при старении и возрастзависимых заболеваниях (в том числе циркулирующих) и длинных некодирующих РНК. Изменение паттернов экспрессии микроРНК можно использовать для оценки эффективности терапевтических и геропротекторных вмешательств [357–359]. В то же время методы анализа модификаций гистонов и оценка экспрессии некодирующих РНК более трудоемкие, дорогие и не полностью стандартизированные. К тому же эпигенетические модификации подвержены быстрым изменениям в течение короткого промежутка времени [141].
Разработаны протеомные часы старения, состоящие из белков, количественное содержание которых в плазме человека изменяются с возрастом. Способность этих белков точно предсказывать возраст человека подтверждена на большой выборке лиц разного возраста [360]. Биомаркеры старения, связанные с состоянием протеома, включают также модификации белков (в частности, под влиянием конечных продуктов гликирования), уровни молекулярных шаперонов (метионин-сульфоксид-редуктаза), активность протеасом (например, 26S) и аутофагосом. Эти показатели можно определить в плазме и клетках крови. Кроме того, показательной может быть оценка модификаций долгоживущих белков внеклеточного матрикса. Однако в настоящее время использование этих параметров в качестве биомаркеров старения либо недостаточно валидировано, либо дорого и сложно [94, 141, 339]. Исключение представляет анализ КПГ, содержание которых можно достаточно точно оценить с помощью совмещенной технологии жидкостной хроматографии и тандемной масс-спектрометрии, а также флуоресценции КПГ, иммуноферментного анализа и Вестерн-блотинга [141, 361]. Еще один биомаркер старения, связанный с нарушением протеостаза (аутофагийно-лизосомной функции) и накоплением окислительных повреждений в клетке – липофусцин [109].
Метаболическую функцию митохондрий (степень окислительного фосфорилирования) можно оценить in vitro методом респирометрии высокого разрешения биопсийных образцов пермеабилизированных волокон скелетных мышц, и in vivo с помощью ЯМР-спектроскопии на ядрах фосфора-31. Однако первый метод инвазивный, а второй достаточно дорогостоящий [141]. Количество копий мтДНК и степень гетероплазмии, оцениваемые в клетках крови и в биопсийных образцах тканей, также являются важным показателями физиологии митохондрий, скорости их репликации и целостности генома [141, 339]. Биомаркером, отражающим стабильность протеостаза митохондрий, может быть экспрессия протеазы Lon [339]. В целом, показатели функции митохондрий представляются удобными маркерами биологического старения, но также нуждаются в стандартизации [141].
Системные биомаркеры старения, старение функциональных систем
Большое внимание привлекают метаболические биомаркеры старения, обнаруживаемые в образцах крови [362], например 1,5-ангидроглюцитол, диметилгуанозин, ацетилкарнозин, карнозин, глицилглицин, UDP-ацетилглюкозамин, N-ацетиларгинин, N6-ацетиллизин, пантотенат, цитруллин, лейцин, изолейцин, NAD+ и NADP+. Метаболизм липидов подвергается возрастной дерегуляции, а повышение содержания триглицеридов жирных кислот позволяет рассматривать их как еще один биомаркер старения. Холестерин и липопротеины высокой и низкой плотности являются предикторами возрастных заболеваний и смертности. Биомаркерами здорового старения признаны фосфо- и сфинголипиды [363, 364]. Маркеры окислительного повреждения широко представлены в стареющем в организме [365]. Такие соединения, как 3-нитротирозин, 3-хлортирозин и о-тирозин, тесно связаны с повреждением белков при старении. Малоновый альдегид, 4-гидрокси-транс-гексеналь, 4-гидрокси-транс-ноненаль, 8-изопростагландин F2α и альдегиды C6-C12 отражают окислительные повреждения липидов. Такие соединения, как 8-гидрокси-2'-дезоксигуанозин и 8-гидроксигуанозин, появляются при повреждении ДНК окислительными стрессорами, поэтому их можно использовать в качестве индикаторов возрастного окислительного повреждения [365]. Сравнительный анализ надежности, информативности и уровня доказательности большинства из приведенных биомаркеров старения приведен в работе Э. Кримминс и соавт. [366].
Возрастные изменения сердечно-сосудистой системы на структурном уровне представлены компенсаторной концентрической гипертрофией левого желудочка [367], связанной с утратой кардиомиоцитов. Дальнейшее развитие гипертрофии приводит к фиброзу миокарда, аритмиям, сердечной недостаточности [368]. В процессе старения гипертрофируются и подвергаются дилатации также предсердия, что связано в значительной степени с ремоделированием миокарда и ведет к прогрессированию сердечной недостаточности с сохраненной фракцией выброса и атриальной фибрилляции [369]. Биомаркерами старения и прогрессирования сердечной недостаточности наряду с предсердным натрийуретическим пептидом (BNP) могут служить серологические маркеры фиброза [370]. Информативны измерения уровня коллагена первого типа в тканях предсердий и желудочков, тканях сосудов [371]. Для гистопатологической картины старения характерно присутствие в тканях сердечно-сосудистой системы амилоида (он формируется из молекул BNP), количество которого коррелирует с возрастом [372]. Известно множество молекулярных маркеров старения сердечно-сосудистой системы, включенных в сигнальный путь кальция (например, изменения уровня RyR2 [373], SERCA, отношения SERCA и фосфоламбана [374]). Для стареющей сердечной ткани характерна избыточная активация нейрогормонального сигнального пути, который считается точкой сопряжения старения сердечно-сосудистой, выделительной и эндокринной системы органов и имеет две ветви: ренин-ангиотензин-альдостероновую систему и β‑адренергический сигнальный путь. Повышение уровня циркулирующих катехоламинов и снижение частоты встречаемости β-адренорецепторов (β‑адренергическая десенситизация) свойственны патологической картине старения [375].
В стареющих сосудах активируются механизмы ответа на окислительные повреждения, главным образом, провоспалительные: активируется NF-κB, подвергается дерегуляции SIRT1, повышается концентрация инфламмасом, наблюдается дисфункция NRF2, продукция SASP-ассоциированных молекул, сверхактивация TLR-accоциированного сигнального пути [376]. В морфологической картине эти молекулярные изменения выражаются в возрастном атерогенезе, аневризмогенезе, апоптотической гибели клеток, вкупе со множественными паракринными эффектами и нарушением эндотелиальных барьеров [376].
В системе циркулирующей крови также выявлены многочисленные биомаркеры старения – внеклеточные везикулы, белки крови и внеклеточные нуклеиновые кислоты [377].
Возрастные изменения легких включают истощение пула легочных стволовых клеток, старение соматических и иммунных клеток, ремоделирование внеклеточного матрикса, изменения состава системы сурфактанта, мукоцилиарной и легочной нервной системы, фиброзные изменения [378]. Немаловажен для дыхательной системы иммунный статус, ассоциированный со старением, в формировании которого участвуют несколько механизмов, подверженных дерегуляции в процессе старения, куда входит сигнальный каскад TLR и DAMP, сниженная активность NRF2 и повышенная – NF-κB [378]. Введение бактериального липополисахарида в дыхательные пути старых мышей вызывало мгновенный подъем концентраций CXCL1, CXCL2, IL-1β и легочную нейтрофилию, у молодых мышей подобной реакции не наблюдалось [379].
Пищеварительная система имеет собственный набор возрастных изменений: для нее характерны атрофические, метапластические, гиперпластические и диспластические явления, нередко обнаруживаются малигнизированные клетки, микроскопически отмечается расширение крипт, макроскопически – образование дивертикулов, птозы желудка и петель кишечника [380]. Во многом к этим последствиям приводит старение стволовых клеток кишечника и ниш [380]. Часто возникают и разнообразные функциональные расстройства [381]. Значимым количественным и качественным изменениям подвергается микробиота кишечника [382]. В качестве системных биомаркеров старения кишечника используют, главным образом, медиаторы воспаления IL-6 и IL-8, рецептор IL-6sR, гормон адипонектин и маркеры микробной инвазии LBP и CD14 [383]. Сама микробиота также может служить биомаркером старения [384], а ее пересадка от молодого здорового донора пожилому или страдающему прогерией реципиенту признана эффективной противовозрастной интеревенцией [385].
ИНТЕРВЕНЦИИ
Нефармакологические интервенции
Наиболее универсальной и изученной интервенцией для увеличения продолжительности жизни и замедления развития возрастзависимых заболеваний является ограничение калорийности пищи. Во-первых, снижение калорийности питания активирует экспрессию регуляторных белков (например, PPARγ, PGC-1α, AMPK, FOXO, сиртуины и других), которые стимулируют адаптивную реакцию организма [69, 355, 386, 387]. Во-вторых, низкокалорийная диета снижает уровень повреждений макромолекул клетки и митохондрий, например, за счет ослабления перекисного окисления липидов [388], а, в-третьих, влияет (как и прерывистое голодание или кетогенная диета) на эпигенетический ландшафт клеток и организма, способствуя выработке бета-гидроксибутирата – ингибитора гистондеацетилаз, и предотвращает многие изменения эпигенома, связанные со старением [69, 355, 386, 389, 390]. В-четвертых, снижение калорийности служит одним из центральных триггеров аутофагии [98, 101, 102], а также благоприятно влияет на другие механизмы обеспечения протеостаза, например, улучшает фолдинг белков в ЭПС [104]. Наконец, данная интервенция ослабляет митохондриальную дисфункцию, стимулирует биогенез митохондрий, уменьшает выработку АФК [391]. Частично эти эффекты могут вызывать фармакологические миметики ограничения калорий, например, активаторы сиртуинов, индукторы аутофагии, рапамицин, метформин [69, 387].
Ограничение питания способствует снижению уровня инсулина и IGF-1, ингибированию активности mTOR [392–394], активации AMPK и SIRT1 [395, 396], что позволяет улучшить состояние здоровья, отсрочить проявление возрастзависимых патологий, а в некоторых случаях увеличить продолжительность жизни. Геропротекторным потенциалом обладает также фармакологическое ингибирование всех компонентов сигнального пути инсулин/IGF-1, специфическое подавление mTOR рапамицином или, напротив, активация AMPK метформином и SIRT1 ресвератролом [339]. На различных модельных организмах (от дрожжей до мышей) и в исследованиях здоровья человека показано, что снижение активности компонентов сигнального пути инсулин/IGF-1 вызывает активацию фактора транскрипции FOXO и повышение стрессоустойчивости, снижение активности mTOR и индукцию аутофагии, что ведет к увеличению продолжительности жизни [147]. Низкие уровни гормона роста и IGF-1 в сыворотке крови млекопитающих и человека в течение жизни связаны с меньшим риском развития возрастных заболеваний и способствуют долголетию [150, 397, 398].
Другая нефармакологическая интервенция – регулярная умеренная физическая нагрузка, которая действует как мягкий фактор стресса и увеличивает выработку АФК, вызывая активацию сигнальных путей, связанных со стрессоустойчивостью и долголетием. Физические упражнения стимулируют активность сиртуинов, модулируют эпигенетические изменения, замедляют укорочение теломер, способствуют аутофагии и устранению стресса ЭПС, снижают митохондриальную дисфункцию, подавляют воспалительные реакции. Эффекты физических нагрузок зависят от типа нагрузки, ее интенсивности и продолжительности, состояния здоровья человека или модельного животного [109, 355, 399, 400].
Геропротекторы
Идентифицировано большое количество фармакологических препаратов и биологически активных веществ, которые снижают повреждение макромолекул, митохондриальную дисфункцию и способствуют замедлению старения в лабораторных экспериментах. Повреждения макромолекул можно предотвратить с помощью экзогенных антиоксидантов и активаторов систем антиоксидантной защиты [108]. Например, многие полифенолы способны перехватывать свободные радикалы, активировать системы антиоксидантной защиты через модуляцию сигнального пути Keap1/NRF2/ARE и экспрессию соответствующих ферментов [401]. Благоприятное воздействие этих соединений может быть обусловлено также эффектом гормезиса (неспецифического стресс-ответа, переводящего систему на более высокий уровень защиты от последующих повреждений). При этом повышенные дозы антиоксидантов могут быть токсичными из-за их прооксидантного действия в высоких концентрациях или в результате снижения полезных концентраций АФК [108, 401]. Данные о геропротекторных эффектах антиоксидантов зачастую противоречат друг другу и указывают на их неэффективность [108, 402].
Содержащиеся в продуктах питания биологически активные вещества и микроэлементы необходимы для синтеза нуклеотидов и репликации ДНК (фолат, витамин B12, магний, цинк, железо), предотвращения окисления ДНК (витамин C, витамин E, цинк, марганец, селен), распознавания и восстановления повреждений ДНК (ниацин, цинк, железо, магний). Дефицит этих соединений увеличивает стресс репликации ДНК и нестабильность генома, усугубляет восприимчивость к повреждению ДНК [403]. Кроме того, ряд веществ природного происхождения может способствовать поддержанию целостности генома. Некоторые полифенольные соединения (например, куркумин, галлат эпигаллокатехин, ресвератрол, нарингенин, хризин, кверцетин и другие) способны снижать уровень повреждений ДНК и стимулировать ответ на повреждение ДНК [401, 404]. Проантоцианидины и их микробные метаболиты повышают экспрессию генов репарации ДНК и активируют белки ATM и ATR [405–407]. Монотерпены способны стимулировать эксцизионную репарацию нуклеотидов [408]. Поддержанию стабильности генома также способствует достаточное потребление витамина D3, который активирует репарацию двухцепочечных повреждений ДНК, поддержание длины теломер, снижает выработку прогерина [409–411]. Некоторые фармакологические препараты, применяемые при возрастзависимых заболеваниях (например, антиангинальное средство никорандил), могут стимулировать репарацию ДНК [412].
Природные вещества (в частности, полиненасыщенные ω3-жирные кислоты) и половые гормоны способны замедлить укорочение теломер и реактивировать теломеразу, но их действие достаточно слабо выражено [400, 413]. Большей эффективностью обладают селективные активаторы теломеразы. Например, TA-65 вызывает умеренное удлинение теломер и улучшает связанные со старением параметры жизнеспособности у мышей и человека, но не влияет на продолжительность жизни [413]. Однако реактивация эндогенной теломеразы может быть связана с риском развития рака [108, 413]. У мышей эту проблему удалось избежать путем введения TERT с помощью аденоассоциированного вируса [414].
Эпигенетические модификации и контролирующие их белки рассматривают в качестве привлекательных мишеней для фармакологических вмешательств, так как потенциально они могут быть полностью или частично переведены в “молодое” состояние и быстро отвечают на воздействие внешних стимулов [73, 108]. За последние годы разработан ряд селективных препаратов, нацеленных на эпигенетические регуляторы и эффективных при определенных заболеваниях [108]. В то же время обнаружен ряд природных соединений, которые могут воздействовать на эпигеном и отменять эпигенетические модификации, связанные со старением.
Состав пищи может влиять на метилирование ДНК, изменяя доступность доноров метильной группы (например, фолиевой кислоты, витаминов B6 и B12, холина, метионина) и активность ДНК-метилтрансфераз (селен, генистеин, кверцетин, куркумин, полифенолы зеленого чая, апигенин, ресвератрол, сульфорафан) [355, 388, 403]. Эти соединения повышают уровень метилирования ДНК, предотвращая активацию нежелательных генов, но не решают проблему гиперметилирования специфических локусов. Существуют и фармакологические ингибиторы ДНК-метилтрансфераз, но они обладают выраженным цитотоксическим действием [108]. Полиамины спермин и спермидин стимулируют активность ДНК-метилтрансфераз и подавляют аберрантное метилирование ДНК [415]. Геропротекторное действие некоторых фармакологических веществ (например, аскорбиновой кислоты и метформина) может быть опосредовано модуляцией метилцитозиндиоксигеназы 2 (TET2) [416, 417].
Известно большое количество соединений, способных предотвращать возникновение возрастных эпигенетических изменений, воздействуя на ферменты, модифицирующие хроматин. К этим соединениям относятся прежде всего активаторы сиртуинов (гистондеацетилазы класса III) и ингибиторы гистондеацетилаз классов I и II [108, 355, 389]. Активация сиртуинов связана с поддержанием структуры хроматина и стимуляцией механизмов стрессоустойчивости. Она может обеспечиваться путем восстановления содержания NAD+, например, при добавлении в пищу никотинамидрибозида [418, 419]. Экспрессия сиртуинов усиливается некоторыми природными соединениями – флавонами, стильбенами, халконами и антоцианидинами. Большая часть этих соединений увеличивает продолжительность жизни модельных организмов и улучшает состояние здоровья пациентов с возрастзависимыми заболеваниями [108, 355, 389, 391]. Наибольшей способностью активировать SIRT1 обладает ресвератрол [108, 391]. В настоящее время разработаны синтетические производные ресвератрола, которые менее токсичны и более эффективно активируют SIRT1. Доказано геропротекторное действие по крайней мере двух производных – SRT1720 и SRT2104 [108, 391].
Ингибиторы гистондеацетилаз классов I и II, к которым относятся циклические пептиды, гидроксамовые кислоты, короткоцепочечные жирные кислоты и синтетические бензамиды [420], подавляют деацетилирование N-концов гистонов и активируют экспрессию генов стрессового ответа. Такие ингибиторы гистондеацетилаз, как 4-фенилбутират, бутират натрия, трихостатин А, субероиланилидгидроксамовая кислота, D-бета-гидроксибутират, вальпроевая кислота и вальпроат натрия увеличивают продолжительность жизни модельных животных и эффективны при связанных с возрастом заболеваниях [420–425]. Способность ингибировать гистондеацетилазы классов I и II проявляют и другие природные соединения (например, спермин, спермидин, сульфорафан, галлат эпигаллокатехина, лютеолин, куркумин, кверцетин и другие), но их действие неселективно. Одновременно они модулируют другие модифицирующие хроматин ферменты, гистон-ацетилтрансферазы и гистон-метилтрансферазы [355, 388, 389]. Кроме того, влиять на экспрессию этих ферментов могут некоторые фармакологические средства с установленным геропротекторным действием, например, аспирин и метформин [416, 426].
Многообещающими терапевтическими мишенями считаются микроРНК. Поскольку микроРНК имеют множество мишеней в клеточных сетях, их регуляция позволяет воздействовать на интегральные пути старения и возрастных заболеваний [108]. В настоящее время для изменения активности микроРНК применяют два основных методических подхода. Первый – модуляция функции микроРНК с помощью избыточной экспрессии на основе вирусного вектора или синтетических двухцепочечных микроРНК; второй – ингибирование микроРНК химически модифицированными антисмысловыми олигонуклеотидами [427]. Кроме того, метформин, а также антибиотик эноксацин способны стимулировать биогенез микроРНК, что опосредует их геропротекторную активность [428–430].
Фармакологические вмешательства для поддержания протеома нацелены, в первую очередь, на активацию механизмов аутофагии. Среди активаторов аутофагии, обладающих способностью увеличивать продолжительность жизни, можно отметить рапамицин, трегалозу, спермидин, уролитин А, вальпроевую кислоту, препараты лития [101, 102, 431]. Стимулировать аутофагию могут активаторы AMPK, например, физетин, бигуаниды (метформин, фенформин, буформин), тиазолидиндионы, салицилаты, агонисты рецепторов глюкагоноподобного пептида-1 [102, 108]. Ресвератрол активирует аутофагию, модулируя сигнальные пути SIRT1 и AMPK [108]. Тем не менее, гиперактивация механизмов аутофагии может вызывать клеточную гибель и усиливать некоторые патологические процессы, связанные с возрастом [102]. Кроме того, рапамицин и другие ингибиторы mTOR могут оказывать благоприятное воздействие на протеостаз путем подавления выработки белков и снижения перегрузки механизмов ответа на протеотоксический стресс [108].
Известен ряд фармакологических препаратов, направленных на предотвращение митохондриальной дисфункции. Разработаны антиоксиданты, действующие на митохондрии, в частности, SkQ и MitoQ. Их потенциальное геропротекторное действие доказано в исследованиях in vivo на различных модельных организмах. Кроме того, эти соединения могут применяться при различных возрастных заболеваниях [432–434]. Снижающуюся с возрастом экспрессию регуляторного белка SIRT3 можно стимулировать с помощью ресвератрола [113, 435]. Однако разработка фармакологических молекул для селективной активации SIRT3 представляет сложную задачу из-за структурных особенностей семейства сиртуинов [113, 391]. Описан также широкий спектр благоприятных эффектов рапамицина в митохондриях, в том числе снижение продукции АФК, предотвращение митохондриальной дисфункции, поддержание стабильности мтДНК, стимуляция митофагии [110, 436, 437]. Кроме того, активировать митофагию способны некоторые другие вещества (спермидин, уролитин А, метформин) [437, 438]. Тем не менее, умеренный митохондриальный стресс (физические упражнения, прием метформина) может вызывать митогормезис, характеризующийся повышенной выработкой АФК при одновременной стимуляции защитных механизмов, и, как следствие, увеличение продолжительности жизни [439–441].
Один из активно развивающихся видов геропротекторных интервенций нацелен на избирательное удаление сенесцентных клеток (сенолитики) и модулирование воспалительного сенесцентного секретома (сеноморфики, или сеностатики) [196, 442]. Потенциальными мишенями действия сенолитиков являются факторы, обеспечивающие устойчивость сенесцентных клеток к апоптозу. К сенолитикам, нацеленным на активацию апоптоза, относятся дазатиниб, кверцетин [219], навитоклакс (ABT263) [443], ABT-737 [444], пайперлонгумин, A1331852, A1155463 [445], дезинтегратор комплекса FOXO4-p53 (пептид FOXO4-DRI) [221], ингибитор HSP90 (17-DMAG) [446], индукторы аутофагии азитромицин и рокситромицин [447]. Действие другой группы сенолитиков основано на повышенной активности лизосомной β-галактозидазы в сенесцентных клетках, что позволяет избирательно элиминировать сенесцентные клетки цитотоксическими препаратами, заключенными в галактоолигосахаридную оболочку [448], или использовать пролекарства, модифицированные остатком галактозы [449]. Также предложен способ элиминации сенесцентных клеток с помощью сенолитиков, нацеленных на белки клеточной поверхности, таких как DPP4 (дипептидилпептидаза-4) [450] и рецепторы CD9 [451].
Применение ряда сенолитиков в качестве геропротекторных препаратов, несмотря на их положительные эффекты на различных моделях возрастзависимых заболеваний, сдерживается нежелательными побочными эффектами и потребностью в определенном количества сенесцентных клеток для поддержания структуры, функционирования и регенерации тканей [196].
Сеностатики лишены неблагоприятных побочных эффектов, поскольку нацелены на подавление секреторного фенотипа сенесцентных клеток. Потенциальными мишенями сеностатиков являются: NF-κB, p38, GATA4, mTOR, BRD4, cGAS/STING и MDM2 [196]. Однако эти мишени выполняют также не связанные со старением функции, что накладывает серьезные ограничения на возможность применения сеностатиков в качестве геропротекторов [442].
Наиболее доступный для современной практической медицины тип интервенций – использование препаратов-геропротекторов, в том числе, перепрофилирование уже лицензированных препаратов. Разработаны принципы их систематизации, концепция поиска и валидации [452], создана курируемая база Geroprotectors.org [453]. Наиболее часто геропротекторным эффектом обладают субстанции, ингибирующие активность ассоциированных со старением сигнальных каскадов [3]. Выделены геропротекторы, специфически воздействующие на: 1) элементы сигнальных путей ростовых факторов; 2) сигнальный путь инсулина, пути метаболизма углеводов и жиров; 3) элементы NAD+-зависимых путей и сиртуины; 4) пути синтеза и метаболизма аминокислот; 5) активаторы аутофагии; 6) механизмы клеточного старения 7) омолаживающие факторы GDF11 и GDF8; 8) эпигенетические ферменты.
Детальный анализ геропротекторного потенциала препаратов с привлечением базы клинических исследований ClinicalTrials.gov проведен М. Гонсалес-Фрейре и соавт. [4]. Результаты этого анализа свидетельствуют о том, что большинство геропротекторов, продлевающих жизнь модельным организмам, представляют собой миметики снижения калорийности питания. Снижение калорийности, периодическое голодание и физические упражнения относятся к вмешательствам с наилучшим образом доказанными эффектами, за ними по степени изученности и доказанности эффектов следуют ресвератрол, метформин и прекурсоры NAD+ [4].
К числу состояний, модифицируемых некоторыми геропротекторами, относится старческая хрупкость [454]. По последним данным течение некоторых возрастных заболеваний может быть облегчено геропротекторами в сочетании со сниженной калорийностью питания [455]. Однако стоит отметить, что применение геропротекторов не гарантирует продления жизни человека и нуждается в проведении клинических исследований, в которых будут проанализированы динамика биомаркеров старения, возрастзависимые заболевания и старческая хрупкость.
Репрограммирование клеток, клеточная терапия старения, переливание плазмы
Наиболее примечательны в этой области работы по омоложению тканеспецифичных стволовых клеток сигнальными молекулами, нацеленными на сигнальные каскады Notch, Wnt, и TGF-β [456–458].
Перспективными считаются технологии эпигенетического перепрограммирования клеток (в частности, с помощью факторов Яманаки, или OSKM), которые позволяют замедлить или обратить вспять возрастные изменения эпигенетических маркеров старения. Положительные результаты получены как на культурах клеток, так и в экспериментах на стареющих и прогероидных мышах (главным образом, при кратковременной циклической или частичной экспрессии факторов Яманаки). Однако в некоторых случаях у грызунов наблюдали повышение опухолеобразования, к тому же проведенные исследования не всегда учитывали долгосрочные эффекты перепрограммирования клеток [459–462].
Пересадка “омоложенных” стволовых клеток костного мозга линии Sca-1+ зарекомендовала себя в качестве средства, облегчающего течение возрастных заболеваний у модельных животных [463].
Отдельно стоит упомянуть генную терапию [464], индуцирующую теломеразную (TERT) активность, и комплексную генотерапию (FGF21 + + αKlotho + sTGFβR2) [465], а также монотерапию Klotho [466], которые способны замедлять течение различных возрастзависимых заболеваний.
Стив Хорват и соавт. обнаружили, что переливание плазмы крови молодых мышей старым значительно уменьшает метиломный биологический возраст образцов различных тканей [467]. В силу малой инвазивности метод переливания плазмы следует признать перспективной потенциальной интервенцией.
ЗАКЛЮЧЕНИЕ
Старение – многофакторный гетерохронный и гетеротопный процесс дерегуляции гомеостатических и гомеодинамических механизмов под влиянием постоянных стохастических изменений внешней и внутренней среды. Оно сочетает в себе стохастические и квазипрограммные черты (рис. 1 ). Квазипрограмма отличается от программы тем, что ее регуляторные элементы изначально не предназначены для ее реализации, а отвечают за другие процессы (клеточный рост, регенерацию, стрессовый ответ, иммунологическую защиту). В то же время, по-видимому, не существует программы старения организма человека, поскольку отсутствуют специфические молекулярные и клеточные подсистемы, берущие на себя функцию управления дезинтеграцией элементов системы целого организма с возрастом, подобные возникающим в процессе раннего индивидуального развития. Приведенные данные показывают, что старение поддается количественной оценке (использование биомаркеров) и модификации под влиянием интервенций, по крайней мере, у модельных животных.
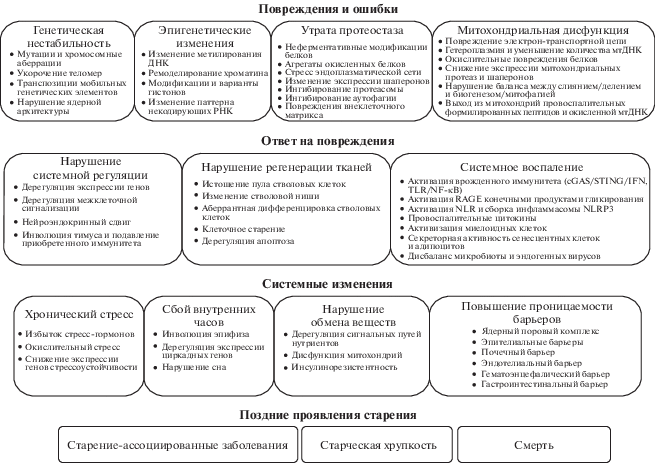
Смещение акцента с лечения конкретных возрастзависимых заболеваний на осуществляемые под контролем биомаркеров интервенции, направленные против старения, – это прежде всего попытка разработать подходы к этиотропному лечению неизлечимых хронических социально значимых возрастзависимых патологий (сахарного диабета второго типа, сердечно-сосудистых и нейродегенеративных заболеваний). Биомаркеры старения отличаются от биомаркеров возрастзависимых патологий, поскольку носят неспецифический и предклинический характер. Не существует одного надежного биомаркера старения, поэтому потребуется создание комплексных панелей биомаркеров (методы оценки биологического возраста).
Таким образом, главной целью современной геронтологии и гериатрии является создание комплексной терапии и модификации не просто отдельных признаков старения, но и вмешательства в механизмы патогенеза возрастных заболеваний на всех уровнях организации живого, рассматриваемых в качестве преморбидных проявлений старения.
Список литературы
Анисимов В.Н. (2010) Медицина антистарения: состояние и перспективы. Российский семейный врач. 14, 4–12.
Анисимов В.Н. (2000) Средства профилактики преждевременного старения (геропротекторы). Успехи геронтологии. 4, 55–74.
Москалев А.А., Шапошников М.В., Соловьёв И.А. (2017) Исследование геропротекторных свойств ингибиторов активности связанных со старением сигнальных каскадов на модельных организмах. Мед. Вест. Сев. Кав. 12, 342–347.
Gonzalez-Freire M., Diaz-Ruiz A., Hauser D., Martinez-Romero J., Ferrucci L., Bernier M., de Cabo R. (2020) The road ahead for health and lifespan interventions. Ageing Res. Rev. 59, 101037.
Moskalev A., Chernyagina E., Tsvetkov V., Fedintsev A., Shaposhnikov M., Krut’ko V., Zhavoronkov A., Kennedy B.K. (2016) Developing criteria for evaluation of geroprotectors as a key stage toward translation to the clinic. Aging Cell. 15, 407–415.
Gompertz B. (1825) On the Nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies. Phil. Transact. Royal Soc. London. 115, 513–583.
Bayersdorf R., Schumacher B. (2019) Recent advances in understanding the mechanisms determining longevity. F1000Res. 8, F1000 Faculty Rev-1403.
Шапошников М.В., Прошкина Е.Н., Шилова Л.А., Москалев А.А. (2015) Роль репарации повреждений ДНК в долголетии. Москва: Товарищество научных изданий КМК.
Moskalev A.A., Shaposhnikov M.V., Plyusnina E.N., Zhavoronkov A., Budovsky A., Yanai H., Fraifeld V.E. (2013) The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 12, 661–684.
Risques R.A., Kennedy S.R. (2018) Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 14, e1007108.
Forsberg L.A., Gisselsson D., Dumanski J.P. (2017) Mosaicism in health and disease – clones picking up speed. Nat. Rev. Genet. 18, 128–142.
Burtner C.R., Kennedy B.K. (2010) Progeria syndromes and ageing: what is the connection? Nat. Rev. Mol. Cell Biol. 11, 567–578.
Kubben N., Misteli T. (2017) Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 18, 595–609.
Keijzers G., Bakula D., Scheibye-Knudsen M. (2017) Monogenic diseases of DNA repair. N. Engl. J. Med. 377, 1868–1876.
Петрусева И.O., Евдокимов A.Н., Лаврик O.И. (2017) Поддержание стабильности генома у Heterocephalus glabe. Acta Naturae. 9, 31–41.
Seim I., Fang X., Xiong Z., Lobanov A.V., Huang Z., Ma S., Feng Y., Turanov A.A., Zhu Y., Lenz T.L., Gerashchenko M.V., Fan D., Hee Yim S., Yao X., Jordan D., Xiong Y., Ma Y., Lyapunov A.N., Chen G., Kulakova O.I., Sun Y., Lee S.G., Bronson R.T., Moskalev A.A., Sunyaev S.R., Zhang G., Krogh A., Wang J., Gladyshev V.N. (2013) Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 4, 2212.
Keane M., Semeiks J., Webb A.E., Li Y.I., Quesada V., Craig T., Madsen L.B., van Dam S., Brawand D., Marques P.I., Michalak P., Kang L., Bhak J., Yim H.S., Grishin N.V., Nielsen N.H., Heide-Jorgensen M.P., Oziolor E.M., Matson C.W., Church G.M., Stuart G.W., Patton J.C., George J.C., Suydam R., Larsen K., Lopez-Otin C., O’Connell M.J., Bickham J.W., Thomsen B., de Magalhaes J.P. (2015) Insights into the evolution of longevity from the bowhead whale genome. Cell Rept. 10, 112–122.
Schmidt H., Malik A., Bicker A., Poetzsch G., Avivi A., Shams I., Hankeln T. (2017) Hypoxia tolerance, longevity and cancer-resistance in the mole rat Spalax – a liver transcriptomics approach. Sci. Rep. 7, 14348.
Wirthlin M., Lima N.C.B., Guedes R.L.M., Soares A.E.R., Almeida L.G.P., Cavaleiro N.P., Loss de Morais G., Chaves A.V., Howard J.T., Teixeira M.d.M., Schneider P.N., Santos F.R., Schatz M.C., Felipe M.S., Miyaki C.Y., Aleixo A., Schneider M.P.C., Jarvis E.D., Vasconcelos A.T.R., Prosdocimi F., Mello C.V. (2018) Parrot genomes and the evolution of heightened longevity and cognition. Curr. Biol. 28, 4001–4008. e4007.
Bhargava V., Goldstein C.D., Russell L., Xu L., Ahmed M., Li W., Casey A., Servage K., Kollipara R., Picciarelli Z., Kittler R., Yatsenko A., Carmell M., Orth K., Amatruda J.F., Yanowitz J.L., Buszczak M. (2020) GCNA preserves genome integrity and fertility across species. Dev. Cell. 52, 38–52. e10.
Zhang L., Dong X., Lee M., Maslov A.Y., Wang T., Vijg J. (2019) Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA. 116, 9014–9019.
García-Nieto P.E., Morrison A.J., Fraser H.B. (2019) The somatic mutation landscape of the human body. Genome Biol. 20, 298.
Zhang L., Vijg J. (2018) Somatic Mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 52, 397–419.
Tiwari V., Wilson D.M., III (2019) DNA damage and associated DNA repair defects in disease and premature aging. Am. J. Hum. Genet. 105, 237–257.
Mendelsohn A.R., Larrick J.W. (2017) The NAD+/PARP1/SIRT1 axis in aging. Rejuvenation Res. 20, 244–247.
Hämäläinen R.H., Landoni J.C., Ahlqvist K.J., Goffart S., Ryytty S., Rahman M.O., Brilhante V., Icay K., Hautaniemi S., Wang L., Laiho M., Suomalainen A. (2019) Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nat. Metab. 1, 958–965.
Cho S., Vashisth M., Abbas A., Majkut S., Vogel K., Xia Y., Ivanovska I.L., Irianto J., Tewari M., Zhu K., Tichy E.D., Mourkioti F., Tang H.-Y., Greenberg R.A., Prosser B.L., Discher D.E. (2019) Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell. 49, 920–935. e925.
Cardoso A.C., Lam N.T., Savla J.J., Nakada Y., Pereira A.H.M., Elnwasany A., Menendez-Montes I., Ensley E.L., Bezan Petric U., Sharma G., Sherry A.D., Malloy C.R., Khemtong C., Kinter M.T., Tan W.L.W., Anene-Nzelu C.G., Foo R.S.-Y., Nguyen N.U.N., Li S., Ahmed M.S., Elhelaly W.M., Abdisalaam S., Asaithamby A., Xing C., Kanchwala M., Vale G., Eckert K.M., Mitsche M.A., McDonald J.G., Hill J.A., Huang L., Shaul P.W., Szweda L.I., Sadek H.A. (2020) Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nat. Metab. 2, 167–178.
Huang W.T., Akhter H., Jiang C., MacEwen M., Ding Q., Antony V., Thannickal V.J., Liu R.M. (2015) Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 61, 62–75.
Soria-Valles C., Lopez-Soto A., Osorio F.G., Lopez-Otin C. (2017) Immune and inflammatory responses to DNA damage in cancer and aging. Mech. Ageing Dev. 165, 10–16.
Hodskinson M.R., Bolner A., Sato K., Kamimae-Lanning A.N., Rooijers K., Witte M., Mahesh M., Silhan J., Petek M., Williams D.M., Kind J., Chin J.W., Patel K.J., Knipscheer P. (2020) Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature. 579, 603–608.
Yoshida K., Gowers K.H.C., Lee-Six H., Chandrasekharan D.P., Coorens T., Maughan E.F., Beal K., Menzies A., Millar F.R., Anderson E., Clarke S.E., Pennycuick A., Thakrar R.M., Butler C.R., Kakiuchi N., Hirano T., Hynds R.E., Stratton M.R., Martincorena I., Janes S.M., Campbell P.J. (2020) Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 578, 266–272.
Cheung V., Yuen V.M., Wong G.T.C., Choi S.W. (2019) The effect of sleep deprivation and disruption on DNA damage and health of doctors. Anaesthesia. 74, 434–440.
Barroso-Vilares M., Macedo J.C., Reis M., Warren J.D., Compton D., Logarinho E. (2020) Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep. 21, e49248.
Janssen A., Colmenares S.U., Karpen G.H. (2018) Heterochromatin: guardian of the genome. Annu. Rev. Cell. Dev. Biol. 34, 265–288.
Qiu G.H., Zheng X., Fu M., Huang C., Yang X. (2019) The protective function of non-coding DNA in DNA damage accumulation with age and its roles in age-related diseases. Biogerontology. 20, 741–761.
Qiu G.H., Huang C., Zheng X., Yang X. (2018) The protective function of noncoding DNA in genome defense of eukaryotic male germ cells. Epigenomics. 10, 499–517.
Akbari M., Kirkwood T.B.L., Bohr V.A. (2019) Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 54, 100940.
Olovnikov A.M. (1973) A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41, 181–190.
Оловников А.М. (1971) Принцип маргинотомии в матричном синтезе полинуклеотидов. Докл. АН СССР. 201, 1496–1499.
Turner K.J., Vasu V., Griffin D.K. (2019) Telomere biology and human phenotype. Cells. 8, 73.
Hewitt G., Jurk D., Marques F.D.M., Correia-Melo C., Hardy T., Gackowska A., Anderson R., Taschuk M., Mann J., Passos J.F. (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708–708.
Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J.M., Bucci G., Dobreva M., Matti V., Beausejour C.M., Herbig U., Longhese M.P., d’Adda di Fagagna F. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365.
Wood J.G., Helfand S.L. (2013) Chromatin structure and transposable elements in organismal aging. Front. Genet. 4, 274.
De Cecco M., Criscione S.W., Peckham E.J., Hillenmeyer S., Hamm E.A., Manivannan J., Peterson A.L., Kreiling J.A., Neretti N., Sedivy J.M. (2013) Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 12, 247–256.
Cardelli M. (2018) The epigenetic alterations of endogenous retroelements in aging. Mech. Ageing Dev. 174, 30–46.
Buzdin A.A., Prassolov V., Garazha A.V. (2017) Friends-enemies: endogenous retroviruses are major transcriptional regulators of human DNA. Front Chem. 5, 35.
Andrenacci D., Cavaliere V., Lattanzi G. (2020) The role of transposable elements activity in aging and their possible involvement in laminopathic diseases. Ageing Res. Rev. 57, 100995.
Mattioli E., Andrenacci D., Garofalo C., Prencipe S., Scotlandi K., Remondini D., Gentilini D., Di Blasio A.M., Valente S., Scarano E., Cicchilitti L., Piaggio G., Mai A., Lattanzi G. (2018) Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell. 17, e12824.
Ashapkin V.V., Kutueva L.I., Kurchashova S.Y., Kireev I.I. (2019) Are there common mechanisms between the Hutchinson–Gilford progeria syndrome and natural aging? Front. Genet. 10, 455.
Worman H.J. (2012) Nuclear lamins and laminopathies. J. Pathol. 226, 316–325.
Romero-Bueno R., de la Cruz Ruiz P., Artal-Sanz M., Askjaer P., Dobrzynska A. (2019) Nuclear organization in stress and aging. Cells. 8, 664.
Niedernhofer L.J., Gurkar A.U., Wang Y., Vijg J., Hoeijmakers J.H.J., Robbins P.D. (2018) Nuclear genomic instability and aging. Annu. Rev. Biochem. 87, 295–322.
Hetz C., Chevet E., Harding H.P. (2013) Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719.
Hampel B., Wagner M., Teis D., Zwerschke W., Huber L.A., Jansen-Durr P. (2005) Apoptosis resistance of senescent human fibroblasts is correlated with the absence of nuclear IGFBP-3. Aging Cell. 4, 325–330.
Zhang L., Yousefzadeh M.J., Suh Y., Niedernhofer L.J., Robbins P.D. (2019) Signal transduction, ageing and disease. In: Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Eds Harris J.R., Korolchuk V.I. Singapore: Springer Singapore, pp. 227–247.
Robinson A.R., Yousefzadeh M.J., Rozgaja T.A., Wang J., Li X., Tilstra J.S., Feldman C.H., Gregg S.Q., Johnson C.H., Skoda E.M., Frantz M.C., Bell-Temin H., Pope-Varsalona H., Gurkar A.U., Nasto L.A., Robinson R.A.S., Fuhrmann-Stroissnigg H., Czerwinska J., McGowan S.J., Cantu-Medellin N., Harris J.B., Maniar S., Ross M.A., Trussoni C.E., LaRusso N.F., Cifuentes-Pagano E., Pagano P.J., Tudek B., Vo N.V., Rigatti L.H., Opresko P.L., Stolz D.B., Watkins S.C., Burd C.E., Croix C.M.S., Siuzdak G., Yates N.A., Robbins P.D., Wang Y., Wipf P., Kelley E.E., Niedernhofer L.J. (2018) Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 17, 259–273.
Nakad R., Schumacher B. (2016) DNA damage response and immune defense: links and mechanisms. Front. Genet. 7, 147.
Goulielmaki E., Ioannidou A., Tsekrekou M., Stratigi K., Poutakidou I.K., Gkirtzimanaki K., Aivaliotis M., Evangelou K., Topalis P., Altmüller J., Gorgoulis V.G., Chatzinikolaou G., Garinis G.A. (2020) Tissue-infiltrating macrophages mediate an exosome-based metabolic reprogramming upon DNA damage. Nat. Commun. 11, 42.
Shanbhag N.M., Evans M.D., Mao W., Nana A.L., Seeley W.W., Adame A., Rissman R.A., Masliah E., Mucke L. (2019) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7, 77.
Kim H.-N., Chang J., Shao L., Han L., Iyer S., Manolagas S.C., O’Brien C.A., Jilka R.L., Zhou D., Almeida M. (2017) DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 16, 693–703.
Walter D., Lier A., Geiselhart A., Thalheimer F.B., Huntscha S., Sobotta M.C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P., Muedder K., Klein C., Jauch A., Schroeder T., Geiger H., Dick T.P., Holland-Letz T., Schmezer P., Lane S.W., Rieger M.A., Essers M.A., Williams D.A., Trumpp A., Milsom M.D. (2015) Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 520, 549–552.
Rodier F., Muñoz D.P., Teachenor R., Chu V., Le O., Bhaumik D., Coppé J.-P., Campeau E., Beauséjour C.M., Kim S.-H., Davalos A.R., Campisi J. (2011) DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 124, 68.
Lu Y., Krishnan A., Brommer B., Tian X., Meer M., Vera D.L., Wang C., Zeng Q., Yu D., Bonkowski M.S., Yang J.-H., Hoffmann E.M., Zhou S., Korobkina E., Davidsohn N., Schultz M.B., Chwalek K., Rajman L.A., Church G.M., Hochedlinger K., Gladyshev V.N., Horvath S., Gregory-Ksander M.S., Ksander B.R., He Z., Sinclair D.A. (2019) Reversal of ageing- and injury-induced vision loss by Tet-dependent epigenetic reprogramming. bioRxiv. https://doi.org/10.1101/710210
Paluvai H., Di Giorgio E., Brancolini C. (2020) The histone code of senescence. Cells. 9, 466.
Vanyushin B.F., Nemirovsky L.E., Klimenko V.V., Vasiliev V.K., Belozersky A.N. (1973) The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia. 19, 138–152.
Vanyushin B.F., Kirnos M.D. (1977) Structure of animal mitochondrial DNA (base composition, pyrimidine clusters, character of methylation). Biochim Biophys Acta. 475, 323–336.
Booth L.N., Brunet A. (2016) The aging epigenome. Mol. Cell. 62, 728–744.
Kane A.E., Sinclair D.A. (2019) Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 54, 61–83.
Morgan A.E., Davies T.J., Mc Auley M.T. (2018) The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 77, 412–422.
Xie W., Baylin S.B., Easwaran H. (2019) DNA methylation in senescence, aging and cancer. Oncoscience. 6, 291–293.
Unnikrishnan A., Hadad N., Masser D.R., Jackson J., Freeman W.M., Richardson A. (2018) Revisiting the genomic hypomethylation hypothesis of aging. Ann. N.Y. Acad. Sci. 1418, 69–79.
Ashapkin V.V., Kutueva L.I., Vanyushin B.F. (2019) Epigenetic clock: just a convenient marker or an active driver of aging? Adv. Exp. Med. Biol. 1178, 175–206.
Michalak E.M., Burr M.L., Bannister A.J., Dawson M.A. (2019) The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 20, 573–589.
Yu R., McCauley B., Dang W. (2020) Loss of chromatin structural integrity is a source of stress during aging. Hum. Genet. 139, 371–380.
Baldensperger T., Eggen M., Kappen J., Winterhalter P.R., Pfirrmann T., Glomb M.A. (2020) Comprehensive analysis of posttranslational protein modifications in aging of subcellular compartments. Sci. Rep. 10, 7596.
Contrepois K., Coudereau C., Benayoun B.A., Schuler N., Roux P.F., Bischof O., Courbeyrette R., Carvalho C., Thuret J.Y., Ma Z., Derbois C., Nevers M.C., Volland H., Redon C.E., Bonner W.M., Deleuze J.F., Wiel C., Bernard D., Snyder M.P., Rube C.E., Olaso R., Fenaille F., Mann C. (2017) Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 8, 14995.
Tvardovskiy A., Schwammle V., Kempf S.J., Rogowska-Wrzesinska A., Jensen O.N. (2017) Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 45, 9272–9289.
Wang Y., Yuan Q., Xie L. (2018) Histone modifications in aging: the underlying mechanisms and implications. Curr. Stem Cell Res. Therapy. 13, 125–135.
Li Y., Jin M., O’Laughlin R., Bittihn P., Tsimring L.S., Pillus L., Hasty J., Hao N. (2017) Multigenerational silencing dynamics control cell aging. Proc. Natl. Acad. Sci. USA. 114, 11253–11258.
Majidinia M., Mir S.M., Mirza-Aghazadeh-Attari M., Asghari R., Kafil H.S., Safa A., Mahmoodpoor A., Yousefi B. (2020) MicroRNAs, DNA damage response and ageing. Biogerontology. 21, 275–291.
Lenart P., Novak J., Bienertova-Vasku J. (2018) PIWI-piRNA pathway: setting the pace of aging by reducing DNA damage. Mech. Ageing Dev. 173, 29–38.
Sousa-Victor P., Ayyaz A., Hayashi R., Qi Y., Madden D.T., Lunyak V.V., Jasper H. (2017) Piwi is required to limit exhaustion of aging somatic stem cells. Cell Rept. 20, 2527–2537.
Kour S., Rath P.C. (2016) Long noncoding RNAs in aging and age-related diseases. Ageing Res. Rev. 26, 1–21.
Greene J., Baird A.M., Brady L., Lim M., Gray S.G., McDermott R., Finn S.P. (2017) Circular RNAs: biogenesis, function and role in human diseases. Front. Mol. Biosci. 4, 38.
Cai H., Li Y., Niringiyumukiza J.D., Su P., Xiang W. (2019) Circular RNA involvement in aging: an emerging player with great potential. Mech. Ageing Dev. 178, 16–24.
Mori M.A., Ludwig R.G., Garcia-Martin R., Brandao B.B., Kahn C.R. (2019) Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. 30, 656–673.
Dluzen D.F., Noren Hooten N., Evans M.K. (2017) Extracellular RNA in aging. Wiley Interdiscip. Rev. RNA. 8, .https://doi.org/10.1002/wrna.1385
Koga H., Kaushik S., Cuervo A.M. (2011) Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215.
Trigo D., Nadais A., da Cruz E.S.O.A.B. (2019) Unravelling protein aggregation as an ageing related process or a neuropathological response. Ageing Res. Rev. 51, 67–77.
Grune T. (2020) Oxidized protein aggregates: formation and biological effects. Free Radic. Biol. Med. 150, 120–124.
Hipp M.S., Kasturi P., Hartl F.U. (2019) The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435.
Fournet M., Bonte F., Desmouliere A. (2018) Glycation damage: a possible hub for major pathophysiological disorders and aging. Aging Dis. 9, 880–900.
Vanhooren V., Navarrete Santos A., Voutetakis K., Petropoulos I., Libert C., Simm A., Gonos E.S., Friguet B. (2015) Protein modification and maintenance systems as biomarkers of ageing. Mech. Ageing Dev. 151, 71–84.
Gorisse L., Pietrement C., Vuiblet V., Schmelzer C.E., Kohler M., Duca L., Debelle L., Fornes P., Jaisson S., Gillery P. (2016) Protein carbamylation is a hallmark of aging. Proc. Natl. Acad. Sci. USA. 113, 1191–1196.
Moldogazieva N.T., Mokhosoev I.M., Mel’nikova T.I., Porozov Y.B., Terentiev A.A. (2019) Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid. Med. Cell Longev. 2019, 3085756.
Birch H.L. (2018) Extracellular matrix and ageing. Subcell. Biochem. 90, 169–190.
Cheon S.Y., Kim H., Rubinsztein D.C., Lee J.E. (2019) Autophagy, cellular aging and age-related human diseases. Exp. Neurobiol. 28, 643–657.
Basisty N., Holtz A., Schilling B. (2020) Accumulation of “old proteins” and the critical need for MS-based protein turnover measurements in aging and longevity. Proteomics. 20, e1800403.
Lee C.W., Wilfling F., Ronchi P., Allegretti M., Mosalaganti S., Jentsch S., Beck M., Pfander B. (2020) Selective autophagy degrades nuclear pore complexes. Nat. Cell Biol. 22, 159–166.
Stead E.R., Castillo-Quan J.I., Miguel V.E.M., Lujan C., Ketteler R., Kinghorn K.J., Bjedov I. (2019) Agephagy – adapting autophagy for health during aging. Front. Cell Dev. Biology. 7, 308.
Wong S.Q., Kumar A.V., Mills J., Lapierre L.R. (2020) Autophagy in aging and longevity. Hum. Genet. 139, 277–290.
Taylor R.C. (2016) Aging and the UPR(ER). Brain Res. 1648, 588–593.
Martinez G., Duran-Aniotz C., Cabral-Miranda F., Vivar J.P., Hetz C. (2017) Endoplasmic reticulum proteostasis impairment in aging. Aging Cell. 16, 615–623.
Brehme M., Voisine C., Rolland T., Wachi S., Soper J.H., Zhu Y., Orton K., Villella A., Garza D., Vidal M., Ge H., Morimoto R.I. (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rept. 9, 1135–1150.
Hansen M., Rubinsztein D.C., Walker D.W. (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593.
Hegde A.N., Smith S.G., Duke L.M., Pourquoi A., Vaz S. (2019) Perturbations of ubiquitin-proteasome-mediated proteolysis in aging and Alzheimer’s disease. Front. Aging Neurosci. 11, 324.
Vaiserman A.M., Lushchak O.V., Koliada A.K. (2016) Anti-aging pharmacology: promises and pitfalls. Ageing Res. Rev. 31, 9–35.
Nilsson M.I., Tarnopolsky M.A. (2019) Mitochondria and aging-the role of exercise as a countermeasure. Biology (Basel). 8, 40.
Zheng Q., Huang J., Wang G. (2019) Mitochondria, telomeres and telomerase subunits. Front. Cell Dev. Biology. 7, 274.
Moro L. (2019) Mitochondrial dysfunction in aging and cancer. J. Clin. Med. 8, 1983.
Hekimi S., Lapointe J., Wen Y. (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576.
Gomes P., Viana S.D., Nunes S., Rolo A.P., Palmeira C.M., Reis F. (2020) The yin and yang faces of the mitochondrial deacetylase sirtuin 3 in age-related disorders. Ageing Res. Rev. 57, 100983.
Tao R., Coleman M.C., Pennington J.D., Ozden O., Park S.H., Jiang H., Kim H.S., Flynn C.R., Hill S., Hayes McDonald W., Olivier A.K., Spitz D.R., Gius D. (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell. 40, 893–904.
Whitehall J.C., Greaves L.C. (2019) Aberrant mitochondrial function in ageing and cancer. Biogerontology. 21, 445–459https://doi.org/10.1007/s10522-10019-09853-y
Reynolds J.C., Bwiza C.P., Lee C. (2020) Mitonuclear genomics and aging. Hum. Genet. 139, 381–399.
Billard P., Poncet D.A. (2019) Replication stress at telomeric and mitochondrial DNa: common origins and consequences on ageing. Int. J. Mol. Sci. 20, 4959.
Gaziev A.I., Abdullaev S., Podlutsky A. (2014) Mitochondrial function and mitochondrial DNA maintenance with advancing age. Biogerontology. 15, 417–438.
Muftuoglu M., Mori M.P., de Souza-Pinto N.C. (2014) Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion. 17, 164–181.
Газиев А.И., Подлуцкий А.Я. (2003) Низкая эффективность репарации ДНК в митохондриях. Цитология. 45, 403–417.
Газиев А.И., Шайхаев Г.О. (2010) Ядерно-митохондриальные псевдогены. Молекуляр. биология. 44, 405–417.
Moehle E.A., Shen K., Dillin A. (2019) Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 294, 5396–5407.
Nakada K., Sato A., Hayashi J. (2009) Mitochondrial functional complementation in mitochondrial DNA-based diseases. Int. J. Biochem. Cell. Biol. 41, 1907–1913.
Sharma A., Smith H.J., Yao P., Mair W.B. (2019) Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 20, e48395.
Garza-Lombo C., Pappa A., Panayiotidis M.I., Franco R. (2020) Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 51, 105–117.
Thomas H.E., Zhang Y., Stefely J.A., Veiga S.R., Thomas G., Kozma S.C., Mercer C.A. (2018) Mitochondrial complex I activity is required for maximal autophagy. Cell Rept. 24, 2404–2417. e2408.
Tan J.X., Finkel T. (2020) Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 219, e202002179.
Melber A., Haynes C.M. (2018) UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295.
McManus M.J., Picard M., Chen H.W., De Haas H.J., Potluri P., Leipzig J., Towheed A., Angelin A., Sengupta P., Morrow R.M., Kauffman B.A., Vermulst M., Narula J., Wallace D.C. (2019) Mitochondrial DNA variation dictates expressivity and progression of nuclear DNA mutations causing cardiomyopathy. Cell Metab. 29, 78–90. e75.
Maniyadath B., Shukla N., Kolthur-Seetharam U. (2018) Gene expression, epigenetics and ageing. Subcell. Biochem. 90, 471–504.
Stegeman R., Weake V.M. (2017) Transcriptional signatures of aging. J. Mol. Biol. 429, 2427–2437.
Solovev I., Shaposhnikov M., Moskalev A. (2020) Multi-omics approaches to human biological age estimation. Mech. Ageing Dev. 185, 111192–111192.
Anisimova A.S., Meerson M.B., Gerashchenko M.V., Kulakovskiy I.V., Dmitriev S.E., Gladyshev V.N. (2020) Multi-faceted deregulation of gene expression and protein synthesis with age. bioRxiv. https://doi.org/10.1101/2020.1101.1119.911404
Wood S.H., Craig T., Li Y., Merry B., de Magalhaes J.P. (2013) Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age (Dordr). 35, 763–776.
Dönertaş H.M., İzgi H., Kamacıoğlu A., He Z., Khaitovich P., Somel M. (2017) Gene expression reversal toward pre-adult levels in the aging human brain and age-related loss of cellular identity. Sci. Rep. 7, 5894–5894.
Rangaraju S., Solis G.M., Thompson R.C., Gomez-Amaro R.L., Kurian L., Encalada S.E., Niculescu A.B., 3rd, Salomon D.R., Petrascheck M. (2015) Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. eLife. 4, e08833–e08833.
Zhavoronkov A., Buzdin A.A., Garazha A.V., Borisov N.M., Moskalev A.A. (2014) Signaling pathway cloud regulation for in silico screening and ranking of the potential geroprotective drugs. Front. Genet. 5, 49.
de Magalhaes J.P., Curado J., Church G.M. (2009) Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 25, 875–881.
Lai R.W., Lu R., Danthi P.S., Bravo J.I., Goumba A., Sampathkumar N.K., Benayoun B.A. (2019) Multi-level remodeling of transcriptional landscapes in aging and longevity. BMB Rept. 52, 86–108.
Stoeger T., Grant R.A., McQuattie-Pimentel A.C., Anekalla K., Liu S.S., Tejedor-Navarro H., Singer B.D., Abdala-Valencia H., Schwake M., Tetreault M.-P., Perlman H., Balch W.E., Chandel N., Ridge K., Sznajder J.I., Morimoto R.I., Misharin A.V., Budinger G.R.S., Amaral L.A.N. (2019) Aging is associated with a systemic length-driven transcriptome imbalance. bioRxiv. https://doi.org/10.1101/691154
Ferrucci L., Gonzalez-Freire M., Fabbri E., Simonsick E., Tanaka T., Moore Z., Salimi S., Sierra F., de Cabo R. (2020) Measuring biological aging in humans: a quest. Aging Cell. 19, e13080.
Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G. (2013) The hallmarks of aging. Cell. 153, 1194–1217.
Everitt A.V., Rattan S.I.S., le Couteur D.G., de Cabo R. (2010) Calorie Restriction, Aging and Longevity. New York: Springer.
Kalache A., de Hoogh A.I., Howlett S.E., Kennedy B., Eggersdorfer M., Marsman D.S., Shao A., Griffiths J.C. (2019) Nutrition interventions for healthy ageing across the lifespan: a conference report. Eur. J.Nutrit. 58, 1–11.
Piper Matthew D.W., Partridge L., Raubenheimer D., Simpson Stephen J. (2011) Dietary restriction and aging: a unifying perspective. Cell Metab. 14, 154–160.
Miyamoto T., Wright G., Amrein H. (2013) Nutrient sensors. Curr. Biol. 23, R369–373.
Kenyon C.J. (2010) The genetics of ageing. Nature. 464, 504–512.
Kim E. (2009) Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr. Res. Pract. 3, 64–71.
Grimaldi P.A. (2007) Peroxisome proliferator-activated receptors as sensors of fatty acids and derivatives. Cell. Mol. Life Sci. 64, 2459–2464.
Johnson S.C. (2018) Nutrient sensing, signaling and ageing: the role of IGF-1 and mTOR in ageing and age-related disease. Subcell. Biochem. 90, 49–97.
Papadopoli D., Boulay K., Kazak L., Pollak M., Mallette F., Topisirovic I., Hulea L. (2019) mTOR as a central regulator of lifespan and aging. F1000Res. 8, F1000 Faculty Rev-1998.
Jeon S.M. (2016) Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48, e245.
Salminen A., Kaarniranta K., Kauppinen A. (2016) Age-related changes in AMPK activation: role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 28, 15–26.
Kosciuk T., Wang M., Hong J.Y., Lin H. (2019) Updates on the epigenetic roles of sirtuins. Curr. Opin. Chem. Biol. 51, 18–29.
Yaribeygi H., Farrokhi F.R., Butler A.E., Sahebkar A. (2019) Insulin resistance: review of the underlying molecular mechanisms. J. Cell. Physiol. 234, 8152–8161.
Ryan A.S. (2000) Insulin resistance with aging. Sports Med. 30, 327–346.
Lann D., LeRoith D. (2007) Insulin resistance as the underlying cause for the metabolic syndrome. Med. Clin. North Am. 91, 1063–1077.
Fontana L., Partridge L., Longo V.D. (2010) Extending healthy life span – from yeast to humans. Science. 328, 321.
Laplante M., Sabatini D.M. (2012) mTOR signaling in growth control and disease. Cell. 149, 274–293.
Filippi B.M., Lam T.K. (2014) Leptin and aging. Aging (Albany NY). 6, 82–83.
Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., Li B., Liu G., Cai D. (2013) Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 497, 211–216.
Bauer M.E. (2005) Stress, glucocorticoids and ageing of the immune system. Stress. 8, 69–83.
Galley J.D., Bailey M.T. (2014) Impact of stressor exposure on the interplay between commensal microbiota and host inflammation. Gut Microbes. 5, 390–396.
Дильман В.М. (1960) Возрастная гиперхолестеринемия как показатель повышенной активности гипоталамических центров. Терапевтический архив. 32, 72–77.
Москалев А.А., Кременцова А.В., Малышева О.А. (2008) Влияние мелатонина на продолжительность жизни Drosophila melanogaster при различных режимах освещения. Экологическая генетика. 6, 24–32.
Tan D.X., Xu B., Zhou X., Reiter R.J. (2018) Pineal calcification, melatonin production, aging, associated health consequences and rejuvenation of the pineal gland. Molecules. 23,
Wang Y., Chen F., Ye L., Zirkin B., Chen H. (2017) Steroidogenesis in Leydig cells: effects of aging and environmental factors. Reproduction (Cambridge, England). 154, R111–R122.
Taneja C., Gera S., Kim S.M., Iqbal J., Yuen T., Zaidi M. (2019) FSH-metabolic circuitry and menopause. J. Mol. Endocrinol. 63, R73–R80.
Jung Y., Brack A.S. (2014) Cellular mechanisms of somatic stem cell aging. Curr. Topics Dev. Biol. 107, 405–438.
Ren R., Ocampo A., Liu G.-H., Izpisua Belmonte J.C. (2017) Regulation of stem cell aging by metabolism and epigenetics. Cell Metab. 26, 460–474.
Sameri S., Samadi P., Dehghan R., Salem E., Fayazi N., Amini R. (2020) Stem cell aging in lifespan and disease: a state-of-the-art review. Curr. Stem Cell Res. Therapy. 15, 362–378. https://doi.org/10.2174/1574888X15666200213105155
Oh J., Lee Y.D., Wagers A.J. (2014) Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat. Med. 20, 870–880.
Москалёв А.А. (2009) Роль стволовой ниши в процессах старения организма. Рос. хим. журн. 53, 83–87.
Carlson M.E., Conboy M.J., Hsu M., Barchas L., Jeong J., Agrawal A., Mikels A.J., Agrawal S., Schaffer D.V., Conboy I.M. (2009) Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 8, 676–689.
Franceschi C., Bonafè M., Valensin S., Olivieri F., De Luca M., Ottaviani E., De Benedictis G. (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 908, 244–254.
Coppé J.P., Desprez P.Y., Krtolica A., Campisi J. (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118.
Elabd C., Cousin W., Upadhyayula P., Chen R.Y., Chooljian M.S., Li J., Kung S., Jiang K.P., Conboy I.M. (2014) Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat. Commun. 5, 4082.
Sinha M., Jang Y.C., Oh J., Khong D., Wu E.Y., Manohar R., Miller C., Regalado S.G., Loffredo F.S., Pancoast J.R., Hirshman M.F., Lebowitz J., Sha-drach J.L., Cerletti M., Kim M.-J., Serwold T., Goodyear L.J., Rosner B., Lee R.T., Wagers A.J. (2014) Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 344, 649–652.
Katsimpardi L., Litterman N.K., Schein P.A., Miller C.M., Loffredo F.S., Wojtkiewicz G.R., Chen J.W., Lee R.T., Wagers A.J., Rubin L.L. (2014) Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 344, 630–634.
Ancel S., Mashinchian O., Feige J.N. (2019) Adipogenic progenitors keep muscle stem cells young. Aging. 11, 7331–7333.
Li H., Hou L. (2018) Regulation of melanocyte stem cell behavior by the niche microenvironment. Pigment Cell Melanoma Res. 31, 556–569.
Ho Y.-H., Méndez-Ferrer S. (2020) Microenvironmental contributions to hematopoietic stem cell aging. Haematologica. 105, 38–46.
Chakkalakal J.V., Jones K.M., Basson M.A., Brack A.S. (2012) The aged niche disrupts muscle stem cell quiescence. Nature. 490, 355–360.
Geiger H., Koehler A., Gunzer M. (2007) Stem cells, aging, niche, adhesion and Cdc42: a model for changes in cell–cell interactions and hematopoietic stem cell aging. Cell Cycle. 6, 884–887.
Stearns-Reider K.M., D’Amore A., Beezhold K., Rothrauff B., Cavalli L., Wagner W.R., Vorp D.A., Tsamis A., Shinde S., Zhang C., Barchowsky A., Rando T.A., Tuan R.S., Ambrosio F. (2017) Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell. 16, 518–528.
Doles J., Storer M., Cozzuto L., Roma G., Keyes W.M. (2012) Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 26, 2144–2153.
Choi J., Artandi S. (2009) Stem cell aging and aberrant differentiation within the niche. Cell Stem Cell. 5, 6–8.
Schultz M.B., Sinclair D.A. (2016) When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 143, 3.
Latchney S.E., Calvi L.M. (2017) The aging hematopoietic stem cell niche: phenotypic and functional changes and mechanisms that contribute to hematopoietic aging. Semin. Hematol. 54, 25–32.
Inomata K., Aoto T., Binh N.T., Okamoto N., Tanimura S., Wakayama T., Iseki S., Hara E., Masunaga T., Shimizu H., Nishimura E.K. (2009) Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 137, 1088–1099.
Brack A.S., Conboy M.J., Roy S., Lee M., Kuo C.J., Keller C., Rando T.A. (2007) Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 317, 807–810.
Campisi J., d’Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740.
Aravinthan A. (2015) Cellular senescence: a hitchhiker’s guide. Human Cell. 28, 51–64.
Herbig U., Ferreira M., Condel L., Carey D., Sedivy J.M. (2006) Cellular senescence in aging primates. Science. 311, 1257.
Chapman J., Fielder E., Passos J.F. (2019) Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. 593, 1566–1579.
Kang C. (2019) Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells. 42, 821–827.
Khosla S., Farr J.N., Tchkonia T., Kirkland J.L. (2020) The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 16, 263–275.
da Silva P.F.L., Ogrodnik M., Kucheryavenko O., Glibert J., Miwa S., Cameron K., Ishaq A., Saretzki G., Nagaraja-Grellscheid S., Nelson G., von Zglinicki T. (2019) The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 18, e12848–e12848.
Freund A., Orjalo A.V., Desprez P.-Y., Campisi J. (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246.
Stout M.B., Tchkonia T., Pirtskhalava T., Palmer A.K., List E.O., Berryman D.E., Lubbers E.R., Escande C., Spong A., Masternak M.M., Oberg A.L., LeBrasseur N.K., Miller R.A., Kopchick J.J., Bartke A., Kirkland J.L. (2014) Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging. 6, 575–586.
del Nogal M., Troyano N., Calleros L., Griera M., Rodriguez-Puyol M., Rodriguez-Puyol D., Ruiz-Torres M.P. (2014) Hyperosmolarity induced by high glucose promotes senescence in human glomerular mesangial cells. Int. J. Biochem. Cell. Biol. 54, 98–110.
Li M., You L., Xue J., Lu Y. (2018) Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front. Pharmacol. 9, 522.
von Zglinicki T., Petrie J., Kirkwood T.B.L. (2003) Telomere-driven replicative senescence is a stress response. Nat. Biotechnol. 21, 229–230.
Anderson R., Lagnado A., Maggiorani D., Walaszczyk A., Dookun E., Chapman J., Birch J., Salmonowicz H., Ogrodnik M., Jurk D., Proctor C., Correia-Melo C., Victorelli S., Fielder E., Berlinguer-Palmini R., Owens A., Greaves L.C., Kolsky K.L., Parini A., Douin-Echinard V., LeBrasseur N.K., Arthur H.M., Tual-Chalot S., Schafer M.J., Roos C.M., Miller J.D., Robertson N., Mann J., Adams P.D., Tchkonia T., Kirkland J.L., Mialet-Perez J., Richardson G.D., Passos J.F. (2019) Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492.
da Silva P.F.L., Schumacher B. (2019) DNA damage responses in ageing. Open Biol. 9, 190168.
Andriani G.A., Almeida V.P., Faggioli F., Mauro M., Tsai W.L., Santambrogio L., Maslov A., Gadina M., Campisi J., Vijg J., Montagna C. (2016) Whole chromosome instability induces senescence and promotes SASP. Sci. Rep. 6, 35218–35218.
Korolchuk V.I., Miwa S., Carroll B., von Zglinicki T. (2017) Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 21, 7–13.
Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 88, 593–602.
Hernandez-Segura A., Nehme J., Demaria M. (2018) Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453.
Hernandez-Segura A., de Jong T.V., Melov S., Guryev V., Campisi J., Demaria M. (2017) Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660.
Wang E. (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 55, 2284–2292.
Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 92, 9363–9367.
Sharpless N.E. (2004) Ink4a/Arf links senescence and aging. Exp. Gerontol. 39, 1751–1759.
Choudhury A.R., Ju Z., Djojosubroto M.W., Schienke A., Lechel A., Schaetzlein S., Jiang H., Stepczynska A., Wang C., Buer J., Lee H.-W., von Zglinicki T., Ganser A., Schirmacher P., Nakauchi H., Rudolph K.L. (2007) Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 39, 99–105.
Georgakopoulou E.A., Tsimaratou K., Evangelou K., Fernandez Marcos P.J., Zoumpourlis V., Trougakos I.P., Kletsas D., Bartek J., Serrano M., Gorgoulis V.G. (2013) Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging. 5, 37–50.
Davalos A.R., Kawahara M., Malhotra G.K., Schaum N., Huang J., Ved U., Beausejour C.M., Coppe J.P., Rodier F., Campisi J. (2013) p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629.
Narita M., Nũnez S., Heard E., Narita M., Lin A.W., Hearn S.A., Spector D.L., Hannon G.J., Lowe S.W. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 113, 703–716.
Basisty N., Kale A., Jeon O.H., Kuehnemann C., Payne T., Rao C., Holtz A., Shah S., Sharma V., Ferrucci L., Campisi J., Schilling B. (2020) A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599.
Zhu Y., Tchkonia T., Pirtskhalava T., Gower A.C., Ding H., Giorgadze N., Palmer A.K., Ikeno Y., Hubbard G.B., Lenburg M., O’Hara S.P., LaRusso N.F., Miller J.D., Roos C.M., Verzosa G.C., LeBrasseur N.K., Wren J.D., Farr J.N., Khosla S., Stout M.B., McGowan S.J., Fuhrmann-Stroissnigg H., Gurkar A.U., Zhao J., Colangelo D., Dorronsoro A., Ling Y.Y., Barghouthy A.S., Navarro D.C., Sano T., Robbins P.D., Niedernhofer L.J., Kirkland J.L. (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell. 14, 644–658.
Kirkland J.L., Tchkonia T. (2017) Cellular senescence: a translational perspective. EBioMedicine. 21, 21–28.
Baar M.P., Brandt R.M.C., Putavet D.A., Klein J.D.D., Derks K.W.J., Bourgeois B.R.M., Stryeck S., Rijksen Y., van Willigenburg H., Feijtel D.A., van der Pluijm I., Essers J., van Cappellen W.A., van I.W.F., Houtsmuller A.B., Pothof J., de Bruin R.W.F., Madl T., Hoeijmakers J.H.J., Campisi J., de Keizer P.L.J. (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 169, 132–147. e116.
Franceschi C., Campisi J. (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl. 1), S4–S9.
Müller L., Di Benedetto S., Pawelec G. (2019) The immune system and its dysregulation with aging. In: Biochem. Cell Biol. Ageing: Part II Clin. Sci. Eds Harris J.R., Korolchuk V.I. Singapore: Springer Singapore, pp. 21–43.
Sempowski G.D., Hale L.P., Sundy J.S., Massey J.M., Koup R.A., Douek D.C., Patel D.D., Haynes B.F. (2000) Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J. Immunol. 164, 2180–2187.
Majumdar S., Nandi D. (2018) Thymic atrophy: experimental studies and therapeutic interventions. Scand. J. Immunol. 87, 4–14.
Chaudhry M.S., Velardi E., Dudakov J.A., van den Brink M.R. (2016) Thymus: the next (re)generation. Immunol. Rev. 271, 56–71.
Crooke S.N., Ovsyannikova I.G., Poland G.A., Kennedy R.B. (2019) Immunosenescence: a systems-level overview of immune cell biology and strategies for improving vaccine responses. Exp. Gerontol. 124, 110632.
Pera A., Caserta S., Albanese F., Blowers P., Morrow G., Terrazzini N., Smith H.E., Rajkumar C., Reus B., Msonda J.R., Verboom M., Hallensleben M., Blasczyk R., Davies K.A., Kern F. (2018) CD28null pro-atherogenic CD4 T-cells explain the link between CMV infection and an increased risk of cardiovascular death. Theranostics. 8, 4509–4519.
Michaud M., Balardy L., Moulis G., Gaudin C., Peyrot C., Vellas B., Cesari M., Nourhashemi F. (2013) Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 14, 877–882.
Feldman N., Rotter-Maskowitz A., Okun E. (2015) DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 24, 29–39.
Ferrucci L., Fabbri E. (2018) Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522.
Franceschi C., Garagnani P., Parini P., Giuliani C., Santoro A. (2018) Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590.
Huang J., Xie Y., Sun X., Zeh H.J., 3rd, Kang R., Lotze M.T., Tang D. (2015) DAMPs, ageing, and cancer: the “DAMP hypothesis”. Ageing Res. Rev. 24, 3–16.
Gong T., Liu L., Jiang W., Zhou R. (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112.
Mangan M.S.J., Olhava E.J., Roush W.R., Seidel H.M., Glick G.D., Latz E. (2018) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 17, 588–606.
De Cecco M., Ito T., Petrashen A.P., Elias A.E., Skvir N.J., Criscione S.W., Caligiana A., Brocculi G., Adney E.M., Boeke J.D., Le O., Beauséjour C., Ambati J., Ambati K., Simon M., Seluanov A., Gorbunova V., Slagboom P.E., Helfand S.L., Neretti N., Sedivy J.M. (2019) L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 566, 73–78.
Vizioli M.G., Liu T., Miller K.N., Robertson N.A., Gilroy K., Lagnado A.B., Perez-Garcia A., Kiourtis C., Dasgupta N., Lei X., Kruger P.J., Nixon C., Clark W., Jurk D., Bird T.G., Passos J.F., Berger S.L., Dou Z., Adams P.D. (2020) Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445.
Wu Y., Wei Q., Yu J. (2019) The cGAS/STING pathway: a sensor of senescence-associated DNA damage and trigger of inflammation in early age-related macular degeneration. Clin. Interv. Aging. 14, 1277–1283.
Ablasser A., Hur S. (2020) Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29.
Lugrin J., Martinon F. (2018) The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol. Rev. 281, 99–114.
Rai V., Maldonado A.Y., Burz D.S., Reverdatto S., Yan S.F., Schmidt A.M., Shekhtman A. (2012) Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem. 287, 5133–5144.
Senatus L.M., Schmidt A.M. (2017) The AGE-RAGE axis: implications for age-associated arterial diseases. Front. Genet. 8, 187.
Enioutina E.Y., Bareyan D., Daynes R.A. (2011) A role for immature myeloid cells in immune senescence. J. Immunol. 186, 697–707.
Ergen A.V., Boles N.C., Goodell M.A. (2012) Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 119, 2500–2509.
Kuilman T., Peeper D.S. (2009) Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer. 9, 81–94.
Rea I.M., Gibson D.S., McGilligan V., McNerlan S.E., Alexander H.D., Ross O.A. (2018) Age and age-related diseases: role of inflammation triggers and cytokines. Front. Immunol. 9, 586.
Watanabe S., Kawamoto S., Ohtani N., Hara E. (2017) Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 108, 563–569.
Huh J.Y., Park Y.J., Ham M., Kim J.B. (2014) Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells. 37, 365–371.
Francisco V., Pino J., Gonzalez-Gay M.A., Mera A., Lago F., Gómez R., Mobasheri A., Gualillo O. (2018) Adipokines and inflammation: is it a question of weight? Br. J. Pharmacol. 175, 1569–1579.
Fain J.N. (2006) Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. In: Vitamins & Hormones. 74. Acad. Press, 443–477.
Forsythe L.K., Wallace J.M., Livingstone M.B. (2008) Obesity and inflammation: the effects of weight loss. Nutr. Res. Rev. 21, 117–133.
Tilg H., Moschen A.R. (2006) Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6, 772–783.
Frasca D., Blomberg B.B. (2016) Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology. 17, 7–19.
Buford T.W. (2017) (Dis)Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome. 5, 80.
Nagpal R., Mainali R., Ahmadi S., Wang S., Singh R., Kavanagh K., Kitzman D.W., Kushugulova A., Marotta F., Yadav H. (2018) Gut microbiome and aging: physiological and mechanistic insights. Nutr. Healthy Aging. 4, 267–285.
Dudakov J.A., Hanash A.M., van den Brink M.R. (2015) Interleukin-22: immunobiology and pathology. Annu. Rev. Immunol. 33, 747–785.
Satoh-Takayama N., Vosshenrich C.A., Lesjean-Pottier S., Sawa S., Lochner M., Rattis F., Mention J.J., Thiam K., Cerf-Bensussan N., Mandelboim O., Eberl G., Di Santo J.P. (2008) Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 29, 958–970.
Rakoff-Nahoum S., Paglino J., Eslami-Varzaneh F., Edberg S., Medzhitov R. (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 118, 229–241.
Ngo V.L., Abo H., Maxim E., Harusato A., Geem D., Medina-Contreras O., Merlin D., Gewirtz A.T., Nusrat A., Denning T.L. (2018) A cytokine network involving IL-36γ, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc. Natl. Acad. Sci. USA. 115, E5076–e5085.
Sugimoto K., Ogawa A., Mizoguchi E., Shimomura Y., Andoh A., Bhan A.K., Blumberg R.S., Xavier R.J., Mizoguchi A. (2008) IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544.
Kühn F., Adiliaghdam F., Cavallaro P.M., Hamarneh S.R., Tsurumi A., Hoda R.S., Munoz A.R., Dhole Y., Ramirez J.M., Liu E., Vasan R., Liu Y., Samarbafzadeh E., Nunez R.A., Farber M.Z., Chopra V., Malo M.S., Rahme L.G., Hodin R.A. (2020) Intestinal alkaline phosphatase targets the gut barrier to prevent aging. JCI Insight. 5, 134049.
Lycke N.Y., Bemark M. (2017) The regulation of gut mucosal IgA B-cell responses: recent developments. Mucosal Immunol. 10, 1361–1374.
Macpherson A.J., Harris N.L. (2004) Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 4, 478–485.
Bourassa M.W., Alim I., Bultman S.J., Ratan R.R. (2016) Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci. Lett. 625, 56–63.
Jeffery I.B., Lynch D.B., O’Toole P.W. (2016) Composition and temporal stability of the gut microbiota in older persons. ISME J. 10, 170–182.
Meier J., Sturm A. (2009) The intestinal epithelial barrier: does it become impaired with age? Dig. Dis. 27, 240–245.
Bai J., Liu F. (2019) The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes. 68, 1099.
Scheid M.M.A., Moreno Y.M.F., Maróstica Junior M.R., Pastore G.M. (2013) Effect of prebiotics on the health of the elderly. Food Res. Int. 53, 426–432.
Stebegg M., Silva-Cayetano A., Innocentin S., Jenkins T.P., Cantacessi C., Gilbert C., Linterman M.A. (2019) Heterochronic faecal transplantation boosts gut germinal centres in aged mice. Nat. Commun. 10, 2443.
Wu H., Esteve E., Tremaroli V., Khan M.T., Caesar R., Mannerås-Holm L., Ståhlman M., Olsson L.M., Serino M., Planas-Fèlix M., Xifra G., Mercader J.M., Torrents D., Burcelin R., Ricart W., Perkins R., Fernàndez-Real J.M., Bäckhed F. (2017) Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858.
Hurez V., Dao V., Liu A., Pandeswara S., Gelfond J., Sun L., Bergman M., Orihuela C.J., Galvan V., Padrón Á., Drerup J., Liu Y., Hasty P., Sharp Z.D., Curiel T.J. (2015) Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell. 14, 945–956.
Qiao Y., Sun J., Xia S., Tang X., Shi Y., Le G. (2014) Effects of resveratrol on gut microbiota and fat storage in a mouse model with high-fat-induced obesity. Food Funct. 5, 1241–1249.
Neil J.A., Cadwell K. (2018) The intestinal virome and immunity. J. Immunol. 201, 1615–1624.
Mukhopadhya I., Segal J.P., Carding S.R., Hart A.L., Hold G.L. (2019) The gut virome: the “missing link” between gut bacteria and host immunity? Therapeut. Adv. Gastroenterol. 12, 1756284819836620.
Bayo J., Molina R., Perez J., Perez-Ruiz E., Aparicio J., Beato C., Berros J.P., Bolanos M., Grana B., Santaballa A. (2019) SEOM clinical guidelines to primary prevention of cancer (2018). Clin. Transl. Oncol. 21, 106–113.
Mancuso R., Sicurella M., Agostini S., Marconi P., Clerici M. (2019) Herpes simplex virus type 1 and Alzheimer’s disease: link and potential impact on treatment. Expert. Rev. Anti Infect. Ther. 17, 715–731.
Readhead B., Haure-Mirande J.-V., Funk C.C., Richards M.A., Shannon P., Haroutunian V., Sano M., Liang W.S., Beckmann N.D., Price N.D., Reiman E.M., Schadt E.E., Ehrlich M.E., Gandy S., Dudley J.T. (2018) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 99, 64–82. e67.
Reinke H., Asher G. (2019) Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 20, 227–241.
Соловьёв И.А., Шапошников М.В., Москалев А.А. (2018) Генетические механизмы влияния света и фототрансдукции на продолжительность жизни Drosophila melanogaster. Вавиловский журн. генет. селекции. 22, 878–886.
Kinouchi K., Magnan C., Ceglia N., Liu Y., Cervan-tes M., Pastore N., Huynh T., Ballabio A., Baldi P., Masri S., Sassone-Corsi P. (2018) Fasting imparts a switch to Alternative daily pathways in liver and muscle. Cell Rept. 25, 3299–3314. e3296.
Ulgherait M., Chen A., McAllister S.F., Kim H.X., Delventhal R., Wayne C.R., Garcia C.J., Recinos Y., Oliva M., Canman J.C., Picard M., Owusu-Ansah E., Shirasu-Hiza M. (2020) Circadian regulation of mitochondrial uncoupling and lifespan. Nat. Commun. 11, 1927.
Solovev I., Dobrovolskaya E., Shaposhnikov M., Sheptyakov M., Moskalev A. (2019) Neuron-specific overexpression of core clock genes improves stress-resistance and extends lifespan of Drosophila melanogaster. Exp. Gerontol. 117, 61–71.
Solovev I., Shegoleva E., Fedintsev A., Shaposhnikov M., Moskalev A. (2019) Circadian clock genes' overexpression in Drosophila alters diet impact on lifespan. Biogerontology. 20, 159–170.
Adler P., Chiang C.K., Mayne J., Ning Z., Zhang X., Xu B., Cheng H.M., Figeys D. (2019) Aging disrupts the circadian patterns of protein expression in the murine hippocampus. Front. Aging Neurosci. 11, 368.
Zhao J., Warman G.R., Cheeseman J.F. (2019) The functional changes of the circadian system organization in aging. Ageing Res. Rev. 52, 64–71.
Xie Y., Tang Q., Chen G., Xie M., Yu S., Zhao J., Chen L. (2019) New insights into the circadian rhythm and its related diseases. Front. Physiol. 10, 682.
McAlpine C.S., Kiss M.G., Rattik S., He S., Vassalli A., Valet C., Anzai A., Chan C.T., Mindur J.E., Kahles F., Poller W.C., Frodermann V., Fenn A.M., Gregory A.F., Halle L., Iwamoto Y., Hoyer F.F., Binder C.J., Libby P., Tafti M., Scammell T.E., Nahrendorf M., Swirski F.K. (2019) Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. 566, 383–387.
Dimitrov S., Lange T., Gouttefangeas C., Jensen A.T.R., Szczepanski M., Lehnnolz J., Soekadar S., Rammensee H.-G., Born J., Besedovsky L. (2019) Gαs-coupled receptor signaling and sleep regulate integrin activation of human antigen-specific T cells. J. Exp. Med. 216, 517–526.
Axelsson J., Rehman J.U., Akerstedt T., Ekman R., Miller G.E., Hoglund C.O., Lekander M. (2013) Effects of sustained sleep restriction on mitogen-stimulated cytokines, chemokines and T helper 1/ T helper 2 balance in humans. PLoS One. 8, e82291.
Chang J., Garva R., Pickard A., Yeung C.-Y.C., Mallikarjun V., Swift J., Holmes D.F., Calverley B., Lu Y., Adamson A., Raymond-Hayling H., Jensen O., Shearer T., Meng Q.J., Kadler K.E. (2020) Circadian control of the secretory pathway maintains collagen homeostasis. Nat. Cell Biol. 22, 74–86.
Zada D., Bronshtein I., Lerer-Goldshtein T., Garini Y., Appelbaum L. (2019) Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat. Commun. 10, 895.
Bellesi M., Bushey D., Chini M., Tononi G., Cirelli C. (2016) Contribution of sleep to the repair of neuronal DNA double-strand breaks: evidence from flies and mice. Sci. Rep. 6, 36804.
Hablitz L.M., Vinitsky H.S., Sun Q., Staeger F.F., Sigurdsson B., Mortensen K.N., Lilius T.O., Nedergaard M. (2019) Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci. Adv. 5, eaav5447.
Lucey B.P., McCullough A., Landsness E.C., Toedebusch C.D., McLeland J.S., Zaza A.M., Fagan A.M., McCue L., Xiong C., Morris J.C., Benzinger T.L.S., Holtzman D.M. (2019) Reduced non–rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 11, eaau6550.
Benedict C., Blennow K., Zetterberg H., Cedernaes J. (2020) Effects of acute sleep loss on diurnal plasma dynamics of CNS health biomarkers in young men. Neurology. 94, e1181.
Lucey B.P., Hicks T.J., McLeland J.S., Toedebusch C.D., Boyd J., Elbert D.L., Patterson B.W., Baty J., Morris J.C., Ovod V., Mawuenyega K.G., Bateman R.J. (2018) Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 83, 197–204.
Carskadon M.A., Chappell K.R., Barker D.H., Hart A.C., Dwyer K., Gredvig-Ardito C., Starr C., McGeary J.E. (2019) A pilot prospective study of sleep patterns and DNA methylation-characterized epigenetic aging in young adults. BMC Res. Notes. 12, 583.
Hardeland R. (2017) Melatonin as a geroprotector: healthy aging vs. extension of lifespan. In: Anti-Aging Drugs: from Basic Research to Clinical Practice. The Royal Soc. Chem. 474–495.
Finkel T. (2015) The metabolic regulation of aging. Nat. Med. 21, 1416–1423.
Cornu M., Albert V., Hall M.N. (2013) mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 23, 53–62.
Schmeisser K., Parker J.A. (2019) Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biology. 7, 192.
Yeo D., Kang C., Gomez-Cabrera M.C., Vina J., Ji L.L. (2019) Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 130, 361–368.
López-Lluch G., Hunt N., Jones B., Zhu M., Jamieson H., Hilmer S., Cascajo M.V., Allard J., Ingram D.K., Navas P., de Cabo R. (2006) Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA. 103, 1768–1773.
Finley L.W.S., Lee J., Souza A., Desquiret-Dumas V., Bullock K., Rowe G.C., Procaccio V., Clish C.B., Arany Z., Haigis M.C. (2012) Skeletal muscle transcriptional coactivator PGC-1α mediates mitochondrial, but not metabolic, changes during calorie restriction. Proc. Natl. Acad. Sci. USA. 109, 2931.
Picca A., Lezza A.M.S. (2015) Regulation of mitochondrial biogenesis through TFAM–mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 25, 67–75.
Aquilano K., Vigilanza P., Baldelli S., Pagliei B., Rotilio G., Ciriolo M.R. (2010) Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J. Biol. Chem. 285, 21590–21599.
Martins R., Lithgow G.J., Link W. (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging cell. 15, 196–207.
López-Lluch G., Hernández-Camacho J.D., Fernández-Ayala D.J.M., Navas P. (2018) Mitochondrial dysfunction in metabolism and ageing: shared mechanisms and outcomes? Biogerontology. 19, 461–480.
Schlachetzki J.C.M., Toda T., Mertens J. (2020) When function follows form: nuclear compartment structure and the epigenetic landscape of the aging neuron. Exp. Gerontol. 133, 110876.
Mathieson T., Franken H., Kosinski J., Kurzawa N., Zinn N., Sweetman G., Poeckel D., Ratnu V.S., Schramm M., Becher I., Steidel M., Noh K.-M., Bergamini G., Beck M., Bantscheff M., Savitski M.M. (2018) Systematic analysis of protein turnover in primary cells. Nat. Commun. 9, 689.
Rempel I.L., Steen A., Veenhoff L.M. (2020) Poor old pores – the challenge of making and maintaining nuclear pore complexes in aging. FEBS J. 287, 1058–1075.
Buchwalter A., Kaneshiro J.M., Hetzer M.W. (2019) Coaching from the sidelines: the nuclear periphery in genome regulation. Nat. Rev. Genet. 20, 39–50.
Parrish A.R. (2017) The impact of aging on epithelial barriers. Tissue Barriers. 5, e1343172.
Surber C., Humbert P., Abels C., Maibach H. (2018) The acid mantle: a myth or an essential part of skin health? Curr. Probl. Dermatol. 54, 1–10.
Shin J.-W., Kwon S.-H., Choi J.-Y., Na J.-I., Huh C.-H., Choi H.-R., Park K.-C. (2019) Molecular mechanisms of dermal aging and antiaging approaches. Int. J. Mol. Sci. 20, 2126.
O’Sullivan E.D., Hughes J., Ferenbach D.A. (2017) Renal aging: causes and consequences. J. Am. Soc. Nephrol. 28, 407–420.
Wiggins J.E. (2012) Aging in the glomerulus. J. Gerontol. Ser. A, Biol. Sci. Med. Sci. 67, 1358–1364.
Menon M.C., Chuang P.Y., He C.J. (2012) The glomerular filtration barrier: components and crosstalk. Int. J. Nephrol. 2012, 749010.
Festa B.P., Chen Z., Berquez M., Debaix H., Tokonami N., Prange J.A., Hoek G.v.d., Alessio C., Raimondi A., Nevo N., Giles R.H., Devuyst O., Luciani A. (2018) Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 9, 161.
Zhang J., Hansen K.M., Pippin J.W., Chang A.M., Taniguchi Y., Krofft R.D., Pickering S.G., Liu Z.H., Abrass C.K., Shankland S.J. (2012) De novo expression of podocyte proteins in parietal epithelial cells in experimental aging nephropathy. Am. J. Physiol. Renal Physiol. 302, F571–F580.
Ungvari Z., Tarantini S., Kiss T., Wren J.D., Giles C.B., Griffin C.T., Murfee W.L., Pacher P., Csiszar A. (2018) Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 15, 555–565.
Silva T.M.D., Li Y., Kinzenbaw D.A., Sigmund C.D., Faraci F.M. (2018) Endothelial PPARγ (Peroxisome Proliferator–Activated Receptor-γ) is essential for preventing endothelial dysfunction with aging. Hypertension. 72, 227–234.
Cho J.H., Kim E.C., Son Y., Lee D.W., Park Y.S., Choi J.H., Cho K.H., Kwon K.S., Kim J.R. (2020) CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death Differ. 27, 2681–2696.https://doi.org/10.1038/s41418-41020-40537-41419
Sepúlveda C., Palomo I., Fuentes E. (2017) Mechanisms of endothelial dysfunction during aging: predisposition to thrombosis. Mech. Ageing Dev. 164, 91–99.
Stamatovic S.M., Martinez-Revollar G., Hu A., Choi J., Keep R.F., Andjelkovic A.V. (2019) Decline in Sirtuin-1 expression and activity plays a critical role in blood–brain barrier permeability in aging. Neurobiol. Dis. 126, 105–116.
Mitchell S.J., Martin-Montalvo A., Mercken E.M., Palacios H.H., Ward T.M., Abulwerdi G., Minor R.K., Vlasuk G.P., Ellis J.L., Sinclair D.A., Dawson J., Allison D.B., Zhang Y., Becker K.G., Bernier M., de Cabo R. (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rept. 6, 836–843.
Doyle K.P., Cekanaviciute E., Mamer L.E., Buckwalter M.S. (2010) TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflammation. 7, 62.
Senatorov V.V., Jr., Friedman A.R., Milikovsky D.Z., Ofer J., Saar-Ashkenazy R., Charbash A., Jahan N., Chin G., Mihaly E., Lin J.M., Ramsay H.J., Moghbel A., Preininger M.K., Eddings C.R., Harrison H.V., Patel R., Shen Y., Ghanim H., Sheng H., Veksler R., Sudmant P.H., Becker A., Hart B., Rogawski M.A., Dillin A., Friedman A., Kaufer D. (2019) Blood–brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 11, eaaw8283.
Erickson M.A., Banks W.A. (2019) Age-associated changes in the immune system and blood–brain barrier functions. Int. J. Mol. Sci. 20, 1632.
Ding F., Yao J., Rettberg J.R., Chen S., Brinton R.D. (2013) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One. 8, e79977.
Johanson C., Flaherty S., Messier A., Duncan J., Silverberg G. (2006) Expression of the beta-amyloid transporter, LRP-1, in aging choroid plexus: implications for the CSF-brain system in NPH and Alzheimer’s disease. Cerebrospinal Fluid Res. 3, S29.
Tejera D., Mercan D., Sanchez-Caro J.M., Hanan M., Greenberg D., Soreq H., Latz E., Golenbock D., Heneka M.T. (2019) Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 38, e101064.
Sartorius T., Peter A., Heni M., Maetzler W., Fritsche A., Häring H.U., Hennige A.M. (2015) The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One. 10, e0126804.
Banks W.A., Farr S.A., Morley J.E. (2000) Permeability of the blood-brain barrier to albumin and insulin in the young and aged SAMP8 mouse. J. Gerontol. A Biol. Sci. Med. Sci. 55, B601–B606.
Montagne A., Nation D.A., Sagare A.P., Barisano G., Sweeney M.D., Chakhoyan A., Pachicano M., Joe E., Nelson A.R., D’Orazio L.M., Buennagel D.P., Harrington M.G., Benzinger T.L.S., Fagan A.M., Ringman J.M., Schneider L.S., Morris J.C., Reiman E.M., Caselli R.J., Chui H.C., Tcw J., Chen Y., Pa J., Conti P.S., Law M., Toga A.W., Zlokovic B.V. (2020) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 581, 71–76.
Branca J.J.V., Gulisano M., Nicoletti C. (2019) Intestinal epithelial barrier functions in ageing. Ageing Res. Rev. 54, 100938.
Rodriguez-Fernandez I.A., Qi Y., Jasper H. (2019) Loss of a proteostatic checkpoint in intestinal stem cells contributes to age-related epithelial dysfunction. Nat. Commun. 10, 1050.
Wilms E., Troost F.J., Elizalde M., Winkens B., de Vos P., Mujagic Z., Jonkers D.M.A.E., Masclee A.A.M. (2020) Intestinal barrier function is maintained with aging – a comprehensive study in healthy subjects and irritable bowel syndrome patients. Sci. Rep. 10, 475.
Фоменко А.Н., Прошкина Е.Н., Фединцев А.Ю., Цветков В.О., Шапошников М.В., Москалев А.А. (2016) Потенциальные геропротекторы. Санкт-Петербург: Европейский Дом.
Whittemore K., Vera E., Martinez-Nevado E., Sanpera C., Blasco M.A. (2019) Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA. 116, 15122–15127.
Epel E.S., Merkin S.S., Cawthon R., Blackburn E.H., Adler N.E., Pletcher M.J., Seeman T.E. (2008) The rate of leukocyte telomere shortening predicts mortality from cardiovascular disease in elderly men. Aging (Albany NY). 1, 81–88.
Lin Y., Damjanovic A., Metter E.J., Nguyen H., Truong T., Najarro K., Morris C., Longo D.L., Zhan M., Ferrucci L., Hodes R.J., Weng N.P. (2015) Age-associated telomere attrition of lymphocytes in vivo is co-ordinated with changes in telomerase activity, composition of lymphocyte subsets and health conditions. Clin. Sci. (Lond). 128, 367–377.
Jiang H., Schiffer E., Song Z., Wang J., Zurbig P., Thedieck K., Moes S., Bantel H., Saal N., Jantos J., Brecht M., Jeno P., Hall M.N., Hager K., Manns M.P., Hecker H., Ganser A., Dohner K., Bartke A., Meissner C., Mischak H., Ju Z., Rudolph K.L. (2008) Proteins induced by telomere dysfunction and DNA damage represent biomarkers of human aging and disease. Proc. Natl. Acad. Sci. USA. 105, 11299–11304.
Wagner W. (2019) The link between epigenetic clocks for aging and senescence. Front. Genet. 10, 303.
Levine M.E., Lu A.T., Quach A., Chen B.H., Assimes T.L., Bandinelli S., Hou L., Baccarelli A.A., Stewart J.D., Li Y., Whitsel E.A., Wilson J.G., Rei-ner A.P., Aviv A., Lohman K., Liu Y., Ferrucci L., Horvath S. (2018) An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 10, 573–591.
Bell C.G., Lowe R., Adams P.D., Baccarelli A.A., Beck S., Bell J.T., Christensen B.C., Gladyshev V.N., Heijmans B.T., Horvath S., Ideker T., Issa J.J., Kelsey K.T., Marioni R.E., Reik W., Relton C.L., Schalkwyk L.C., Teschendorff A.E., Wagner W., Zhang K., Rakyan V.K. (2019) DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249.
Lu A.T., Quach A., Wilson J.G., Reiner A.P., Aviv A., Raj K., Hou L., Baccarelli A.A., Li Y., Stewart J.D., Whitsel E.A., Assimes T.L., Ferrucci L., Horvath S. (2019) DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 11, 303–327.
Ashapkin V.V., Kutueva L.I., Vanyushin B.F. (2020) Quantitative analysis of DNA methylation by bisulfite sequencing. Methods Mol. Biol. 2138, 297–312.
Marioni R.E., Shah S., McRae A.F., Chen B.H., Colicino E., Harris S.E., Gibson J., Henders A.K., Redmond P., Cox S.R., Pattie A., Corley J., Murphy L., Martin N.G., Montgomery G.W., Feinberg A.P., Fallin M.D., Multhaup M.L., Jaffe A.E., Joehanes R., Schwartz J., Just A.C., Lunetta K.L., Murabito J.M., Starr J.M., Horvath S., Baccarelli A.A., Levy D., Visscher P.M., Wray N.R., Deary I.J. (2015) DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16, 25.
Bocklandt S., Lin W., Sehl M.E., Sanchez F.J., Sinsheimer J.S., Horvath S., Vilain E. (2011) Epigenetic predictor of age. PLoS One. 6, e14821.
Koch C.M., Wagner W. (2011) Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY). 3, 1018–1027.
Lim U., Song M.A. (2018) DNA methylation as a biomarker of aging in epidemiologic studies. Methods Mol. Biol. 1856, 219–231.
Horvath S., Erhart W., Brosch M., Ammerpohl O., von Schonfels W., Ahrens M., Heits N., Bell J.T., Tsai P.C., Spector T.D., Deloukas P., Siebert R., Sipos B., Becker T., Rocken C., Schafmayer C., Hampe J. (2014) Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA. 111, 15538–15543.
Quach A., Levine M.E., Tanaka T., Lu A.T., Chen B.H., Ferrucci L., Ritz B., Bandinelli S., Neuhouser M.L., Beasley J.M., Snetselaar L., Wallace R.B., Tsao P.S., Absher D., Assimes T.L., Stewart J.D., Li Y., Hou L., Baccarelli A.A., Whitsel E.A., Horvath S. (2017) Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY). 9, 419–446.
Gadecka A., Bielak-Zmijewska A. (2019) Slowing down ageing: the role of nutrients and microbiota in modulation of the epigenome. Nutrients. 11, 1251.
Borkowska J., Domaszewska-Szostek A., Kolodziej P., Wicik Z., Polosak J., Buyanovskaya O., Charzewski L., Stanczyk M., Noszczyk B., Puzianowska-Kuznicka M. (2020) Alterations in 5hmC level and genomic distribution in aging-related epigenetic drift in human adipose stem cells. Epigenomics. 12, 423–437.
Dhahbi J.M. (2014) Circulating small noncoding RNAs as biomarkers of aging. Ageing Res. Rev. 17, 86–98.
Olivieri F., Capri M., Bonafe M., Morsiani C., Jung H.J., Spazzafumo L., Vina J., Suh Y. (2017) Circulating miRNAs and miRNA shuttles as biomarkers: perspective trajectories of healthy and unhealthy aging. Mech. Ageing Dev. 165, 162–170.
Zhang X., Hong R., Chen W., Xu M., Wang L. (2019) The role of long noncoding RNA in major human disease. Bioorg. Chem. 92, 103214.
Johnson A.A., Shokhirev M.N., Wyss-Coray T., Lehallier B. (2020) Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Ageing Res. Rev. 60, 101070.
Mayer O., Gelzinsky J., Seidlerova J., Materankova M., Mares S., Svobodova V., Trefil L., Cifkova R., Filipovsky J. (2020) The role of advanced glycation end products in vascular aging: which parameter is the most suitable as a biomarker? J. Hum. Hypertens.https://doi.org/10.1038/s41371-41020-40327-41373
Chaleckis R., Murakami I., Takada J., Kondoh H., Yanagida M. (2016) Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. USA. 113, 4252–4259.
Xia X., Chen W., McDermott J., Han J.-D.J. (2017) Molecular and phenotypic biomarkers of aging. F1000Res. 6, 860.
Solovev I.A., Shaposhnikov M.V., Moskalev A. (2019) An overview of the molecular and cellular biomarkers of aging. In: Biomarkers of Human Aging. Ed. Moskalev A. Cham: Springer Int. Publ., pp. 67–78.
Moskalev A. (2019) Biomarkers of Human Aging. Cham: Springer Internat. Publ.
Crimmins E., Vasunilashorn S., Kim J.K., Alley D. (2008) Biomarkers related to aging in human populations. Adv. Clin. Chem. 46, 161–216.
Olivetti G., Melissari M., Capasso J.M., Anversa P. (1991) Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ. Res. 68, 1560–1568.
Steenman M., Lande G. (2017) Cardiac aging and heart disease in humans. Biophys. Rev. 9, 131–137.
Santhanakrishnan R., Wang N., Larson M.G., Magnani J.W., McManus D.D., Lubitz S.A., Ellinor P.T., Cheng S., Vasan R.S., Lee D.S., Wang T.J., Levy D., Benjamin E.J., Ho J.E. (2016) Atrial fibrillation begets heart failure and vice versa: temporal associations and differences in preserved versus reduced ejection fraction. Circulation. 133, 484–492.
Martos R., Baugh J., Ledwidge M., O’Loughlin C., Murphy N.F., Conlon C., Patle A., Donnelly S.C., McDonald K. (2009) Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover. Eur. J. Heart Fail. 11, 191–197.
Ling L.H., Kistler P.M., Ellims A.H., Iles L.M., Lee G., Hughes G.L., Kalman J.M., Kaye D.M., Taylor A.J. (2012) Diffuse ventricular fibrosis in atrial fibrillation: noninvasive evaluation and relationships with aging and systolic dysfunction. J. Am. Coll. Cardiol. 60, 2402–2408.
Tanskanen M., Peuralinna T., Polvikoski T., Notkola I.L., Sulkava R., Hardy J., Singleton A., Kiuru-Enari S., Paetau A., Tienari P.J., Myllykangas L. (2008) Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann. Med. 40, 232–239.
Xie W., Santulli G., Reiken S.R., Yuan Q., Osborne B.W., Chen B.X., Marks A.R. (2015) Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 5, 11427.
Feridooni H.A., Dibb K.M., Howlett S.E. (2015) How cardiomyocyte excitation, calcium release and contraction become altered with age. J. Mol. Cell. Cardiol. 83, 62–72.
Najafi A., Sequeira V., Kuster D.W., van der Velden J. (2016) β-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur. J. Clin. Invest. 46, 362–374.
Ungvari Z., Tarantini S., Donato A.J., Galvan V., Csiszar A. (2018) Mechanisms of vascular aging. Circ. Res. 123, 849–867.
Zhang H., Wang B., Jin K. (2019) Circulating biomarkers of aging. In: Biomarkers of Human Aging. Ed. Moskalev A. Cham: Springer Internat. Publ., 349–371.
Brandenberger C., Mühlfeld C. (2017) Mechanisms of lung aging. Cell Tissue Res. 367, 469–480.
Lowery E.M., Brubaker A.L., Kuhlmann E., Kovacs E.J. (2013) The aging lung. Clin. Intervent. Aging. 8, 1489–1496.
Sperka T., Rudolph K.L. (2010) Intestinal stem cell aging. In: Mol. Mech. Adult Stem Cell Aging, 1. Ed. Rudolph K.L. Karger, 63–78.
Saffrey M.J. (2014) Aging of the mammalian gastrointestinal tract: a complex organ system. Age (Dordrecht, Netherlands). 36, 9603–9603.
Xu C., Zhu H., Qiu P. (2019) Aging progression of human gut microbiota. BMC Microbiol. 19, 236.
Kavanagh K., Hsu F.-C., Davis A.T., Kritchevsky S.B., Rejeski W.J., Kim S. (2019) Biomarkers of leaky gut are related to inflammation and reduced physical function in older adults with cardiometabolic disease and mobility limitations. GeroScience. 41, 923–933.
Vaiserman A.M., Koliada A.K., Marotta F. (2017) Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res. Rev. 35, 36–45.
Bárcena C., Valdés-Mas R., Mayoral P., Garabaya C., Durand S., Rodríguez F., Fernández-García M.T., Salazar N., Nogacka A.M., Garatachea N., Bossut N., Aprahamian F., Lucia A., Kroemer G., Freije J.M.P., Quirós P.M., López-Otín C. (2019) Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 25, 1234–1242.
Zhang W., Qu J., Liu G.H., Belmonte J.C.I. (2020) The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 21, 137–150.
Madeo F., Carmona-Gutierrez D., Hofer S.J., Kroemer G. (2019) Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 29, 592–610.
Evans L.W., Stratton M.S., Ferguson B.S. (2020) Dietary natural products as epigenetic modifiers in aging-associated inflammation and disease. Nat. Product Repts. https://doi.org/10.1039/c1039np00057g
Molina-Serrano D., Kyriakou D., Kirmizis A. (2019) Histone modifications as an intersection between diet and longevity. Front. Genet. 10, 192.
Gensous N., Franceschi C., Santoro A., Milazzo M., Garagnani P., Bacalini M.G. (2019) The impact of caloric restriction on the epigenetic signatures of aging. Int. J. Mol. Sci. 20, 2022.
Dai H., Sinclair D.A., Ellis J.L., Steegborn C. (2018) Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol. Ther. 188, 140–154.
Lettieri-Barbato D., Giovannetti E., Aquilano K. (2016) Effects of dietary restriction on adipose mass and biomarkers of healthy aging in human. Aging (Albany NY). 8, 3341–3355.
Rahmani J., Kord Varkaneh H., Clark C., Zand H., Bawadi H., Ryan P.M., Fatahi S., Zhang Y. (2019) The influence of fasting and energy restricting diets on IGF-1 levels in humans: a systematic review and meta-analysis. Ageing Res. Rev. 53, 100910.
Vitale G., Pellegrino G., Vollery M., Hofland L.J. (2019) Role of IGF-1 system in the modulation of longevity: controversies and new insights from a centenarians’ perspective. Front. Endocrinol. (Lausanne). 10, 27.
Solon-Biet S.M., Mitchell S.J., de Cabo R., Raubenheimer D., Le Couteur D.G., Simpson S.J. (2015) Macronutrients and caloric intake in health and longevity. J. Endocrinol. 226, R17–R28.
Martin-Montalvo A., Mercken E.M., Mitchell S.J., Palacios H.H., Mote P.L., Scheibye-Knudsen M., Gomes A.P., Ward T.M., Minor R.K., Blouin M.J., Schwab M., Pollak M., Zhang Y., Yu Y., Becker K.G., Bohr V.A., Ingram D.K., Sinclair D.A., Wolf N.S., Spindler S.R., Bernier M., de Cabo R. (2013) Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192.
Guevara-Aguirre J., Balasubramanian P., Guevara-Aguirre M., Wei M., Madia F., Cheng C.W., Hwang D., Martin-Montalvo A., Saavedra J., Ingles S., de Cabo R., Cohen P., Longo V.D. (2011) Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 3, 70ra13.
van der Spoel E., Jansen S.W., Akintola A.A., Ballieux B.E., Cobbaert C.M., Slagboom P.E., Blauw G.J., Westendorp R.G.J., Pijl H., Roelfsema F., van Heemst D. (2016) Growth hormone secretion is diminished and tightly controlled in humans enriched for familial longevity. Aging Cell. 15, 1126–1131.
Estebanez B., de Paz J.A., Cuevas M.J., Gonzalez-Gallego J. (2018) Endoplasmic reticulum unfolded protein response, aging and exercise: an update. Front. Physiol. 9, 1744.
Erusalimsky J.D. (2020) Oxidative stress, telomeres and cellular senescence: what non-drug interventions might break the link? Free Radic. Biol. Med. 150, 87–95.
Majidinia M., Bishayee A., Yousefi B. (2019) Polyphenols: major regulators of key components of DNA damage response in cancer. DNA Repair. 82, 102679.
Pomatto L.C.D., Davies K.J.A. (2018) Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 124, 420–430.
Fenech M. (2020) The role of nutrition in DNA replication, DNA damage prevention and DNA repair. In: Principles of Nutrigenetics and Nutrigenomics: Fundamentals for Individualized Nutrition. Eds Caterina R.D.E., Martinez J.A., Kohlmeier M. Acad. Press, 27–32.
Nagasaka M., Hashimoto R., Inoue Y., Ishiuchi K., Matsuno M., Itoh Y., Tokugawa M., Ohoka N., Morishita D., Mizukami H., Makino T., Hayashi H. (2018) Anti-tumorigenic activity of chrysin from Oroxylum indicum via pathway. Molecules. 23, 1394.
Vaid M., Prasad R., Singh T., Katiyar S.K. (2017) Dietary grape seed proanthocyanidins inactivate regulatory T cells by promoting NER-dependent DNA repair in dendritic cells in UVB-exposed skin. Oncotarget. 8, 49625–49636.
Vaid M., Sharma S.D., Katiyar S.K. (2010) Proanthocyanidins inhibit photocarcinogenesis through enhancement of DNA repair and xeroderma pigmentosum group A-dependent mechanism. Cancer Prev. Res. (Phila). 3, 1621–1629.
Thilakarathna W., Rupasinghe H.P.V. (2019) Microbial metabolites of proanthocyanidins reduce chemical carcinogen-induced DNA damage in human lung epithelial and fetal hepatic cells in vitro. Food Chem. Toxicol. 125, 479–493.
Nikolic B., Mitic-Culafic D., Vukovic-Gacic B., Knezevic-Vukcevic J. (2011) Modulation of genotoxicity and DNA repair by plant monoterpenes camphor, eucalyptol and thujone in Escherichia coli and mammalian cells. Food Chem. Toxicol. 49, 2035–2045.
Graziano S., Johnston R., Deng O., Zhang J., Gonzalo S. (2016) Vitamin D/vitamin D receptor axis regulates DNA repair during oncogene-induced senescence. Oncogene. 35, 5362–5376.
Драпкина О.М., Шепель Р.Н., Фомин В.В., Свистунов А.А. (2018) Место витамина D в профилактике преждевременного старения и развитии заболеваний, ассоциированных с возрастом. Терапевтический архив. 90, 69–75.
Kreienkamp R., Croke M., Neumann M.A., Bedia-Diaz G., Graziano S., Dusso A., Dorsett D., Carlberg C., Gonzalo S. (2016) Vitamin D receptor signaling improves Hutchinson–Gilford progeria syndrome cellular phenotypes. Oncotarget. 7, 30018–30031.
Georgiadis M.M., Chen Q., Meng J., Guo C., Wireman R., Reed A., Vasko M.R., Kelley M.R. (2016) Small molecule activation of apurinic/apyrimidinic endonuclease 1 reduces DNA damage induced by cisplatin in cultured sensory neurons. DNA Repair. 41, 32–41.
Martinez P., Blasco M.A. (2017) Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 216, 875–887.
Ramunas J., Yakubov E., Brady J.J., Corbel S.Y., Holbrook C., Brandt M., Stein J., Santiago J.G., Cooke J.P., Blau H.M. (2015) Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 29, 1930–1939.
Soda K. (2020) Spermine and gene methylation: a mechanism of lifespan extension induced by polyamine-rich diet. Amino Acids. 52, 213–224.
Bridgeman S.C., Ellison G.C., Melton P.E., Newsholme P., Mamotte C.D.S. (2018) Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes. Metab. 20, 1553–1562.
Agathocleous M., Meacham C.E., Burgess R.J., Piskounova E., Zhao Z., Crane G.M., Cowin B.L., Bruner E., Murphy M.M., Chen W., Spangrude G.J., Hu Z., DeBerardinis R.J., Morrison S.J. (2017) Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 549, 476–481.
Scheibye-Knudsen M., Mitchell S.J., Fang E.F., Iyama T., Ward T., Wang J., Dunn C.A., Singh N., Veith S., Hasan-Olive M.M., Mangerich A., Wilson M.A., Mattson M.P., Bergersen L.H., Cogger V.C., Warren A., Le Couteur D.G., Moaddel R., Wilson D.M., 3rd, Croteau D.L., de Cabo R., Bohr V.A. (2014) A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855.
Gomes A.P., Price N.L., Ling A.J., Moslehi J.J., Montgomery M.K., Rajman L., White J.P., Teodoro J.S., Wrann C.D., Hubbard B.P., Mercken E.M., Palmeira C.M., de Cabo R., Rolo A.P., Turner N., Bell E.L., Sinclair D.A. (2013) Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 155, 1624–1638.
Pasyukova E.G., Vaiserman A.M. (2017) HDAC inhibitors: a new promising drug class in anti-aging research. Mech. Ageing Dev. 166, 6–15.
Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R.C., Cota P., Wittnam J.L., Gogol-Doering A., Opitz L., Salinas-Riester G., Dettenhofer M., Kang H., Farinelli L., Chen W., Fischer A. (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 328, 753–756.
Krishnan V., Chow M.Z., Wang Z., Zhang L., Liu B., Liu X., Zhou Z. (2011) Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA. 108, 12325–12330.
Singh P., Thakur M.K. (2018) Histone deacetylase 2 inhibition attenuates downregulation of hippocampal plasticity gene expression during aging. Mol. Neurobiol. 55, 2432–2442.
Narayanan B.A., Narayanan N.K., Re G.G., Nixon D.W. (2003) Differential expression of genes induced by resveratrol in LNCaP cells: P53-mediated molecular targets. Int. J. Cancer. 104, 204–212.
Edwards C., Canfield J., Copes N., Rehan M., Lipps D., Bradshaw P.C. (2014) D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging (Albany NY). 6, 621–644.
Pietrocola F., Castoldi F., Markaki M., Lachkar S., Chen G., Enot D.P., Durand S., Bossut N., Tong M., Malik S.A., Loos F., Dupont N., Marino G., Abdelkader N., Madeo F., Maiuri M.C., Kroemer R., Codogno P., Sadoshima J., Tavernarakis N., Kroemer G. (2018) Aspirin recapitulates features of caloric restriction. Cell Rept. 22, 2395–2407.
van Rooij E., Kauppinen S. (2014) Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 6, 851–864.
Noren Hooten N., Martin-Montalvo A., Dluzen D.F., Zhang Y., Bernier M., Zonderman A.B., Becker K.G., Gorospe M., de Cabo R., Evans M.K. (2016) Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell. 15, 572–581.
Pinto S., Sato V.N., De-Souza E.A., Ferraz R.C., Camara H., Pinca A.P.F., Mazzotti D.R., Lovci M.T., Tonon G., Lopes-Ramos C.M., Parmigiani R.B., Wurtele M., Massirer K.B., Mori M.A. (2018) Enoxacin extends lifespan of C. elegans by inhibiting miR-34-5p and promoting mitohormesis. Redox Biol. 18, 84–92.
Gioia U., Francia S., Cabrini M., Brambillasca S., Michelini F., Jones-Weinert C.W., d’Adda di Fagagna F. (2019) Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci. Rep. 9, 6460.
Eisenberg T., Knauer H., Schauer A., Buttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L., Fussi H., Deszcz L., Hartl R., Schraml E., Criollo A., Megalou E., Weiskopf D., Laun P., Heeren G., Breitenbach M., Grubeck-Loebenstein B., Herker E., Fahrenkrog B., Frohlich K.U., Sinner F., Tavernarakis N., Minois N., Kroemer G., Madeo F. (2009) Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314.
Скулачев М.В., Скулачев В.П. (2017) Доказательство запрограммированности старения млекопитающих и перспективы биохимического подхода в борьбе со старостью. Биохимия. 82, 1747–1770.
Feniouk B.A., Skulachev V.P. (2017) Cellular and molecular mechanisms of action of mitochondria-targeted antioxidants. Curr. Aging Sci. 10, 41–48.
Lukashev A.N., Skulachev M.V., Ostapenko V., Savchenko A.Y., Pavshintsev V.V., Skulachev V.P. (2014) Advances in development of rechargeable mitochondrial antioxidants. Progr. Mol. Biol. Translat. Sci. 127, 251–265.
Bagul P.K., Katare P.B., Bugga P., Dinda A.K., Banerjee S.K. (2018) SIRT-3 Modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells. 7, 235.
Martinez-Cisuelo V., Gomez J., Garcia-Junceda I., Naudi A., Cabre R., Mota-Martorell N., Lopez-Torres M., Gonzalez-Sanchez M., Pamplona R., Barja G. (2016) Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle-aged mice. Exp. Gerontol. 83, 130–138.
Vernucci E., Tomino C., Molinari F., Limongi D., Aventaggiato M., Sansone L., Tafani M., Russo M.A. (2019) Mitophagy and oxidative stress in cancer and aging: focus on sirtuins and nanomaterials. Oxid. Med. Cell Longev. 2019, 6387357.
Ryu D., Mouchiroud L., Andreux P.A., Katsyuba E., Moullan N., Nicolet-Dit-Felix A.A., Williams E.G., Jha P., Lo Sasso G., Huzard D., Aebischer P., Sandi C., Rinsch C., Auwerx J. (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888.
Barcena C., Mayoral P., Quiros P.M. (2018) Mitohormesis, an antiaging paradigm. Int. Rev. Cell Mol. Biol. 340, 35–77.
Palmeira C.M., Teodoro J.S., Amorim J.A., Steegborn C., Sinclair D.A., Rolo A.P. (2019) Mitohormesis and metabolic health: the interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 141, 483–491.
Klaus S., Ost M. (2020) Mitochondrial uncoupling and longevity – a role for mitokines? Exp. Gerontol. 130, 110796.
Thoppil H., Riabowol K. (2019) Senolytics: a translational bridge between cellular senescence and organismal aging. Front. Cell Dev. Biol. 7, 367.
Zhu Y., Tchkonia T., Fuhrmann-Stroissnigg H., Dai H.M., Ling Y.Y., Stout M.B., Pirtskhalava T., Giorgadze N., Johnson K.O., Giles C.B., Wren J.D., Niedernhofer L.J., Robbins P.D., Kirkland J.L. (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 15, 428–435.
Yosef R., Pilpel N., Tokarsky-Amiel R., Biran A., Ovadya Y., Cohen S., Vadai E., Dassa L., Shahar E., Condiotti R., Ben-Porath I., Krizhanovsky V. (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190.
Zhu Y., Doornebal E.J., Pirtskhalava T., Giorgadze N., Wentworth M., Fuhrmann-Stroissnigg H., Niedernhofer L.J., Robbins P.D., Tchkonia T., Kirkland J.L. (2017) New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 9, 955–963.
Fuhrmann-Stroissnigg H., Ling Y.Y., Zhao J., McGowan S.J., Zhu Y., Brooks R.W., Grassi D., Gregg S.Q., Stripay J.L., Dorronsoro A., Corbo L., Tang P., Bukata C., Ring N., Giacca M., Li X., Tchkonia T., Kirkland J.L., Niedernhofer L.J., Robbins P.D. (2017) Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422.
Ozsvari B., Nuttall J.R., Sotgia F., Lisanti M.P. (2018) Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging (Albany NY). 10, 3294–3307.
Muñoz-Espín D., Rovira M., Galiana I., Giménez C., Lozano-Torres B., Paez-Ribes M., Llanos S., Chaib S., Muñoz-Martín M., Ucero A.C., Garaulet G., Mulero F., Dann S.G., VanArsdale T., Shields D.J., Bernardos A., Murguía J.R., Martínez-Máñez R., Serrano M. (2018) A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 10, e9355.
Guerrero A., Guiho R., Herranz N., Uren A., Withers D.J., Martínez-Barbera J.P., Tietze L.F., Gil J. (2020) Galactose‐modified duocarmycin prodrugs as senolytics. Aging Cell. 19, e13133. https://doi.org/10.1111/acel.13133
Kim K.M., Noh J.H., Bodogai M., Martindale J.L., Yang X., Indig F.E., Basu S.K., Ohnuma K., Morimoto C., Johnson P.F., Biragyn A., Abdelmohsen K., Gorospe M. (2017) Identification of senescent cell surface targetable protein DPP4. Genes Dev. 31, 1529–1534.
Thapa R.K., Nguyen H.T., Jeong J.-H., Kim J.R., Choi H.-G., Yong C.S., Kim J.O. (2017) Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci. Rep. 7, 43299.
Moskalev A., Chernyagina E., Kudryavtseva A., Shaposhnikov M. (2017) Geroprotectors: a unified concept and screening approaches. Aging Dis. 8, 354–363.
Moskalev A., Chernyagina E., de Magalhães J.P., Barardo D., Thoppil H., Shaposhnikov M., Budovsky A., Fraifeld V.E., Garazha A., Tsvetkov V., Bronovitsky E., Bogomolov V., Scerbacov A., Kuryan O., Gurinovich R., Jellen L.C., Kennedy B., Mamoshina P., Dobrovolskaya E., Aliper A., Kaminsky D., Zhavo-ronkov A. (2015) Geroprotectors.org: a new, structured and curated database of current therapeutic interventions in aging and age-related disease. Aging. 7, 616–628.
Trendelenburg A.U., Scheuren A.C., Potter P., Müller R., Bellantuono I. (2019) Geroprotectors: a role in the treatment of frailty. Mech. Ageing Dev. 180, 11–20.
Figueira I., Fernandes A., Mladenovic Djordjevic A., Lopez-Contreras A., Henriques C.M., Selman C., Ferreiro E., Gonos E.S., Trejo J.L., Misra J., Rasmussen L.J., Xapelli S., Ellam T., Bellantuono I. (2016) Interventions for age-related diseases: shifting the paradigm. Mech. Ageing Dev. 160, 69–92.
Conboy I.M., Conboy M.J., Smythe G.M., Rando T.A. (2003) Notch-mediated restoration of regenerative potential to aged muscle. Science. 302, 1575.
Carlson M.E., Hsu M., Conboy I.M. (2008) Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 454, 528–532.
Brack A.S., Conboy M.J., Roy S., Lee M., Kuo C.J., Keller C., Rando T.A. (2007) Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 317, 807.
Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676.
Sogabe Y., Seno H., Yamamoto T., Yamada Y. (2018) Unveiling epigenetic regulation in cancer, aging, and rejuvenation with in vivo reprogramming technology. Cancer Sci. 109, 2641–2650.
Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E., Araoka T., Vazquez-Ferrer E., Donoso D., Roman J.L., Xu J., Rodriguez Esteban C., Nuñez G., Nuñez Delicado E., Campistol J.M., Guillen I., Guillen P., Izpisua Belmonte J.C. (2016) In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell. 167, 1719–1733. e1712.
Gowing G., Svendsen S., Svendsen C.N. (2017) Ex vivo gene therapy for the treatment of neurological disorders. Prog. Brain Res. 230, 99–132.
Brooks R.W., Robbins P.D. (2018) Treating age-related diseases with somatic stem cells. In: Exosomes, Stem Cells and MicroRNA: Aging, Cancer and Age Related Disorders. Eds Mettinger K.L., Rameshwar P., Kumar V. Springer Internat. Publ. 29–45.
Hong J., Yun C.-O. (2019) Telomere gene therapy: polarizing therapeutic goals for treatment of various diseases. Cells. 8, 392.
Davidsohn N., Pezone M., Vernet A., Graveline A., Oliver D., Slomovic S., Punthambaker S., Sun X., Liao R., Bonventre J.V., Church G.M. (2019) A single combination gene therapy treats multiple age-related diseases. Proc. Natl. Acad. Sci. USA. 116, 23505–23511.
Kim J.-H., Hwang K.-H., Park K.-S., Kong I.D., Cha S.-K. (2015) Biological role of anti-aging protein Klotho. J. Lifestyle Med. 5, 1–6.
Horvath S., Singh K., Raj K., Khairnar S., Sanghavi A., Shrivastava A., Zoller J.A., Li C.Z., Herenu C.B., Canatelli-Mallat M., Lehmann M., Solberg Woods L.C., Martinez A.G., Wang T., Chiavellini P., Levine A.J., Chen H., Goya R.G., Katcher H.L. (2020) Reversing age: dual species measurement of epigenetic age with a single clock. bioRxiv. https://doi.org/10.1101/2020.1105.1107.082917
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология