

Молекулярная биология, 2020, T. 54, № 6, стр. 1006-1017
Опосредованная shРНК супрессия γ-синуклеина приводит к подавлению фосфорилирования p38/ERK/JNK и задержке клеточного цикла в клетках рака эндометрия
D. Sun a, W.-Y. Li b, S.-H. Chen c, Z.-F. Zhi a, H.-S. Lin a, J.-T. Fan a, *, Y.-J. Fan d, **
a Department of Gynaecology and Obstetrics, The First Affiliated Hospital of Guangxi Medical University
530021 Nanning, People’s Republic of China
b Department of Gynaecology and Obstetrics, Liuzhou General Hospital
545006 Liuzhou, People’s Republic of China
c Department of Gynaecology, Liuzhou Maternity and Child Healthcare Hospital
545001 Liuzhou, People’s Republic of China
d Department of Gynaecology and Obstetrics, University of Chinese Academy of Sciences, Shenzhen Hospital
518106 Shenzhen, People’s Republic of China
* E-mail: yjfan530@163.com
** E-mail: jiangtaofan1969@163.com
Поступила в редакцию 23.11.2019
После доработки 18.03.2020
Принята к публикации 23.03.2020
Аннотация
Исследованы эффекты лечения раковых клеток эндометрия человека с помощью γ-синуклеинспецифической образующей шпильки малой РНК (shRNA) и связанные с этим механизмы, действующие in vitro и in vivo через сигнальные пути p38, внеклеточной киназы (ERK) и N-терминальной киназы c-Jun (JNK). Пролиферацию и миграцию клеток оценивали с помощью тестов CCK8, Transwell и залечивания царапины. Для оценки изменений клеточного цикла использовали проточную цитометрию и лазерную сканирующую конфокальную микроскопию. Относительные уровни фосфорилированных и нефосфорилированных р38, ERK1/2 и JNK1/2/3 определяли in vitro и in vivo с помощью иммуноблотинга. В трансфицированных shРНК клетках значительно снизилась пролиферация, а также скорость миграции (Р < 0.05). Уровни фосфорилированных (р) белков p-p38, p-ERK1/2 и p‑JNK1/2/3 были понижены в экспериментальной группе in vitro и in vivo. Объем и масса опухоли в опытной группе были достоверно ниже (Р < 0.05). Время образования опухоли в группе отрицательного контроля было достоверно короче (Р < 0.05). Проточная цитометрия показала, что после подавления экспрессии гена SNCG количество клеток в G1- и митотической фазах увеличивается, а в S-фазе уменьшается (Р < 0.05). Конфокальная микроскопия показала, что процент клеток в митотической фазе увеличивается после подавления экспрессии гена SNCG (Р < 0.05). На основании полученных результатов можно предполагать, что shRNA-опосредованное подавление γ-синуклеина снижает пролиферацию, миграцию и опухолевую активность раковых клеток эндометрия за счет снижения регуляции фосфорилирования р38, ERK и JNK. Высокая экспрессия SNCG тесно связана с циклом роста раковых клеток эндометрия.
Синуклеины представляют собой семейство небольших растворимых нейрональных белков с высококонсервативной последовательностью и включают альфа-синуклеин (SNCA), бета-синуклеин (SNCB) и гамма-синуклеин (SNCG). Ген SNCG человека расположен в хромосомной области 10q23.2-q23.3, имеет длину около 5.0 тыс.н. и содержит четыре интрона и пять экзонов [1]. Митогенактивированные протеинкиназы (MAPKs) формируют сложную систему сетей, которая играет важную роль в пролиферации клеток, апоптозе, инвазии, метастазировании, формировании сосудов и передаче сигналов в опухолевых клетках. В клетках млекопитающих известно пять параллельных сигнальных путей MAPK, а именно: путь регулируемой внеклеточными сигналами киназы 1 (ERK1) и 2 (ERK2), путь N-концевой киназы c-Jun (JNK)/стрессактивируемой протеинкиназы, путь изофермента р38-киназы и пути ERK3/ERK4 и ERK5. Выживание клеток и апоптоз тесно связаны с сигнальными путями ERK и JNK [2]. В последние годы изучена взаимосвязь между SNCG и опухолями в зависимости от регуляторных факторов p38, ERK и JNK. Показано, что SNCG способствует миграции клеток рака молочной железы MCF7, влияя на активацию ERK, а ингибитор передачи ERK‑сигналов в значительной степени препятствует SNCG-индуцированной миграции [3]. Сайленсинг SNCG подавляет миграцию и пролиферацию клеток, возможно, за счет снижения уровней фосфорилированных (р) белков: p-AKT и p-ERK [4]. Гиперэкспрессия γ‑синуклеина придает клеткам колоректальной аденокарциномы LS174T как проинвазивные, так и доксорубицинопосредованные проапоптотические свойства, причем первые опосредуются последующими нисходящими сигнальными каскадами, включая ERK1/2, p38α и pan-JNK (pan ‒ общий белок), а также сигнальными трансдукторами и активаторами передачи транскрипционных сигналов [5]. На субклеточном уровне γ-синуклеин преимущественно располагается в цитоплазме, а на поздних стадиях митоза ‒ в промежуточном и крайне ассиметричном веретене деления и взаимодействует с центросомными белками. В условиях стресса γ-синуклеин перемещается из цитоплазмы в ядро. Эти динамические изменения в локализации γ-синуклеина позволяют предположить, что он может быть тесно связан с митозом и регуляцией клеточного цикла [6].
Однако пока не было сообщений по экспрессии SNCG в зависимости от пути MAPK/ERK/JNK и клеточного цикла в клетках карцины эндометрия. Экспрессия SNCG в ткани карциномы эндометрия человека значительно выше, чем в нормальном эндометрии. В исследованиях Международной федерация гинекологии и акушерства (International Federation of Gynaecology and Obstetrics) стадий рака эндометрия выявлено увеличение глубины инфильтрации миометрия и метастазирования лимфатических узлов в клеточных линиях рака эндометрия с высокой экспрессией SNCG [7]. Ранее мы идентифицировали линии клеток рака эндометрия I типа ‒ Ishikawa и HEC-1A ‒ как сверхэкспрессирующие SNCG [8]. Теперь, как в экспериментах in vivo, так и in vitro, мы предварительно оценили влияние повышенной экспрессии SNCG на онкогенез и биологические характеристики клеток рака эндометрия и на молекулярном уровне исследовали взаимосвязь между повышением экспрессии SNCG и ключевыми изменениями в путях передачи сигналов MAPK/ERK/JNK. Для выявления изменений в клеточном цикле использовали окрашивание акридиновым оранжевым, проточную цитометрию и лазерную конфокальную микроскопию.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клеточные линии. Линии клеток рака эндометрия человека I типа HEC-1A и Ishikawa были получены от Китайской академии наук (the Chinese Academy of Sciences, Shanghai, Китай). Клетки культивировали в среде Игла в модификации Дульбекко (DMEM; “Gibco”, США) с добавлением 10% инактивированной теплом эмбриональной сыворотки крупного рогатого скота (FBS), 100 ед/мл пенициллина и 100 мг/л стрептомицина в инкубаторе с 5% CO2 при 37°C.
Образующие шпильки короткие РНК (shРНК). Полную последовательность гена SNCG (NM_003087) извлекали из базы данных GeneBank и с использованием программного обеспечения Invitrogen Designer design service BLOCK-iT RNAi (“Invitrogen”, США) идентифицировали последовательности-мишени для интерференции с помощью shРНК. Используя BLAST для сравнения с базой данных генома, гомологичные кодирующие последовательности исключали, чтобы в итоге идентифицировать три последовательности, которые можно использовать как мишени для интерференции shRNA:
SNCG-KD1 (5'-CCGGGACCAA GGAGAATGTT GTACACTCGA GTGTACAACA TTCTCCTTGG TCTTTTTGAA TTCAAAAAGA CCAAGGAGAA TGTTGTACAC TCGAGTGTAC AACATTCTCC TTGGTC-3');
SNCG-KD2 (5'-CGGCCAAGGA GAATGTTGTA CAGACTCGAG TCTGTACAAC ATTCTCCTTG GTTTTTGAAT TCAAAAACCA AGGAGAATGT TGTACAGACT CGAGTCTGTAC AACATTCTCC TTGG-3');
SNCG-KD3 (5'-CCGGCAAGAC CAAGGAGAAT GTTGTCTCGA GACAACATTC TCCTTGGTCT TGTTTTTGAA TTCAAAAACA AGACCAAGGA GAATGTTGTC TCGAGACAAC ATTCTCCTTG GTCTTG-3').
Плазмиды были синтезированы в компании “Shanghai Genechem Biotech Co., Ltd.” (Китай).
Трансфекция. Клетки Ishikawa и HEC-1A пассировали и инокулировали в 6-луночные планшеты в количестве 2 × 105 клеток/лунка и культивировали в течение 2 суток. Каждую клеточную линию разделяли на группы: SNCG-KD1, SNCG-KD2, SNCG-KD3 (экспериментальные группы SNCG-KD), отрицательного контроля (группа NC) и контрольную (группа CON). Группы SNCG-KD1, SNCG-KD2, SNCG-KD3 и группу отрицательного контроля трансдуцировали плазмидами соответственно с лентивирусным вектором и SNCG-KD1, SNCG-KD2 или SNCG-KD3 и пустым вектором (лентивирусный вектор без короткой интерферирующей (si) РНК) в течение 24 ч в 100 мкл полибрена, инкубировали в среде DMEM, содержащей 10% FBS, в течение 72 ч, а затем культивировали в свежей среде, содержащей 2 г/мл пуриномицина, в течение 24 ч. В качестве контрольных клеток (CON-группа) использовали нормальные клетки HEC-1A и Ishikawa с высокой экспрессией SNCG.
Количественная ПЦР с обратной транскрипцией (ОТ-ПЦР). Уровни экспрессии мРНК SNCG в клетках HEC-1A и Ishikawa определяли с помощью количественной ОТ-ПЦР. Тотальную РНК выделяли из обеих клеточных линий с использованием реагента TRIzol (“Invitrogen Life Technologies”, США) и транскрибировали в комплементарную ДНК (кДНК) с использованием Fast Start Universal SYBR Green Master Mix (“Genview”, США) в реакционной смеси, содержащей 10 мкл РНК, 1 мкл раствора oligo(dT)18 с конценрацией 0.5 мкг/мкл, 1 мкл не содержащей РНКазы H2O, 4 мкл 5× буфера, 2 мкл 10 мМ смеси dNTP, 1 мкл диэтилпирокарбоната(“Sigma”, США) и 1 мкл фермента Revert Aid M-MLV RT (200 ед/мкл). ПЦР в реальном времени проводили с использованием кДНК в следующих условиях: начальная денатурация при 95°С в течение 2 мин с последующими 40 циклами при 95°С в течение 10 с и при 60°С в течение 30 с. Использованы следующие последовательности праймеров для ПЦР:
прямой SNCG ‒ 5'-CAAGAAGGGCTTCTCCATCG-3';
обратный SNCG ‒ 5'-GGTCACGCTCTGTACAACAT-3';
прямой GAPDH ‒ 5'-GAAGGTGAAGGTCGGAGT-3';
обратный GAPDH ‒ 5'-GAAGATGGTGATGGGATTTC-3'.
Ген GAPDH использован в качестве референс-гена. Метод 2–ΔΔCt использовали для измерения относительной экспрессии генов среди разных образцов. Полученные данные анализировали с помощью программного обеспечения 7300 RT-PCR System.
Анализ CCK8. После обработки трипсином клетки в группах SNCG-KD1, CON и NC расщепляли в полной среде, а затем центрифугировали при 800 об/мин в течение 6 мин. После удаления супернатанта клетки высевали в 96-луночные планшеты (“Neuroprobe”, США) при плотности 5 × 103 клеток/лунка и инкубировали при 37°С в атмосфере 5%-ного CO2. Начиная со второго дня после посева, за 2‒4 ч до окончания культивирования, в лунку добавляли 10 мкл реагента CCK8 (“Sigma”). Значения поглощения измеряли при 450 нм (OD450). Оптическую плотность пяти лунок каждой группы клеток измеряли ежедневно в течение 5 суток и определяли среднее значение поглощения. Значения OD450 наносили на график зависимости от времени инкубации и получали кривые роста для каждой группы клеток.
Анализ методом зарастания царапины (скретч-анализ). Клетки HEC-1A культивировали в 6-луночных планшетах (4 × 104 клеток/лунка), промывали бессывороточной средой 2‒3 раза и добавляли среду, содержащую 0.5% FBS, с последующей инкубацией при 37°С в атмосфере 5% CO2. Лунки помещали под микроскоп так, чтобы царапина находилась в центре изображения. Подвижность клеток оценивали по размеру разрыва на клеточном слое до и после рассечения клеточного монослоя.
Анализ в системе Transwell. Клетки высевали в новый 24-луночный планшет. Верхнюю и нижнюю камеры заполняли 500 мкл бессывороточной среды и помещали в инкубатор при 37°С на 2 ч для завершения формирования регидратационного слоя субстрата Matrigel. После этого среду в верхней камере заменяли на 500 мкл клеточной суспензии, и 750 мкл среды, содержащей 30% FBS, добавляли в нижнюю камеру. Каждое поле зрения было случайно выбрано для изображения под микроскопом; четыре изображения были получены при увеличении 100× и девять при увеличении 200×.
Анализ клеточного цикла. Распределение клеток HEC-1A и Ishikawa в трех группах анализировали методом проточной цитометрии до и после сайленсинга SNCG. После указанной выше обработки клетки трипсинизировали, промывали PBS и фиксировали 70%-ным этанолом при 4°C в течение ночи. Фиксированные клетки промывали PBS и суспендировали в 500 мкл окрашивающего раствора, содержащего йодид пропидия/тритон X‑100/РНКазу, в течение 30 мин при 37°C в темноте. Анализ клеточного цикла проводили с использованием проточного цитометра MACSQuant™ Analyzer (“Miltenyi Biotec”, США). Гистограммы ДНК анализировали с помощью программы MACSQuantify™, версия 2.1.
Проточная цитометрия. Клетки HEC-1A и Ishikawa помещали в инкубатор с постоянной температурой 37°C в атмосфере 5%-ного CO2 и культивировали в полной культуральной среде, содержащей 10% FBS. Три группы клеток на логарифмической фазе роста собирали и инокулировали в 6-луночные планшеты при плотности 5 × 105 клеток/мл (с тремя повторами для каждой группы). Клетки фиксировали и обрабатывали пропидий йодидом (PI; “Sigma-Aldrich”, США) и РНКазой A (“Fermentas”, Канада). Проточную цитометрию использовали для определения соотношения клеток в G1-, S- и М-фазах в трех группах. Детекцию проводили при длине волны 488 нм.
Лазерная конфокальная микроскопия. В логарифмической фазе роста клетки трех групп в концентрации 5 × 104 клеток/мл инокулировали в культуральную чашку для конфокальной микроскопии и помещали в бокс для культивирования на 24 ч. Клетки трижды промывали PBS, а затем добавляли 4%-ный параформальдегид для фиксации клеток в течение 30 мин, после чего трижды промывки PBS. В клетки добавляли 0.01%-ный раствор акридинового оранжевого и инкубировали в темноте в течение 10 мин и трижды промывали PBS. Для получения наилучшего томографического изображения клеток использовали лазерную конфокальную микроскопию (FV3000, версия 2.1.1.98; “Olympus”, Япония) при длине волны 488 нм, с блокирующим светофильтром на 515 нм. Количество клеток в М-фазе регистрировали после наблюдения каждого из четырех квадрантов культуральной чашки. Для лазерной конфокальной микроскопии использовали программу FV31S-SW Olympus.
Модель опухолевого ксенотрансплантата мыши. Двадцать самок мышей BALB/c nude (средний возраст 4 недели) были приобретены в Центре экспериментальных животных Медицинского университета Гуанси (Experimental Animal Center of Guangxi Medical University, Китай). Мышей случайным образом разделяли на две группы ‒ по 10 мышей в каждой, ‒ чтобы сформировать группу SNCG-KD1 и группу NC. Мышам вводили стабильные клетки HEC-IA с сайленсингом SNCG (группы SNCG-KD1) и клетки HEC-IA, экспрессирующие пустой вектор (группа NC). Суспензии клеток HEC-1A готовили в концентрации 2 × 107 клеток/мл. В общей сложности 4 × 106 клеток (200 мкл) инокулировали подкожно в правую подмышечную область nude-мышей. Ежедневно проводили осмотр nude-мышей по следующим параметрам: развитие подкожной опухоли, пищевое поведение, активность, питание и кахексия. У nude-мышей наблюдали и регистрировали видимый рост и текстуру подкожных опухолей. Вес тела, самый длинный диаметр (L) и самый короткий диаметр (W) подкожных опухолей у nude-мышей измеряли через неделю после подкожной инокуляции, в дальнейшем данные собирали два раза в неделю. Через 4 недели после подкожной инокуляции nude-мышей забивали путем введения 2%-ного пентобарбитала натрия, после чего извлекали шейку матки. Опухоль выделяли хирургическим путем и измеряли ее длину, ширину диаметр и вес. После замораживания в резервуаре с жидким азотом в течение одних суток опухоль хранили в холодильнике при ‒80°С. Данные сопоставляли, рассчитывали объем опухоли (V = 1/6 PI × L × W2), строили кривую роста подкожной опухоли и анализировали данные с помощью статистического программного обеспечения. Все процедуры на животных выполняли в соответствии с требованиями руководства Национального института здравоохранения по уходу и использованию лабораторных животных (the National Institutes of Health guide for the care and use of laboratory animals, Китай). Это исследование, включая использование животных, было одобрено Комитетом по этике лабораторных животных Первой академической клиники Медицинского университета Гуанси (the Lab Animal Ethical Committee of The First Affiliated Hospital of Guangxi Medical University, Китай).
Иммуноблотинг. Культивируемые клетки в фазе пролиферативного роста и опухолевые ткани от nude-мышей собирали для выделения общего белка. Соответствующее количество буфера для лизиса готовили за 5 мин до использования и добавляли к клеткам или тканям вместе с коктейлем ингибиторов протеаз, не содержащим EDTA (cOmplete™; “Sigma-Aldrich”). После центрифугирования 150‒200 мкл буфера для лизиса клеток, содержащего ингибиторы протеаз, добавляли к клеточному осадку в каждой пробирке. Образцы центрифугировали с использованием многоцелевой высокоскоростной центрифуги при 4°С, 14 000 об/мин в течение 15 мин. Супернатант использовали в качестве общего белкового экстракта, а концентрацию белка в образце определяли в тесте с бицинхониновой кислотой (“Sigma-Aldrich”). Экспрессию белка анализировали с использованием новой системы анализа белка на основе капиллярного электрофореза Nanovolume, как описано ранее [9]. В качестве специфических первичных антител использовали моноклональные кроличьи антитела против p38 MAPK, р-p38 MAPK, JNK и p-JNK человека, мышиные моноклональные антитела против ERK и p-ERK человека (“Abcam”, Великобритания) и мышиные моноклональные антитела против β-актина человека (“Santa Cruz Biotechnology, Inc.”, США). В качестве вторичных антител использовали мышиные моноклональные антитела против кроличьего IgG и кроличьи моноклональные антитела против мышиного IgG (“Abcam” и “Santa Cruz Biotechnology, Inc.”).
Статистический анализ. Для статистического анализа использовали SPSS 21.0 (“SPSS Inc.”, США). Все данные выражены как среднее значение ± стандартное отклонение. Статистические различия между группами оценивали с использованием одностороннего дисперсионного анализа. Две группы сравнивали с использованием независимого простого t-критерия Стьюдента. Статистически значимыми считались значения P < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Трансдукция клеток лентивирусными векторами с shRNA к SNCG
Ранее нами показано [8], что по сравнению с группой NC экспрессия мРНК SNCG в клетках HEC-1A и Ishikawa, трансдуцированных SNCG-KD1, SNCG-KD2 и SNCG-KD3, снижалась в разной степени. По сравнению с группой NC уровень экспрессии белка в обеих клетках в трех группах также снизился в разной степени. Среди них в группе SNCG-KD1 ингибирование было наиболее эффективным. Экспрессия белка SNCG в группе NC существенно не отличалась от экспрессии в группе CON.
Сайленсинг SNCG снижает пролиферацию клеток
Пролиферацию клеток в трех группах анализировали методом CCK8. Скорость роста в группе SNCG-KD1 была значительно ниже, чем в группах CON и NC (P < 0.05; рис. 1a). Это свидетельствует о том, что скорость роста клеток HEC-1A карциномы эндометрия человека, трансдуцированных лентивирусным вектором SNCG-KD, значительно ниже, чем в ложнотрансдуцированных и контрольных клетках. Ингибирование экспрессии SNCG снижало пролиферативную активность клеток HEC-1A по сравнению с интактными контрольными клетками.
Сайленсинг SNCG подавляет миграцию клеток
Микроскопическое исследование миграции клеток в группах CON, NC и SNCG-KD1 проводили через 0, 8 и 24 ч после разрыва монослоя. Как показано на рис. 1б, через 0 или 8 ч явных различий в зонах разрывов между тремя группами не выявлено. Через 24 ч после ранения клетки групп CON и NC явно мигрировали в область разрыва, причем не наблюдалось статистически значимых различий между ними (P > 0.05). В группе SNCG-KD1 обнаружено совсем небольшое число клеток, мигрирующих вследствие ранения.
Рис. 1.
Биологические характеристики клеток HEC-1A. a ‒ Кривые роста клеток, полученные с использованием результатов анализа CCK8. б ‒ Оценка миграционной способности клеток в трех группах с использованием тестов на заживление царапин. Миграционную способность клеток анализировали через 0, 8 и 24 ч после ранения. в ‒ Скорость заживления царапин (мкм/мин) сравнивали среди трех групп через 24 ч после ранения. *Р > 0.05 по сравнению с контрольной группой, **Р < 0.05 по сравнению с контрольной группой.
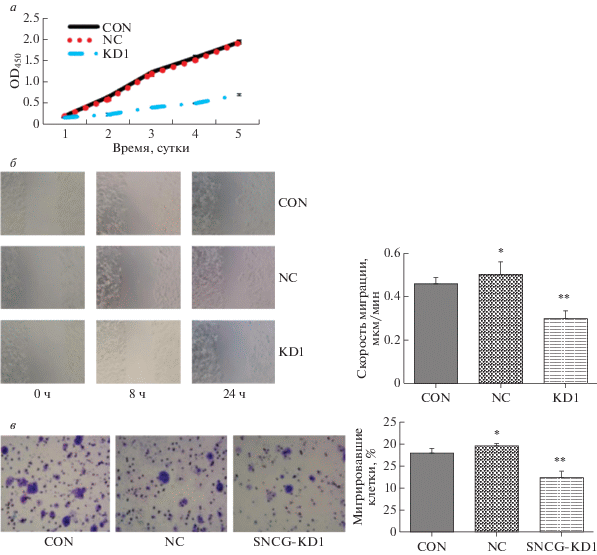
Способность клеток SNCG-KD1 мигрировать была ниже по сравнению с таковой в группах CON и NC, частота заживления разрыва через 24 ч составила 46, 50 и 28% в группах CON, NC и SNCG-KD1 соответственно (рис. 1б). Различия между SNCG-KD1 и группами CON и NC были статистически значимыми (P < 0.05).
Сайленсинг SNCG подавляет инвазию
Мигрировавшие клетки в группах CON, NC и SNCG-KD1 наблюдали под инвертированным микроскопом после 72 ч инкубации и подсчитывали. Среднее число клеток, прошедших через мембрану, в поле зрения для групп CON, NC и SNCG-KD1 составляло 18 ± 18, 20.00 ± 0.50 и 12.00 ± 1.53 соответственно (рис. 1в). Количество клеток, прошедших через мембрану, в группе SNCG-KD1 было ниже, чем в группах CON и NC, причем это снижение инвазии было статистически значимым (P < 0.05).
Изучение фаз клеточного цикла методом проточной цитометрии
Для изучения распределения фаз клеточного цикла в клетках трех групп использовали метод проточной цитометрии. В экспериментальной, CON и NC группах доля клеток в фазе G1 составляла соответственно 27.75 ± 0.23, 23.95 ± 0.74 и 22.70 ± 0.34% в клетках HEC-1A и 45.83 ± 2.08, 37.68 ± 3.51 и 39.82 ± 2.45% в клетках Ishikawa. Соотношение клеток в S-фазе было таким: 40.40 ± 1.69, 55.36 ± 0.89 и 52.21 ± 1.05% в клетках HEC-1A и 48.44 ± 5.47, 58.82 ± 4.01 и 56.6 ± 2.28% в клетках Ishikawa соответственно. Доля клеток в митотической фазе составила 31.86 ± 1.87, 20.69 ± ± 0.34 и 25.09 ± 0.73 в клетках HEC-1A и 5.74 ± 1.41, 3.5 ± 0.5 и 3.58 ± 0.8 в клетках Ishikawa соответственно. По сравнению с группами CON и NC доля клеток экспериментальной группы в обеих клеточных линиях в G1- и митотической фазах была выше, а в S-фазе ‒ ниже (P < 0.05). В то же время между группами CON и NC не обнаружено никакой разницы (P > 0.05). Эти результаты показывают, что сайленсинг SNCG под действием соответствующей shRNA в клетках HEC-1A и Ishikawa вызывает остановку клеточного цикла в G1- и митотической фазах (рис. 2 и 3).
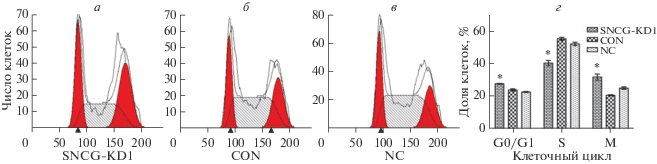
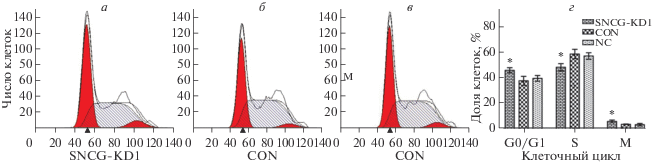
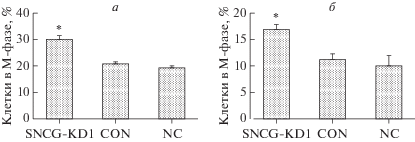
Анализ клеток в митотической фазе
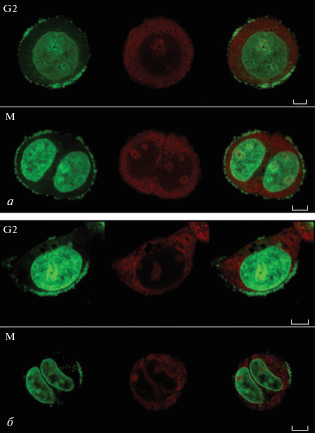
Для наблюдения клеток в митотической фазе, окрашенных акридиновым оранжевым, использовали лазерную конфокальную микроскопию (рис. 4 и 5). В экспериментальной группе клеток HEC-1A доля клеток в митотической фазе (30.17 ± ± 1.13%) была выше по сравнению с таковой в группах CON (20.77 ± 0.72%) и NC (19.32 ± 0.72%), как и в экспериментальной группе клеток Ishikawa (23.35 ± 2.02% против 16.25 ± 1.02 и 15.02 ± 1.96% соответственно). Эти различия были статистически значимыми (P < 0.05). Не было различий между группами CON и NC (P > 0.05).
Рис. 4.
Клетки HEC-1A (а) и Ishikawa (б) экспериментальной группы в митотической фазе после окрашивания акридиновым оранжевым. Зеленая и красная флуоресценция соответствуют свечению ДНК и РНК. Клетки в фазе G2 имели больший объем. По мере того, как клетки переходили в фазу М, происходило перераспределение зеленой флуоресценции от середины к полюсам, что в соответствует образованию двух клеток. Шкала ‒ 5 мкм.
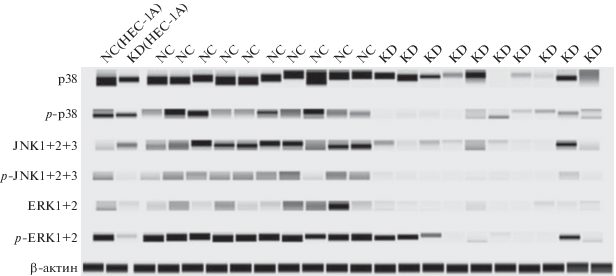
Экспрессия белков p38, ERK и JNK in vitro и in vivo
Для выяснения влияния экспрессии SNCG на регуляцию экспрессии молекул-мишеней в сигнальном пути MAPK/JNK/ERK мы проанализировали экспрессию белков p38, ERK и JNK in vitro и in vivo, используя метод иммуноблотинга. В группе SNCG-KD1 по сравнению с группой NC была значительно снижена экспрессия p-p38, p‑JNK1/2/3 и p-ERK1/2 (рис. 6).
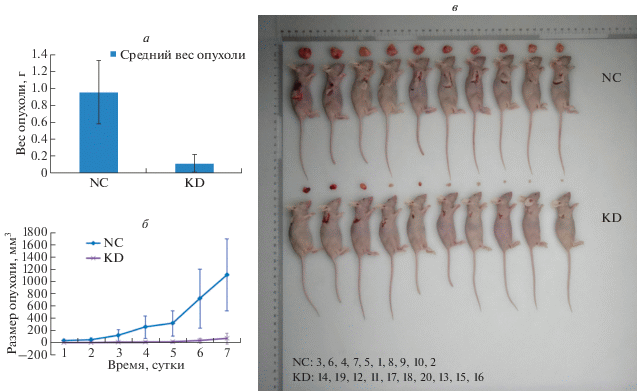
Нокдаун SNCG подавляет онкогенез
У мышей, задействованных в эксперименте, не наблюдали снижения аппетита. Не было никаких существенных изменений в массе тела мышей и не было случаев смерти ни в одной группе. Уровень онкогенеза у nude-мышей в обеих группах достигал 100%. Результаты показали, что скорость роста подкожных ксенотрансплантатов карциномы эндометрия человека в группе SNCG-KD1 была значительно ниже, чем в группе NC, а время до образования опухоли в группе NC было значительно короче, чем в группе SNCG-KD1 (P < 0.05; рис. 7б). Объем опухоли (75.06 ± 85.46 мм3) в группе SNCG-KD1 был значительно ниже, чем в группе NC (1112.58 ± 590.90 мм3), и масса опухоли (0.106 ± 0.105 г) в экспериментальной группе была значимо ниже, чем в группе NC (0.951 ± 0.377 г) (P < 0.05; рис. 7а, в).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящее время считается, что SNCG играет важную роль в передаче сигнала по пути ERK/MAPK [2]. Путь передачи сигнала MAPK играет ключевую роль в инвазии и миграции клеток злокачественной опухоли в окружающее пространство [2]. О существовании порогов, необходимых для трансформации в таких путях, сообщалось ранее [10]. Нами показано, что для поддержания пути передачи сигнала необходим тонкий баланс в активации MAPK. С другой стороны, большинство исследователей рассматривает конститутивную передачу сигналов ERK как важный медиатор в развитии рака, несмотря на то, что высокая активность ERK отмечается далеко не во всех опухолевых клетках [11‒13]. ERK функционально тесно связан с MAPK и играет аналогичную роль в регуляции злокачественного поведения опухолевых клеток [14]. Вполне возможно, что относительно небольшое увеличение активности ERK может быть достаточным для поддержания трансформации, потому что разные уровни и длительности передачи сигналов транслируются в различные результаты выбора субстрата и передачи сигналов в путях MAPK [15]. Фосфорилирование остатков Ser/Thr и Tyr может активировать ERK в сигнальных путях трансдукции. Активация ERK ‒ важный сигнал к метастазированию и инвазии опухолевых клеток в окружающие ткани. Считается, что сигнальный путь MAPK индуцирует пролиферацию опухолевых клеток [16]. γ-Синуклеин активирует путь ERK, который способствует подвижности раковых клеток и их выживанию [2, 16]. Так, в клетках рака молочной железы наблюдали аномально высокую экспрессию гена SNCG, продукт которого опосредует передачу эстрогеновых сигналов рецептором-36 эстрогена и способствует пролиферации клеток через активацию ERK1/2 [17]. В другом исследовании показано, что сайленсинг гена SNCG в клетках рака молочной железы снижал способность клеток к миграции и пролиферации, а также блокировал клеточный цикл со снижением уровней белков p-Akt и p-ERK [4]. Из этого можно предположить, что высокая экспрессия SNCG при раке молочной железы способствует пролиферации и миграции клеток путем активации путей AKT и ERK. Кроме того, обнаружена аномальная экспрессия SNCG при раке яичников, что способствовало выживанию клеток путем активации передачи сигнала через путь ERK1/2 [18]. Ранее показано, что ингибитор митогенактивируемой протеинкиназы, U0126, ингибирует фосфорилирующую активность ERK1/2 и снижает пролиферативную активность раковых клеток [19]. В проведенном нами исследовании показано, что избыточная экспрессия SNCG может быть одним из возможных механизмов, лежащих в основе образования опухоли посредством активации пути передачи сигнала MAPK/ERK, что способствует пролиферации, инвазии и миграции опухоли.
JNK ‒ еще один важный член семейства MAPK [20], который в разных тканях играет разные роли. Его активация приводит к инициации и прогрессированию рака печени, а его гиперактивация наблюдается в поврежденных гепатоцитах [21‒23]. Активация JNK-пути также может ингибировать развитие опухоли при раке печени [24]. Аномальная экспрессия SNCG может блокировать запрограммированную смерть клеток, вызванную противоопухолевыми препаратами паклитакселом и винкристином, через путь передачи сигналов JNK, способствуя тем самым проявлению злокачественных свойств опухолевых клеток [2]. Нами показано, что активность p-JNK1/2/3 в группе NC выше, чем в экспериментальной группе SNCG-KD1, причем экспрессия p-JNK1/2/3 значительно снижена при ингибировании экспрессии SNCG, как in vitro, так и in vivo. При избыточной экспрессии SNCG в раковых клетках эндометрия усиливался онкогенез. Можно предположить, что избыточная экспрессия SNCG играет специфическую роль в активации пути JNK, а активация JNK связана со злокачественным прогрессированием рака эндометрия. Считается, что сигнальный путь JNK играет ключевую роль в возникновении, развитии, инвазии и метастазировании опухолей и рассматривается как возможная новая мишень для лечения онкологических заболеваний [25]. На основании этих данных мы предполагаем, что гиперэкспрессия SNCG усиливает пролиферацию опухолевых клеток эндометрия, стимулируя антиапоптотические механизмы за счет повышения уровня фосфорилирования молекул пути передачи сигналов JNK, которые способствуют возникновению и развитию рака эндометрия.
Нами показано, что экспрессия гена SNCG лежит в основе цикла роста раковых клеток эндометрия, а специфическое подавление его экспрессии блокирует клеточный цикл в фазах G1 и митоза. Репликация ДНК и деление клеток относятся к двумя важнейшим процессам роста клеток. Точное копирование ДНК, ее разделение на дочерние цепи при отсутствии повреждений или после устранения любого повреждения ‒ эти механизмы регулируются контрольными точками клеточного цикла [26]. Мы предполагаем, что SNCG взаимодействует с белками, связанными с контрольными точками, в фазе G1, G2 или M и при необходимости блокирует клетки. В тканях рака молочной железы BubR1 и SNCG экспрессированы на высоком уровне. BubR1 относится к ключевым белкам в контрольной точке М-фазы, а взаимодействие между сверхэкспрессированными SNCG и BubR1 активирует контрольную точку М-фазы, в результате чего митоз клеток останавливается в метафазе [27]. Подавление экспрессии гена SNCG в клетках рака молочной железы может блокировать клетки в фазе G0/G1 [28] в результате того, что SNCG останавливает фазу G1 и подавляет контрольные точки митоза, связанные с белком BubR1. Полученные нами результаты вполне согласуются с этими данными: по-видимому, SNCG блокирует как рост клеток рака молочной железы, так и эндометрия.
Один из ключевых белков в регуляции митоза ‒ BubR1, который препятствует участию аномально делящихся клеток в цикле деления, например, в тех случаях, когда клетки серьезно повреждены, или из-за мутаций их генетического материала. По литературным данным, белок SNCG в опухолевых клетках взаимодействует с BubR1. Gupta с соавт. [29] подробно описали этот механизм для клеток рака молочной железы. Ими показано, что белки BubR1 и SNCG тесно взаимодействуют, что способствует аномальной пролиферации опухолевых клеток. В углубленных исследованиях механизма действия сайленсинга гена SNCG выявлено, что его аномально высокая экспрессия может приводить к разрушению 26S-протеазы и, как следствие, снижать экспрессию BubR1 в клетках, а применение специфического ингибитора 26S-протеазы, mg-132, устраняет эти изменения. С уменьшением экспрессии BubR1 аномально высокая экспрессия гена SNCG приводит к дальнейшему нарушению функции контроля митоза. Этот механизм позволяет понять, почему опухолевые клетки становятся устойчивыми к ингибиторам формирования веретена и микротрубочек. Таким образом, аномально высокая экспрессия гена SNCG может отключить точку контроля клеточного цикла в клетках рака молочной железы, позволяя опухолевым клеткам непрерывно продуцировать аномальные анеуплоидные клетки в ходе клеточного цикла, что приводит к увеличению продолжительности фазы деления ядра и другим патологическим клеточным процессам [30‒32].
В заключение следует отметить, что при супрессии экспрессии гена SNCG через снижение фосфорилирования p38, ERK и JNK снижается миграция, пролиферация, инвазия и онкогенез в клетках HEC-1A, тогда как аномально высокая экспрессия SNCG в клетках рака эндометрия может приводить к нарушениям контроля митоза.
Исследование профинансировано Национальным фондом естественных наук Китая (the National Natural Science Foundation of China) в рамках гранта [81460396].
Все процедуры на животных выполнены в соответствии с требованиями руководства Национального института здравоохранения использованию лабораторных животных и уходу за ними (the National Institutes of Health guide for the care and use of laboratory animals). Это исследование, включая использование животных, было одобрено Комитетом по этике лабораторных животных Первой академической клиники Медицинского университета Гуанси (the Lab Animal Ethical Committee of The First Affiliated Hospital of Guangxi Medical University, Китай).
Список литературы
Marsh J.A., Singh V.K., Jia Z., Forman-Kay J.D. (2006) Sensitivity of secondary structure propensities to sequence differences now between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804.
Pan Z.Z., Bruening W., Giasson B. I., Lee V.M., Godwin A.K. (2002) Gamma-synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J. Biol. Chem. 277, 35050–35060.
Zhuang Q., Liu C., Qu L., Shou C. (2015) Synuclein-γ promotes migration of MCF7 breast cancer cells by activating extracellular-signal regulated kinase pathway and breaking cell-cell junctions. Mol. Med. Rep. 12, 3795–3800.
He J.S., Xie X., Yang J.B., Guan H., Chen W.C. (2018) BCSG1 siRNA delivered by lentiviral vector suppressed proliferation and migration of MDA-MB-231 cells. Int. J. Mol. Med. 41, 1659–1664.
Goh G.W., Say Y.H. (2015) γ-Synuclein confers both pro-invasive and doxorubicin-mediated pro-apoptotic properties to the colon adenocarcinoma LS 174T cell line. Tumour Biol. 36, 7947–7960.
Surgucheva I., McMahon B., Surguchov A. (2006) Gamma-synuclein has a dynamic intracellular localization. Cell Motil. Cytoskeleton. 63, 447–458.
Zou J., Fan Y. J., Meng Y.Q., Xu H., Fan J. (2012) An exploratory analysis of γ-synuclein expression in endometrioid endometrial cancer. BMJ Open. 2, e000611.
Li W.Y., Fan Y.J., Xu H., Fan J.T., Sun D. (2016) Construction and identification of shRNA lentivirus vector plasmid of human SNCG gene. Shandong Med. J. 56, 13–16.
Liu S.B., Sardi S., Sonom B., Zocco D., McSweeney R. (2013) The application of a novel nanovolume capillary electrophoresis-based protein analysis system in personalized & translational medicine research. J. Bioanal. Biomed. S3, 004.
Suzukawa K., Weber T.J., Colburn N.H. (2002) AP-1, NF-κB, and ERK activation thresholds for promotion of neoplastic transformation in the mouse epidermal JB6 model. Environ. Health Perspect. 110, 865–870.
Doillon C.J., Gagnon E., Paradis R., Koutsilieris M. 2004. Three-dimensional culture system as a model for studying cancer cell invasion capacity and anticancer drug sensitivity. Anticancer Res. 24, 2169–2177.
Tsavachidou D., Coleman M.L., Athanasiadis G., Li S., Licht J.D. (2004) SPRY2 is an inhibitor of the ras/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599E mutant. Cancer Res. 64, 5556–5559.
Houben R., Vetter-Kauczok C.S., Ortmann S., Rapp U.R., Broecker E.B. (2008) Phospho-ERK staining is a poor indicator of the mutational status of BRAF and NRAS in human melanoma. J. Invest. Dermatol. 128, 2003–2012.
Murphy L.O., Blenis J. (2006) MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 31, 268–275.
Surgucheva I.G., Sivak J.M., Fini M.E., Palazzo R.E., Surguchov A.P. (2003) Effect of gamma-synuclein over-expression on matrix metalloproteinases in retinoblastoma Y79 cells. Arch. Biochem. Biophys. 410, 167–176.
Pan Z.Z., Bruening W., Godwin A.K. (2006) Involvement of RHO GTPases and ERK in synuclein-gamma enhanced cancer cell motility. Int. J. Oncol. 29, 1201–1205.
Shi Y.E., Chen Y., Dackour R., Potters L., Wang S. (2010) Synuclein gamma stimulates membrane-initiated estrogen signaling by chaperoning estrogen receptor (ER)-alpha36, a variant of ER-alpha. Am. J. Pathol. 177, 964–973.
Zeng P., Wagoner H.A., Pescovitz O.H., Steinmetz R. (2005) RNA interference (RNAi) for extracellular signal-regulated kinase 1 (ERK1) alone is sufficient to suppress cell viability in ovarian cancer cells. Cancer Biol. Ther. 4, 961–967.
Thomas R.S., Sarwar N., Phoenix F., Coombes R.C., Ali S. (2008) Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J. Mol. Endocrinol. 40, 173–184.
Johnson G.L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 298, 1911–1912.
Farazi P.A., DePinho R.A. (2006) Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer. 6, 674–687.
Papa S., Bubici C., Zazzeroni F., Franzoso G. (2009) Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biol. Chem. 390, 965–976.
Seki E., Brenner D.A., Karin M. (2012) A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 143, 307–320.
Cellurale C., Girnius N., Jiang F., Cavanagh-Kyros J., Lu S. (2012) Role of JNK in mammary gland development and breast cancer. Cancer. Res. 72, 472–481.
Bubici C., Papa S. (2014) JNK signaling in cancer: in need of new, smarter therapeutic targets. Br. J. Pharmacol. 171, 24–37.
Gerard C., Goldbeter A. (2011) A skeleton model for the network of cyclin-dependent kinases driving the mammalian cell cycle. Interface Focus. 1, 24–35.
Cirak Y., Furuncuoglu Y., Yapicier O., Alici S., Argon A. (2015) Predictive and prognostic values of BubR1 and synuclein-gamma expression in breast cancer. Int. J. Clin. Exp. 8, 5345‒5353
He J., Xie N., Yang J., Guan H., Chen W., Wu H., Yuan Z., Wang K., Li G., Sun J., Yu L. (2014) siRNA-mediated suppression of synuclein gamma inhibits MDA-MB-231 cell migration and proliferation by downregulating the phosphorylation of AKT and ERK. J. Breast Cancer. 17, 200–206.
Gupta A., Inaba S., Wong O.K., Fang G., Liu J. (2003) Breast cancer-specific gene 1 interacts with the mitotic checkpoint kinase Bub R1. Oncogene. 22, 7593–7599.
Saloustres E., Mavrandis D., Georgouslias V. (2008) Paclitaxel and docetaxel in the treatment of breastcancer. Expert Opin. Pharmacother. 9, 2603–2616.
Perez E.A. (2009) Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 8, 2086–2095.
Lanzi C., Cassinelli G., Cuccuru G., Supino R., Zuco V., Ferlini C., Scambia G., Zunino F. (2001) Cell cycle checkpoint efficiency and cellular response to paclitaxel in prostate cancer cells. Prostate. 48, 254–264.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология