

Молекулярная биология, 2021, T. 55, № 1, стр. 139-151
Изменение экспрессии гена CASC5 коррелирует с таргетными мутациями при лейкозе
К. В. Богданов a, *, О. В. Мерзликина a, Ю. В. Миролюбова a, Л. Л. Гиршова a, Э. Г. Ломаиа a, А. Ю. Зарицкий a
a Научно-исследовательский институт онкологии и гематологии Национального медицинского исследовательского центра им. В.А. Алмазова
197341 Санкт-Петербург, Россия
* E-mail: kvbogdanov@yandex.ru
Поступила в редакцию 21.02.2020
После доработки 31.07.2020
Принята к публикации 13.08.2020
Аннотация
Нарушение функции генов, контролирующих митоз и ответственных за правильное расхождение сестринских хроматид в анафазе, часто сопровождается анеуплоидией, нередко выявляемой при лейкозах. Важным фактором, обеспечивающим корректное связывание прицентромерной области хромосом с микротрубочками веретена деления, является один из компонентов кинетохорного комплекса, а именно, белок AF15q14/KNL1/CASC5. Как показано недавно, при некоторых лейкозах ген этого белка может вовлекаться в образование хромосомной транслокации t(11;15)(q23;q14) или варианта химерного онкогена MLL-AF15Q14, что служит биомаркером плохого прогноза. Несмотря на участие мРНК гена CASC5 в онкогенезе солидных опухолей, экспрессия этого гена при злокачественных новообразованиях кроветворной системы не изучена. Нами проведен анализ уровней экспрессии гена CASC5 и ближайшего круга регуляторных генов, включая WT1, APOBEC3A (A3A), N-MYC. Обнаружено выраженное снижение экспрессии CASC5 в клетках костного мозга первичных больных лейкозом по сравнению со здоровыми донорами. Показано также, что пониженная экспрессия гена CASC5 коррелирует с выявлением таргетных мутаций у пациентов, относящихся к двум прогностическим подгруппам (благоприятная, неблагоприятная), с уровнем значимости (p < 0.05). Отмечено, что изменение уровня экспрессии гена CASC5 при остром миелоидном лейкозе ассоциировано со сверхэкспрессией генов WT1, A3A, а в некоторых случаях с N-MYC и SPT16, что согласуется с устойчивостью к полихимиотерапии и прогрессией лейкоза. Однако вопрос о том, какой из генов-регуляторов инициирует лейкемогенез, остается открытым.
ВВЕДЕНИЕ
Причины развития лейкоза и его прогрессии остаются во многих случаях неизвестными. Тем не менее, за последнее десятилетие появились данные о вовлеченности в эти процессы не только известных таргетных онкогенов, мутации в которых часто ассоциированы с прогнозом течения и исходом заболевания, но и менее изученных генов-регуляторов, комплексная роль которых в опухолевом процессе только начинает проясняться. К генам-регуляторам относятся гены факторов, контролирующих различные процессы, в частности митоз, а также предшествующие митозу события, такие как репликация, транскрипция, репарация, программированная клеточная гибель и др. В число генов-регуляторов входит ген AF15Q14/CASC5/KNL1, белок-продукт которого связывает прицентромерные области хромосом и входит в состав кинетохорного комплекса. Белок AF15Q14/CASC5/KNL1 принимает участие в присоединении микротрубочек веретена деления к центромерам сестринских хроматид в метафазе и способствует их правильному расхождению в дочерние клетки в анафазе митоза. Впервые вклад гена AF15Q14/CASC5/KNL1 в возникновение злокачественных новообразований кроветворной системы показали после обнаружения слитого онкогена MLL-AF15Q14, который образуется в результате хромосомной транслокации t(11;15)(q23;q14) при миелодиспластическом синдроме (МДС), а также при некоторых лейкозах, включая острый миелоидный лейкоз (ОМЛ (М2, M4, М5)) и Т-клеточный острый лимфобластный лейкоз (T-ОЛЛ) как у детей, так и у взрослых [1–4]. Наличие клеток, несущих эту перестройку, свидетельствует, как правило, о плохом прогнозе заболевания.
Кроме того, в гене CASC5 найдены точечные мутации CASC5c.6125G>A и CASC5c.6560A>G, первоначально обнаруженные при наследственной микроцефалии (MCPH4), а также мутация CASC5T142R, вовлеченная (вместе с соматическими изменениями в других генах) в клональную эволюцию хронического миелоидного лейкоза (ХМЛ) [5, 6]. Эти мутации способствуют нарушению локализации белков ZWINT1, MIS12, BUB3 в составе кинетохорного комплекса, что коррелирует с потерей контроля над присоединением микротрубочек веретена деления к центромерам и приводит к неправильной сегрегации хромосом в митозе.
Вместе с тем, об экспрессии гена AF15Q14/ CASC5/KNL1 или кодируемого им белка AF15q14/ CASC5/KNL1 известно не слишком много. Так, недавно показано, что изменение экспрессии гена CASC5 и/или белка CASC5 при некоторых солидных опухолях, включая рак легкого, цервикальный рак и рак толстой кишки, ассоциировано с повышением клеточной пролиферации [7–9]. Одновременно с этим отмечено увеличение экспрессии CASC5 в клеточной линии Jurkat (Т-ОЛЛ) [7]. Тем не менее, изменение экспрессии гена CASC5 при онкогематологических заболеваниях до сих пор не изучали. Именно поэтому основная задача нашей работы состояла в проведении скрининга мутаций таргетных онкогенов у больных лейкозом и в одновременной оценке экспрессии некоторых регуляторных генов, включая CASC5, WT1, A3A, N-MYC.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Культивирование клеток, выделение белков, иммуноблотинг. Клетки Jurkat (Т-ОЛЛ) выращивали в среде RPMI-1640 с добавлением 10% эмбриональной сыворотки теленка при 37°C в атмосфере 5% CO2. Клетки собирали и отмывали от культуральной среды в 1 ×PBS (150 мМ NaCl, 27 мМ KCl, 1.5 мМ KH2PO4 и 8.1 мМ Na2HPO4, pH 7.5). Экстракцию белков проводили на льду в лизирующем буфере, содержащем 50 мМ Трис-HCl, 150 мМ NaCl, 0.1 мМ EDТА, 50 мМ NaF, 1 мМ Na2VO5, 1 мМ фенилметилсульфонилфторид (PMSF), 0.5% NP-40, c добавлением 10 мкг/мл апротинина, леупептина и пепстатина, pH 7.5. Иммуноблотинг проводили с использованием антител к CASC5 (“ABclonal”, США), N-MYC (“Calbiochem”, США), β-актину (“Santa Cruz Biotechnology”, Канада).
Пациенты. В группу больных вошли 40 первичных онкогематологических больных (НМИЦ им. В.А. Алмазова), у которых диагностировали ОМЛ (n = 18), ХМЛ (n = 9), В-клеточный острый лимфобластный лейкоз (В-ОЛЛ, n = 13). Средний возраст пациентов составил 37 лет. Со всеми пациентами и здоровыми донорами (n = 10) подписано добровольное информированное согласие на участие в исследовании. Образцы костного мозга (КМ) отбирали в дебюте заболевания в пробирки с напылением EDТА, а в случае мониторинга – согласно проводимому лечению.
Иммунофенотипирование и селекция клеток CD34. Для определения поверхностных дифференцировочных антигенов (CD-маркеров) и содержания (%) бластных клеток в КМ использовали проточную цитометрию (цитометр FacsCanto, “Becton Dickinson”, США) и коммерческие моноклональные антитела. Клетки-предшественники CD34 из КМ больного ОМЛ выделяли согласно [10]. После сортировки клетки центрифугировали (4000 g, 10 мин), к осадку добавляли 0.1 мл 1 × × PBS/20 мМ EDТА. Полученную суспензию клеток, содержащих ядро, выдерживали при 4°C в течение короткого времени до начала экстракции РНК.
Выделение РНК и синтез кДНК. РНК выделяли из 0.1 мл клеток линии Jurkat, 0.1 мл содержащих ядра клеток CD34, 1 мл клеток КМ с использованием коммерческого набора реактивов: QIAamp RNA Blood Mini Kit (“Qiagen”, Германия). Перед выделением РНК из клеток КМ проводили лизис эритроцитов с последующим центрифугированием. К суспензии клеток CD34 добавляли лизирующий раствор, дальнейшее выделение проводили согласно протоколу. РНК элюировали в 30 мкл буфера. Образцы РНК хранили при –70°C до момента постановки обратной транскрипции (ОТ). Реакцию ОТ проводили согласно методике для синтеза кДНК и набора реактивов RevertAid H Minus First Strand cDNA Synthesis Kit (“Fermentas”, Литва).
Выделение ДНК. ДНК выделяли из 0.2 мл КМ с использованием коммерческого набора реактивов: QIAamp DNA Mini Kit (“Qiagen”) согласно протоколу фирмы-производителя. ДНК элюировали в 50 мкл буфера. Образцы ДНК хранили при –20°C до проведения ПЦР.
Скрининг таргетных мутаций онкогенов. кДНК с целью выявления 15 разных онкогенных мутаций амплифицировали с помощью ПЦР на микрочипах в режиме реального времени (ПЦР-РВ) [11]. Мутации в экзонах 14, 15, 20 гена FLT3, в экзоне 12 гена NPM1 и в экзонах 4–9 гена ABL выявляли с помощью прямого секвенирования по Сэнгеру с использованием секвенатора ABI PRISM 310 Genetic Analyzer (“Applied Biosystems”, США), как описано ранее [12–14].
Количественный анализ генов. Для количественной оценки уровня мРНК регуляторных генов (CASC5, N-MYC, A3A, WT1, SPT16), таргетных онкогенов (CBFβ-MYH11/A, AML1-ETO, NPM1/A, BCR-ABL/P210, BCR-ABL/P190, TEL-AML1), а также референсного гена GAPDH образец кДНК амплифицировали методом ПЦР-РВ согласно протоколу, опубликованному ранее [15, 16]. Для определения экспрессии генов WT1, CBFβ-MYH11/A, AML1-ETO, NPM1/A, BCR-ABL/ P210, BCR-ABL/P190, TEL-AML1 выполняли ПЦР с использованием коммерческих наборов Fusion Quant Kit for RT-QPCR (“Qiagen”) и Universal PCR Master Mix (“Applied Biosystems”). Экспрессию генов CASC5, N-MYC, A3A, SPT16 оценивали методом ПЦР с использованием набора Universal PCR Master Mix (“Applied Biosystems”) и коммерчески синтезированных олигонуклеотидных праймеров, в том числе олигонуклеотидных зондов, несущих на 3'-конце флуоресцентный краситель FAM, подобранных через систему NCBI (нуклеотидные последовательности праймеров могут быть предоставлены по требованию). Результаты ПЦР анализировали с помощью программного обеспечения к термоциклеру “RotorGene 6000” (“Corbet Research”, Австралия) после построения калибровочных кривых со стандартными разведениями плазмидных ДНК (101–106). Пороговый уровень количественной экспрессии гена WT1 в клетках КМ здоровых доноров (n = 10) не превышал 250 копий/GAPDH. Превышение этого показателя рассматривали как повышение экспрессии [17]. Пороговые уровни экспрессии генов CASC5, N-MYC, A3A, SPT16 определяли с использованием кДНК, синтезированной по РНК из клеточной линии Jurkat на второй день культивирования.
Цитогенетическое исследование. Культивирование клеток КМ больных лейкозом, приготовление препаратов хромосом и последующее дифференциальное окрашивание хромосом выполняли согласно [18]. Нарушения кариотипа оценивали по результатам анализа 20 митозов методом стандартного кариотипирования и/или 200 интерфазных ядер после проведения флуоресцентной гибридизации in situ (FISH).
Статистический анализ. Корреляционную зависимость между уровнем экспрессии гена CASC5 и таргетными онкогенными мутациями, выявленными у больных разных прогностических подгрупп, оценивали путем построения четырехпольных таблиц Фишера и с использованием критерия χ2 [19].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Первоначально провели скрининг таргетных мутаций онкогенов у больных лейкозом (n = 40). В зависимости от прогностической значимости выявленных мутаций каждая группа пациентов с известной нозологией (ОМЛ, В-ОЛЛ, ХМЛ) была разделена на две подгруппы (относительно благоприятную и неблагоприятную) (табл. 1). Так, в группе ОМЛ (n = 18) подгруппу неблагоприятного прогноза составили пациенты (n = 6), у которых обнаружили мутацию в тандемном домене онкогена FLT3/ITD. Остальные пациенты (n = 12) относились к подгруппе относительно благоприятного прогноза – мутация FLT3/ITD у них отсутствовала, но обнаружена одна из трех других мутаций: NPM1/A (n = 4), CBFβ-MYH11/A [inv16(p13;q21)] (n = 6), AML1-ETO [t(8;21)(q22;q22)] (n = 2). Среди больных В-ОЛЛ (n = 13) в подгруппу неблагоприятного прогноза (n = 7) вошли носители онкогенной мутации BCR-ABL/P190 [t(9;22)(q34;q11)], а подгруппу относительно благоприятного прогноза (n = 6) составили носители мутации TEL-AML1 [t(12;21)(p13;q22)], большинство из которых были моложе 16 лет. Группа больных ХМЛ (n = 9) состояла из шести человек с подтвержденной хронической фазой (ХФ) и трех пациентов (бласты 30, 57, 90%) с терминальной фазой заболевания или бластным кризом (БК). К подгруппе неблагоприятного прогноза (n = 5) отнесены три пациента с БК и двое больных с ХФ. У всех пациентов этой подгруппы обнаружены оба варианта химерного онкогена BCR-ABL [t(9;22)(q34;q11)]: BCR-ABL/P210 и BCR-ABL/P190, а у одного из них (с подтвержденной ХФ) дополнительно выявлена точечная мутация T315I в тирозинкиназном домене (TKD) гена ABL. К относительно благоприятному прогнозу (n = 4) отнесены остальные больные ХМЛ (ХФ), у которых определен вариант онкогена BCR-ABL/P210. При этом вариант слитого онкогена BCR-ABL/P190, как и точечные мутации в гене ABL, не обнаружены.
Таблица 1.
Таргетные мутации, характерные для относительно благоприятного и неблагоприятного прогноза при лейкозе (ХМЛ, ОМЛ, В-ОЛЛ), и относительная экспрессия гена CASC5
| Таргетная мутация, количество, больные с неблагоприятным прогнозом* |
Таргетная мутация, количество, больные с благоприятным прогнозом** |
Уровень относительной экспрессии регуляторного гена (N/GAPDH) | χ2, F, p |
|---|---|---|---|
| Группа больных В-ОЛЛ (n = 13) | |||
| BCR-ABL/P190 | TEL-AML1 | CASC5 | |
| 6 | 1 | <4 × 103/GAPDH | 6.198, 0.029, p < 0.05 |
| 1 | 5 | >4 × 103/GAPDH | |
| Группа больных ХМЛ (n = 9) | |||
| BCR-ABL/P190, BCR-ABL/P210 |
BCR-ABL/P210 (не выявлено мутаций в гене ABL) |
CASC5 | |
| 4 | 0 | <5 × 103/GAPDH | 5.760, 0.047, p < 0.05 |
| 1 | 4 | >5 × 103/GAPDH | |
| Группа больных ОМЛ (n = 18) | |||
| FLT3/ITD | NPM1/A, AML1-ETO, CBFβ-MYH11/A |
CASC5 | |
| 5 | 1 | <4 × 103/GAPDH | 10.125, 0.003, р <0.05 |
| 1 | 11 | >4 × 103/GAPDH | |
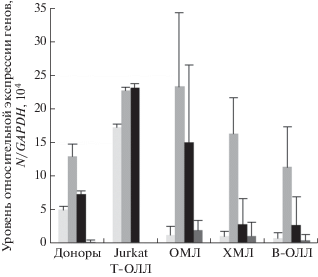
В настоящей работе требовалось определить уровни экспрессии гена CASC5 и ближайшего круга регуляторных генов, включая WT1, A3A, N-MYC, в клетках КМ больных лейкозом и здоровых доноров, а также в контрольной клеточной линии Jurkat, в которой сверхэкспрессируются CASC5 и N-MYC (рис. 1). Результаты количественного анализа генной экспрессии представлены на рис. 2. Нами обнаружена общая тенденция к снижению уровней экспрессии генов CASC5, WT1, A3A, N-MYC в клетках КМ у больных лейкозом. По уровню снижения экспрессии группы расположились в следующем порядке: ОМЛ, ХМЛ, В-ОЛЛ. При этом средние уровни экспрессии гена CASC5 у пациентов оказались значительно ниже, чем у здоровых доноров, а именно, в 4.9 (ОМЛ), 6.9 (ХМЛ) и 11.3 раза (В-ОЛЛ). Средние уровни экспрессии N-MYC и A3A при ОМЛ были выше, при В-ОЛЛ – ниже, при ХМЛ более высокой оказалась только экспрессия A3A (N-MYC – более низкой) по сравнению со здоровыми донорами. При этом средние уровни экспрессии WT1 у всех пациентов были выше, чем у здоровых доноров: в 45.7 (ОМЛ), 22.0 (ХМЛ) и 9.4 раза (В-ОЛЛ).
Рис. 1.
Экспрессия кинетохорного белка CASC5/KNL1 и онкобелка N-MYC в клеточной линии Jurkat (Т-ОЛЛ). Клетки Jurkat на 2, 4, 6-е сутки культивирования (2, 4, 6 соответственно). Иммуноблотинг клеточного экстракта с использованием антител к CASC5, N-MYC, β-актину.
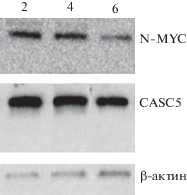
Рис. 2.
Профили относительной экспрессии регуляторных генов в клеточной линии Jurkat (Т-ОЛЛ)
у здоровых доноров (n = 10) и у больных лейкозом (n = 40). N – Количество копий анализируемого гена.  –CASC5,
–CASC5,  –A3A,
–A3A,  –N-MYC,
–N-MYC,  – WT1.
– WT1.
Не обнаружив статистически значимых различий (p > 0.05) между уровнем экспрессии регуляторных генов, включая CASC5, и количеством бластных клеток (>70% vs ≤70%) у больных лейкозом, мы провели сравнительный анализ экспрессии этих генов в разных подгруппах с учетом нозологии и прогностической значимости таргетных мутаций. Показано, что понижение экспрессии гена CASC5 коррелирует с выявлением у больных лейкозом таргетных мутаций онкогенов (табл. 1). Так, у шести из семи больных В-ОЛЛ (рис. 3а), носителей мутации BCR-ABL/P190 (подгруппа неблагоприятного прогноза), уровни экспрессии CASC5 были ниже (<4 × 103/GAPDH), чем у пяти из шести носителей мутации TEL-AML1 (подгруппа относительно благоприятного прогноза), уровни экспрессии CASC5 у которых были, напротив, выше (>4 × 103/GAPDH). С помощью статистического анализа определены значимые различия между уровнем экспрессии гена CASC5 и вариантами таргетных мутаций (р < 0.05, табл. 1) в обеих прогностических подгруппах В-ОЛЛ. Определение экспрессии других регуляторных генов в подгруппах больных В-ОЛЛ выявило сверхэкспрессию A3A (>2.3 × 105/GAPDH) только у одного индивида (бласты 90%), носителя мутации TEL-AML1. При этом у этого же больного количество копий онкогена TEL-AML1 оказалось выше, чем у других пациентов этой подгруппы. И, наконец, еще у одного больного В-ОЛЛ (бласты 67%), носителя мутации BCR-ABL/P190, обнаружена сверхэкспрессия WT1 (>2 × 103/GAPDH). При этом количество копий онкогена BCR-ABL/P190 у этого больного оказалось больше, чем у других членов подгруппы.
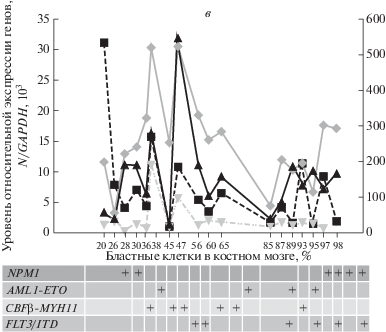
Рис. 3.
Профили относительной экспрессии регуляторных генов, количество бластных клеток (%)
и вариант онкогенной мутации у больных лейкозом. a – Группа В-ОЛЛ; б – группа ХМЛ; в – группа ОМЛ. N – количество копий анализируемого гена; на оси слева:  – CASC5,
– CASC5,  – WT1. на оси справа:
– WT1. на оси справа:  – A3A,
– A3A, 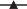 – N-MYC.
– N-MYC.
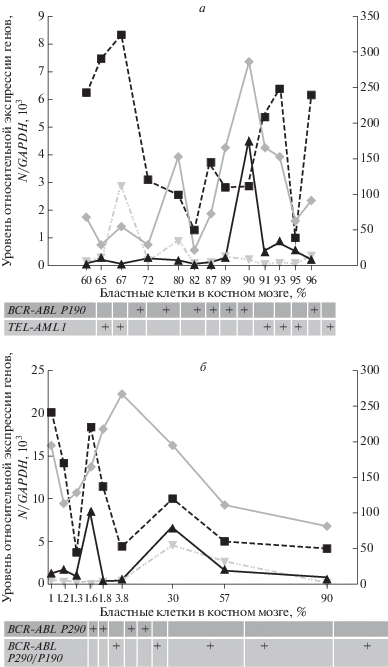
Рис. 3.
Окончание.
В группе больных ХМЛ (рис. 3б) оба варианта BCR-ABL: BCR-ABL/P210 и BCR-ABL/P190 (подгруппа неблагоприятного прогноза) обнаружены у пяти пациентов (БК, n = 3; ХФ, n = 2), и у четырех из них уровни экспрессии CASC5 оказались ниже (<5 × 103/GAPDH), чем у остальных четырех пациентов (ХФ), несущих только один вариант онкогена BCR-ABL/P210 (подгруппа относительно благоприятного прогноза). В последней подгруппе больных, у которых обнаружили мутацию BCR-ABL/P210 и не выявили точечные мутации гена ABL, уровни экспрессии CASC5 оказались, напротив, выше (>5 × 103/GAPDH). Различия между уровнем экспрессии гена CASC5 и вариантами таргетных мутаций в обеих прогностических погруппах были статистически значимыми (р < 0.05, табл. 1). В то же время определение экспрессии других регуляторных генов у больных ХМЛ показало, что только у одного из них (ХФ, бласты 3.8%) одновременное присутствие слитых онкогенов BCR-ABL/P210 и BCR-ABL/P190 сопровождалось сверхэкспрессией A3A (>2.6 × 105/GAPDH) и превышением порогового уровня экспрессии WT1. Сверхэкспрессия WT1 (>2 × 103/GAPDH) обнаружена у двух пациентов с ХМЛ (БК, бласты 30, 57%), у которых выявили оба варианта онкогена BCR-ABL: BCR-ABL/P210 и BCR-ABL/P190.
В группе больных ОМЛ, состоящей из 18 человек (рис. 3в), у пяти из шести носителей мутации FLT3/ITD (подгруппа неблагоприятного прогноза) уровни экспрессии CASC5 были ниже (<4 × × 103/GAPDH), чем у остальных 11 из 12 носителей одной из трех других мутаций: NPM1/A, AML1-ETO, CBFβ-MYH11/A (подгруппа относительно благоприятного прогноза), у которых уровни экспрессии CASC5 были, напротив, выше (>4 × 103/GAPDH). Различия между уровнем экспрессии гена CASC5 и вариантами таргетных мутаций в обеих прогностических подгруппах больных ОМЛ были статистически значимыми (р < 0.05, табл. 1). Интересно, что в подгруппах больных ОМЛ сверхэкспрессия регуляторных генов WT1, A3A и N-MYC выявлена только у двух пациентов (бласты 47, 38%), один из которых — носитель мутации FLT3/ITD, а другой — мутации CBFβ-MYH11/A. Первоначально сверхэкспрессию N-MYC обнаружили у взрослых больных ОМЛ, у детей, получающих лечение при Т-ОЛЛ и нейробластоме, что коррелировало с плохим прогнозом [27–29]. Кроме того, оказалось, что сверхэкспрессия онкогена N-MYC при нейробластоме поддерживалась при участии фактора транскрипции SPT16 [30]. Тем не менее, экспрессию гена SPT16 при ОМЛ не определяли, в том числе и у пациентов с повышенной экспрессией N-MYC.
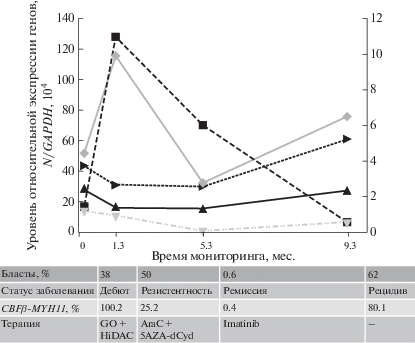
Дополнительно мы провели количественный анализ уровней экспрессии гена SPT16 в группе больных ОМЛ (n = 18). В качестве контроля использовали клеточную линию Jurkat (T-ОЛЛ) со сверхэкспрессией SPT16 (2.9 × 104/GAPDH). Только у двух больных ОМЛ (бласты 47, 38%) со сверхэкспрессией N-MYC повышенным оказался также уровень экспрессии гена SPT16 (>3.4 × × 104/GAPDH). В дальнейшем этим двум пациентам, получающим лечение, потребовалось провести мониторинг экспрессии регуляторных и таргетных генов. Следует отметить, что у одного из них (бласты 47%), носителя таргетной мутации FLT3/ITD, после проведения курса ПХТ выполнили гаплоидентичную трансплантацию гемопоэтических стволовых клеток. Однако мониторинг биомаркеров у этого пациента не был проведен (вскоре после достижения полной клинико-гематологической ремиссии у больного развился рецидив, который привел к летальному исходу). Одновременный анализ таргетных и регуляторных генов проведен у другого больного ОМЛ (бласты 38%), носителя мутации CBFβ-MYH11/A, который получал только ПХТ (трансплантацию гемопоэтических стволовых клеток ему не рекомендовали по кардиологическим показателям) (рис. 4). С помощью количественной ПЦР в КМ обнаружено повышенное количество копий слитого онкогена CBFβ-MYH11/A. В то же время проведенный цитогенетический анализ, включая FISH, подтвердил наличие инверсии хромосомы 16 [inv(16)(p13;q22)] в 88% клеток КМ, приводящей к слиянию генов CBFβ и MYH11. Иммунофенотипирование поверхностных CD-маркеров позволило обнаружить в КМ пациента популяцию бластных клеток с аберрантным фенотипом (Cd45dimCD34+CD38+CD117+CD13+CD33+), что характерно для ОМЛ. Дополнительный анализ уровней экспрессии генов N-MYC, SPT16, A3A, CASC5 в КМ выявил увеличение экспрессии первых трех генов в 5.2, 3.2, 26.3 раза, соответственно, и, напротив, снижение экспрессии CASC5 в 1.7 раза по сравнению с контролем. В дальнейшем (спустя 1 мес после начала ПХТ (GO + HiDAC)) у пациента появилась резистентность к лечению, и популяция бластных клеток в КМ увеличилась до 50%. При этом не обнаружены мутации в гене DNMT3A, кодирующем ДНК-метилтрансферазу, дисфункция которой часто связана с гипометилированием генома и устойчивостью к терапии (HiDAC). В то же время у больного выявили повышение экспрессии генов WT1, N-MYC, SPT16, A3A, CASC5 в клетках CD34, выделенных с использованием проточной цитометрии – в 17.0, 10.6, 4.7, 1.2, 10.4 раза, соответственно, по сравнению с тотальным КМ. Таким образом, превышение уровней экспрессии упомянутых генов, включая SPT16, в клетках-предшественниках CD34 указывает на преобладание в КМ популяции резистентных к лечению клеточных элементов с активной пролиферацией. В дальнейшем смена терапии (малые дозы Ara-c и азацитидина) привела к снижению количества бластных клеток (0.6%) и уровней экспрессии этих генов, что согласуется с наступлением ремиссии. Однако через 4 мес у больного развился рецидив (бласты: 62%). При этом уровень экспрессии генов N-MYC, SPT16, A3A повысился в 1.2, 1.8, 3.3 раза, соответственно, а CASC5 понизился по сравнению с контролем.

ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящем исследовании у всех первичных больных лейкозом (В-ОЛЛ, ОМЛ, ХМЛ) определены таргетные онкогенные мутации, а также оценена экспрессия регуляторных генов, включая CASC5, WT1, A3A, N-MYC, влияющих на область центромеры (перицентромеры) (табл. 2). Так, у больных В-ОЛЛ выявлены мутации онкогенов BCR-ABL/P190, TEL-AML, у больных ХМЛ – разные варианты онкогена BCR-ABL (BCR-ABL/ P210, BCR-ABL/P190), тогда как больные ОМЛ были носителями мутаций NPM1/A, AML1-ETO, CBFβ-MYH11/A, FLT3/ITD. Согласно опубликованным данным, мутации, обнаруженные у больных лейкозом, имеют разную прогностическую значимость (табл. 1). Так, больные В-ОЛЛ, у которых обнаружили мутации онкогена BCR-ABL/P190, больные ХМЛ с обоими вариантами онкогена BCR-ABL (BCR-ABL/P190 и BCR-ABL/P210) и больные ОМЛ с мутациями гена FLT3/ITD были отнесены к подгруппе неблагоприятного прогноза. Остальные пациенты, включая больных В-ОЛЛ (носители мутации TEL-AML1), ХМЛ (носители онкогена BCR-ABL/P210, при отсутствии мутаций в гене ABL), ОМЛ (носители одной из трех мутаций генов NPM1/A, AML1-ETO, CBFβ-MYH11/A), составили подгруппу относительно благоприятного прогноза.
Таблица 2.
Влияние регуляторных генов, включая CASC5, WT1, A3A, N-MYC, на область центромеры (перицентромеры)
| Ген, хромосома | Кодируемый белок и его функция* |
|---|---|
| СASC5, 15q15.1 | Кинетохорный белок CASC5/KNL1, локализуется в центромере, взаимодействует с BUBR1, с которым ассоциирован MAD2 (белок контрольной точки сборки веретена деления) |
| WT1, 11p13 | Белок опухоли Вильмса, онкоген WT1, взаимодействует с MAD2, способствует стабилизации тетрамеров MAD1–MAD2, повышает точность работы контрольной точки сборки веретена деления |
| APOBEC3A(A3A), 22q13.1 | Дезаминаза А3А, высоко экспрессируется в прицентромерном районе хромосомы (α‑сателлитная ДНК), участвует в C/U- редактировании нуклеотидов ДНК; способствует супрессии транспозонов LINE1, локализованных по соседству с α-сателлитной ДНК и микротрубочками веретена деления |
| N-MYC, 2p24.3 | Онкоген N-MYC, высоко экспрессируется в нейробластомах, ассоциирован с AURKA/AURKB, локализуемыми в центросомах/центромерах, что способствует стабилизации и накоплению N-MYC |
Дальнейший анализ экспрессии регуляторных генов, включая CASC5, WT1, A3A, N-MYC, у первичных больных лейкозом позволил обнаружить общую тенденцию к снижению экспрессии этих генов. По уровню снижения экспрессии группы больных расположились в следующем порядке: ОМЛ, ХМЛ, В-ОЛЛ. При этом в клеточной линии Jurkat (T-ОЛЛ) экспрессия генов CASC5 и N-MYC оказалась выше, чем в исследуемых группах больных. Такое различие в уровнях экспрессии регуляторных генов у пациентов и в клеточной линии Jurkat может объясняться несколькими причинами: разной нозологией, соседством лейкозных клеток в КМ с клетками стромального микроокружения, которые регулируют их пролиферацию и дифференцировку, а также с определением экспрессии генов в клетках линии Jurkat на второй день культивирования. Следует отметить, что у всех пациентов экспрессия гена CASC5 была значительно ниже, чем у здоровых доноров. Кроме того, оказалось, что чрезмерное снижение экспрессии гена CASC5 коррелирует с выявлением таргетных мутаций, характерных для неблагоприятного прогноза при лейкозе. Угнетение экспрессии гена CASC5 может приводить к нарушению взаимодействия кодируемого им белка CASC5 с другими белками прицентромерной области, включая кинетохорный комплекс KMN (KNL1/CASC5, MIS12, NDC80), обеспечивающий присоединение микротрубочек веретена деления к соответствующим центромерам до наступления анафазы митоза. Одной из причин изменения экспрессии CASC5 при лейкозе может быть влияние таргетных онкогенных мутаций.
К настоящему времени известно несколько случаев, когда онкогенные мутации оказывают влияние на экспрессию некоторых компонентов кинетохора при лейкозе. Так, при ОМЛ снижается экспрессия BUBR1, белка контрольной точки сборки веретена деления, который вступает в ассоциацию с CASC5 [38]. Полагают, что снижение экспрессии BUBR1 контролируется онкогеном AML1-ETO. Это способствует ослаблению контрольной точки сборки веретена деления и может приводить к развитию анеуплоидии. Однако при В-ОЛЛ слитый онкоген TEL-AML1 участвует в регуляции экспрессии гена MAD2, кодирующего белок контрольной точки сборки веретена деления, который взаимодействует с BUBR1 [39]. Кроме того, недавно обнаружили, что слитый онкоген BCR-ABL может снижать экспрессию MAD2, в частности, в клеточной линии мышей (32D), экспрессирующей экзогенный ген Bcr-Abl. В свою очередь, это коррелирует с нарушением функции митотического веретена деления и развитием анеуплоидии [40]. В двух последних случаях изменение экспрессии MAD2, по-видимому, способствует несбалансированной экспрессии белков кинетохорного комплекса, не исключая CASC5, что стимулирует ослабление их функции и, в конечном итоге, повышает пролиферацию лейкозных клеток. В норме MAD2 контролирует сцепление между сестринскими хроматидами в анафазе митоза, а в случае неправильного присоединения микротрубочек к центромерам блокирует расхождение хромосом и препятствует развитию анеуплоидии. Интересно отметить, что только недавно стало известно, что онкобелок WT1 опухоли Вильмса взаимодействует с белком MAD2 при митозе [33]. Это способствует стабилизации тетрамеров MAD1–MAD2 и повышает точность работы контрольной точки сборки веретена деления (табл. 2).
Повышенная экспрессия гена WT1 (≥1 × × 103/GAPDH) обнаружена нами в одном случае В-ОЛЛ, у двух больных ХМЛ и 13 больных ОМЛ (рис. 3а–в). Сверхэкспрессия WT1 (>2.6 × 103/ GAPDH) выявлена только у двух больных ХМЛ (бласты 30, 57%) с подтвержденной стадией БК, у одного из которых отмечена повышенная экспрессия гена A3A, кодирующего дезаминазу А3А. Ранее при ХМЛ (БК) наблюдали повышенную экспрессию еще одной дезаминазы, ADAR1, отвечающей за редактирование A/I в РНК, что ассоциировано с плохим прогнозом и устойчивостью к терапии [41, 42]. Следует отметить, что ADAR1 контролирует экспрессию CASC5 в клетках глиобластомы (А172), а также ближайшего гомолога A3A, дезаминазы A3С, в клетках цервикального рака (HeLa) [43, 44]. Известно, что экспрессия дезаминазы A3A ассоциирована с активным дезаминированием, повышением редактирования G/A гена WT1с.1303G>A и может быть одной из причин мутационных изменений онкогена WT1 и его сверхэкспрессии [45]. Скрининг мутаций онкогена WT1 в настоящей работе не проводили, однако анализ средних уровней экспрессии WT1 показал, что в группе больных ОМЛ экспрессия WT1 была выше, чем в группах ХМЛ и В-ОЛЛ (рис. 2). Известно, что повышенный уровень экспрессии онкогена WT1 в дебюте ОМЛ является важным прогностическим показателем. Количественный мониторинг экспрессии этого онкогена позволяет оценить минимальную остаточную болезнь у больных ОМЛ, получающих лечение, и предсказать риск развития рецидива [17].
И, наконец, только в двух случаях ОМЛ нами выявлена сверхэкспрессия онкогена N-MYC, что коррелирует с повышением экспрессии WT1, A3A и понижением экспрессии CASC5 (рис. 3в). Ранее было показано, что повышение экспрессии онкогена N-MYC, обнаруженное у взрослых пациентов с ОМЛ и у детей с Т-ОЛЛ, коррелирует с плохим прогнозом [27, 28]. Известно, что онкоген N‑MYC способствует самообновлению стволовых клеток в КМ и является одним из ключевых генов, трансфекция которого здоровым мышам приводит к развитию у них ОМЛ [27]. Сверхэкспрессию N-MYC обнаружили недавно у детей с нейробластомой [29]. Позже показали, что в клетках нейробластомы сверхэкспрессия онкогена N-MYC поддерживается при участии фактора транскрипции SPT16 [30]. Как SPT16, так и SSRP1 входят в состав белкового комплекса FACT (facilitates chromatin transcription) и участвуют в ремоделировании хроматина [46, 47]. К настоящему времени известно, что для выживания и поддержания потенциала самообновления клеток ОМЛ (клеточные линии мышей, несущие мутацию Mll-Af9 или Cbfβ-Myh11) требуется, в частности, фактор транскрипции BRG1 (SMARCA4), который связывается с хроматином и входит в состав комплекса SWI/SNF [48, 49]. Белок BRG1, участвующий в ремоделировании нуклеосомной ДНК, поддерживает повышенную транскрипцию онкогена c-Myc и способствует сохранению пула лейкозных клеток. Повышение экспрессии с-MYC в нашей работе не обнаружено. При этом у двух упомянутых первичных пациентов (ОМЛ) выявлена сверхэкспрессия гена N-MYC и повышение экспрессии SPT16. У одного из этих больных (бласты 38%), носителя мутации CBFβ-MYH11/A, получающего ПХТ, проведен мониторинг экспрессии всех биомаркеров. Повышение экспрессии онкогена N-MYC коррелировало с увеличением экспрессии генов SPT16, WT1, A3A и снижением экспрессии CASC5, что согласуется с устойчивостью к лечению и прогрессией ОМЛ (рис. 4).
Недавно стало известно, что повышению экспрессии N-MYC может способствовать не только SPT16, но и белок AURKA/B. По крайней мере, сверхэкспрессия AURKA при нейробластоме ингибирует протеолиз N-MYC, что приводит к стабилизации этого белка и его накоплению [36]. Повышенная экспрессия гена AURKA/B и кодируемого им белка выявлена при ОМЛ [50]. Если предположить, что AURKA/B вовлекается во взаимодействие с фракцией N-MYC, связанной с микротрубочками веретена деления, то участие обоих белков в регуляции биполярного присоединения микротрубочек до наступления анафазы митоза можно рассматривать как наиболее вероятное событие. И, наконец, благодаря обнаружению ассоциации белков AURKA/B с NDC80 и CASC5 – компонентами комплекса KMN (KNL1/CASC5, MIS12, NDC80), вступающими в контакт с CENPT и CENPA (CenH3), с которыми взаимодействует SPT16, можно предположить возможность взаимодействия AURKA/B, MYCN и SPT16. Однако проверка этой гипотезы требует проведения дополнительных экспериментов.
Таким образом, у всех первичных больных лейкозом (n = 40) обнаружено снижение экспрессии гена CASC5 в клетках КМ – в 4.9 (ОМЛ), 6.9 (ХМЛ) и 11.3 раза (В-ОЛЛ) по сравнению со здоровыми донорами. Показано также, что снижение экспрессии гена CASC5 при ОМЛ, ХМЛ и В-ОЛЛ коррелирует с выявлением таргетных мутаций в обеих прогностических подгруппах (относительно благоприятная и неблагоприятная), которые статистически значимо различаются между собой (p < 0.05). И, наконец, изменение экспрессии гена CASC5 ассоциировано со сверхэкспрессией генов WT1, A3A, а в некоторых случаях и N-MYC, SPT16, что сопровождается устойчивостью к терапии и прогрессией ОМЛ. Какой из генов является драйверным, инициирующим процесс лейкемогенеза, и в чем заключается роль фактора транскрипции SPT16, сверхэкспрессируемого одновременно с онкогеном N-MYC при лейкозе? Ответ на этот вопрос позволит лучше понять механизмы лейкемогенеза.
Написание настоящей статьи не потребовало специального финансирования.
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики. Всеми пациентами и здоровыми донорами подписано добровольное информированное согласие на участие в исследовании.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Hayette S., Tigaud I., Vanier A., Martel S., Corbo L., Charrin C., Beillard E., Deleage G., Magaud J.P., Rimokh R. (2000) AF15q14, a novel partner gene fused to the MLL gene in an acute myeloid leukaemia with a t(11;15)(q23;q14). Oncogene. 19, 4446–4450.
Kuefer M.U., Chinwalla V., Zeleznik-Le N.J., Behm F.G., Naeve C.W., Rakestraw K.M., Mukatira S.T., Raimondi S.C., Morris S.W. (2003) Characterization of the MLL partner gene AF15q14 involved in t(11;15)(q23;q14). Oncogene. 22, 1418–1424.
Chinwalla V., Chien A., Odero M., Neilly M.B., Zeleznik-Le N.J., Rowley J.D. (2003) A t(11;15) fuses MLL to two different genes, AF15q14 and a novel gene MPFYVE on chromosome 15. Oncogene. 22, 1400–1410.
Yang J.J., Park T.S., Lee S.T., Seo J.Y., Oh S.H., Cho E.H., Strehl S., Mühlegger N., Dworzak M.N., Zuna J., Pospisilova D., Meyer C., Marschalek R., Kim H.J., Kim S.H. (2014) Molecular characterization and clinical impact of t(11;15)(q23;q14-15) MLL-CASC5 rearrangement. Haematologica. 99, e11–3. https://doi.org/10.3324/haematol.2013.095638
Genin A., Desir J., Lambert N., Biervliet M., Van Der Aa N., Pierquin G., Killian A., Tosi M., Urbina M., Lefort A., Libert F., Pirson I., Abramowicz M. (2012) Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum. Mol. Genet. 21, 5306–5317. https://doi.org/10.1093/hmg/dds386
Sloma I., Mitjavila-Garcia M.T., Feraud O., Griscelli F., Oudrhiri N., Marsafy S., Gobbo E., Divers D., Proust A., Smadja D.M., Desterke C., Carles A., Ma Y., Hirst M., Marra M.A., Eaves C.J., Bennaceur-Griscelli A., Turhan A.G. (2017) Whole-genome analysis reveals unexpected dynamics of mutant subclone development in a patient with JAK2-V617F-positive chronic myeloid leukemia. Exp. Hematol. 53, 48–58. https://doi.org/10.1016/j.exphem.2017.05.007
Богданов К.В., Такимото М. (2008) Регуляция клеточной пролиферации в норме и при опухолевом росте. Цитология. 50, 590–596.
Takimoto M., Wei G., Dosaka-Akita H., Mao P., Kondo S., Sakuragi N., Chiba I., Miura T., Itoh N., Sasao T., Koya R.C., Tsukamoto T., Fujimoto S., Katoh H., Kuzumaki N. (2002) Frequent expression of new cancer/testis gene D40/AF15q14 in lung cancers of smokers. Br. J. Cancer. 86, 1757–1762.
Bai T., Zhao Y., Liu Y., Cai B., Dong N., Li B. (2019) Effect of KNL1 on the proliferation and apoptosis of colorectal cancer cells. Technol. Cancer Res. Treat. 18, 1533033819858668. https://doi.org/10.1177/1533033819858668
Bornhäuser M., Oelschlaegel U., Platzbecker U., Bug G., Lutterbeck K., Kiehl M.G., Schetelig J., Kiani A., Illmer T., Schaich M., Theuser C., Mohr B., Brendel C., Fauser A.A., Klein S., Martin H., Ehninger G., Thiede C. (2009) Monitoring of donor chimerism in sorted CD34+ peripheral blood cells allows the sensitive detection of imminent relapse after allogeneic stem cell transplantation. Haematologica. 94, 1613–1617. https://doi.org/10.3324/haematol.2009.007765
Богданов К.В., Никулина Т.С., Ломаиа Е.Г., Сляднев М.Н., Зарицкий А.Ю. (2017) Выявление мутаций онкогенов у больных лейкозом с использованием микрочиповой ПЦР. Биоорган. химия. 43, 523–531.
Murphy K.M., Levis M., Hafez M.J., Geiger T., Cooper L.C., Smith B.D., Small D., Berg K.D. (2003) Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J. Mol. Diagn. 5, 96–102.
Döhner K., Schlenk R.F., Habdank M., Scholl C., Rücker F.G., Corbacioglu A., Bullinger L., Fröhling S., Döhner H. (2005) Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 106, 3740–3746.
Bennour A., Beaufils N., Sennana H., Meddeb B., Saad A., Gabert J. (2010) E355G mutation appearing in a patient with e19a2 chronic myeloid leukaemia resistant to imatinib. J. Clin. Pathol. 8, 737–740.
Gabert J., Beillard E., van der Velden V.H., Bi W., Grimwade D., Pallisgaard N., Barbany G., Cazzaniga G., Cayuela J.M., Cavé H., Pane F., Aerts J.L., De Micheli D., Thirion X., Pradel V., González M., Viehmann S., Malec M., Saglio G., van Dongen J.J. (2003) Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a Europe Against Cancer program. Leukemia. 17, 2318–2357.
Beillard E., Pallisgaard N., van der Velden V.H., Bi W., Dee R., van der Schoot E., Delabesse E., Macintyre E., Gottardi E., Saglio G., Watzinger F., Lion T., van Dongen J.J., Hokland P., Gabert J. (2003) Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using 'real-time' quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) – a Europe against cancer program. Leukemia. 17, 2474–2486.
Cilloni D., Renneville A., Hermitte F., Hills R.K., Daly S., Jovanovic J.V., Gottardi E., Fava M., Schnittger S., Weiss T., Izzo B., Nomdedeu J., van der Heijden A., van der Reijden B.A., Jansen J.H., van der Velden V.H., Ommen H., Preudhomme C., Saglio G., Grimwade D. (2009) Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European Leukemia Net study. J. Clin. Oncol. 27, 5195–5201. https://doi.org/10.1200/JCO.2009.22.4865
Verma R., Babu A. (1989) in: Human chromosomes: manual of basic techniques. N.Y.: Pergamon Press, 45–67.
Гублер Е.В., Генкин А.А. (1969) Применение критериев непараметрической статистики для оценки различий двух групп наблюдений в медико-биологических исследованиях. М.: Медицина.
Gleissner B., Gökbuget N., Bartram C.R., Janssen B., Rieder H., Johannes W.G., Janssen J.W.G., Fonatsch C., Heyll A, Voliotis D., Beck J., Lipp T., Munzert G., Maurer J., Hoelzer D. (2002) Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood. 99, 1536–1543. https://doi.org/10.1182/blood.v99.5.1536
Molica M., Zacheo I., Diverio D., Alimena G., Breccia M. (2015) Long-term outcome of chronic myeloid leukaemia patients with p210 and p190 co-expression at baseline. Br. J. Haematol. 169, 148–150. https://doi.org/10.1111/bjh.13184
Sallmyr A., Fan J., Datta K., Kim K., Grosu D., Sha-piro P., Smal D., Rassool F. (2007) Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 111, 3173–3182. https://doi.org/10.1182/blood-2007-05-092510
McLean T.W., Ringold S., Neuberg D., Stegmaier K., Tantravahi R., Ritz J., Koeffler H.P., Takeuchi S., Janssen J. W., Seriu T., Bartram C.R., Sallan S.E., Gilliland D.G., Golub T.R. (1996) TEL/AML-1 dimerizes and is associated with a favorable outcome in childhood acute lymphoblastic leukemia. Blood. 88, 4252–4258.
Verma D., Kantarjian H.M., Jones D., Luthra R., Borthakur G., Verstovsek S., Rios M.B., Cortes J. (2009) Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 114, 2232–2235. https://doi.org/10.1182/blood-2009-02-204693
Grimwade D., Walker H., Oliver F., Wheatley K., Harrison C., Harrison G., Rees J., Hann I., Stevens R., Burnett A., Goldstone A. (1998) The importance of diagnostic cytogenetics on outcome in AML: analysis of 1.612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 92, 2322–2333.
Becker H., Marcucci G., Maharry K., Margeson D., Whitman S.P., Wu Y., Schwind S., Paschka P., Powell B.L., Carter T.H., Kolitz J.E., Wetzler M., Carroll A.J., Baer M.R., Caligiuri M.A., Larson R.A., Bloomfield C.D. (2010) Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J. Clin. Oncol. 28, 596–604. https://doi.org/10.1200/JCO.2009.25.1496
Kawagoe H., Kandilci A., Kranenburg T.A., Gros-veld G.C. (2007) Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res. 67, 10677–10685.
Astolfi A., Vendemini F., Urbini M., Melchionda F., Masetti R., Franzoni M., Libri V., Serravalle S., Togni M., Paone G., Montemurro L., Bressanin D., Chiarini F., Martelli A.M., Tonelli R., Pession A. (2014) MYCN is a novel oncogenic target in pediatric T-cell acute lymphoblastic leukemia. Oncotarget. 5, 120–130.
Alaminos M., Mora J., Cheung N.K., Smith A., Qin J., Chen L., Gerald W.L. (2003) Genome-wide analysis of gene expression associated with MYCN in human neuroblastoma. Cancer Res. 63, 4538–4546.
Carter D.R., Murray J., Cheung B.B., Gamble L., Koach J., Tsang J., Sutton S., Kalla H., Syed S., Gifford A.J., Issaeva N., Biktasova A., Atmadibrata B., Sun Y., Sokolowski N., Ling D., Kim P.Y., Webber H., Clark A., Ruhle M., Liu B., Oberthuer A., Fischer M., Byrne J., Saletta F., Thwe le M., Purmal A., Haderski G., Burkhart C., Speleman F., De Preter K., Beckers A., Ziegler D.S., Liu T., Gurova K.V., Gudkov A.V., Norris M.D., Haber M., Marshall G.M. (2015) Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 7, 312ra176. https://doi.org/10.1126/scitranslmed.aab1803
Kiyomitsu T., Obuse C., Yanagida M. (2007) Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell. 13, 663–676. https://doi.org/10.1016/j.devcel.2007.09.005
Tipton A.R., Wang K., Link L., Bellizzi J.J., Huang H., Yen T., Liu S. (2011) BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. J. Biol. Chem. 286, 21173–21179. https://doi.org/10.1074/jbc.M111.238543
Shandilya J., Toska E., Richard D.J., Medler K.F., Roberts S.G. (2014) WT1 interacts with MAD2 and regulates mitotic checkpoint function. Nat. Commun. 5, 4903. https://doi.org/10.1038/ncomms5903
Shu X., Liu M., Lu Z., Zhu C., Meng H., Huang S., Zhang X., Yi C. (2018) Genome-wide mapping reveals that deoxyuridine is enriched in the human centromeric DNA. Nat. Chem. Biol. 14, 680–687. https://doi.org/10.1038/s41589-018-0065-9
Richardson S.R., Narvaiza I., Planegger R.A., Weitzman M.D., Moran J.V. (2014) APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. Elife. 3, e02008. https://doi.org/10.7554/eLife.02008
Otto T., Horn S., Brockmann M., Eilers U., Schüttrumpf L., Popov N., Kenney A.M., Schulte J.H., Beijersbergen R., Christiansen H., Berwanger B., Eilers M. (2009) Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 15, 67–78. https://doi.org/10.1016/j.ccr.2008.12.005
Bogen D., Wei J.S., Azorsa D.O., Ormanoglu P., Buehler E., Guha R., Keller J.M., Griner L.A.M., Ferrer M., Song Y.K., Liao H., Mendoza A., Gryder B.E., Sindri S., He J., Wen X., Zhang S., Shern J.F., Yohe M.E., Taschner-Mandl S., Shohet J.M., Thomas C.J., Martin S.E., Ambros P.F., Khan J. (2015) Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. 6, 35247–35262. https://doi.org/10.18632/oncotarget.6208
Schnerch D., Schmidts A., Follo M., Udi J., Felthaus J., Pfeifer D., Engelhardt M., Wäsch R. (2013) BubR1 is frequently repressed in acute myeloid leukemia and its re-expression sensitizes cells to antimitotic therapy. Haematologica. 98, 1886–1895. https://doi.org/10.3324/haematol.2013.087452
Krapf G., Kaindl U., Kilbey A., Fuka G., Inthal A., Joas R., Mann G., Neil J.C., Haas O.A., Panzer-Grümayer E.R. (2010) ETV6/RUNX1 abrogates mitotic checkpoint function and targets its key player MAD2L1. Oncogene. 29, 3307–3312. https://doi.org/10.1038/onc.2010.53
Wolanin K., Magalska A., Kusio-Kobialka M., Podszywalow-Bartnicka P., Vejda S., McKenna S.L., Mosieniak G., Sikora E., Piwocka K. (2010) Expression of oncogenic kinase Bcr-Abl impairs mitotic checkpoint and promotes aberrant divisions and resistance to microtubule-targeting agents. Mol. Cancer Ther. 9, 1328–1338. https://doi.org/10.1158/1535-7163.MCT-09-0936
Zipeto M.A., Court A.C., Sadarangani A., Delos Santos N.P., Balaian L., Chun H.J., Pineda G., Morris S.R., Mason C.N., Geron I., Barrett C., Goff D.J., Wall R., Pellecchia M., Minden M., Frazer K.A., Marra M.A., Crews L.A., Jiang Q., Jamieson C.H.M. (2016) ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell. 19, 177–191. https://doi.org/10.1016/j.stem.2016.05.004
Jiang Q., Crews L.A., Barrett C.L., Chun H.J., Court A.C., Isquith J.M., Zipeto M.A., Goff D.J., Minden M., Sadarangani A., Rusert J.M., Dao K.H., Morris S.R., Goldstein L.S., Marra M.A., Frazer K.A., Jamieson C.H. (2013) ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA. 110, 1041–1046. https://doi.org/10.1073/pnas.1213021110
Galipon J., Ishii R., Suzuki Y., Tomita M., Ui-Tei K. (2017) Differential binding of three major human ADAR isoforms to coding and long non-coding transcripts. Genes (Basel). 8, pii: E68. https://doi.org/10.3390/genes8020068
Sakurai M., Shiromoto Y., Ota H., Song C., Kossenkov A.V., Wickramasinghe J., Showe L.C., Skordalakes E., Tang H.Y., Speicher D.W., Nishikura K. (2017) ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 24, 534–543. https://doi.org/10.1038/nsmb.3403
Niavarani A., Currie E., Reyal Y., Anjos-Afonso F., Horswell S., Griessinger E., Luis Sardina J., Bonnet D. (2015) APOBEC3A is implicated in a novel class of G-to-A mRNA editing in WT1 transcripts. PLoS One. 10, e0120089. https://doi.org/10.1371/journal.pone.0120089
Orphanides G., Wu W.H., Lane W.S., Hampsey M., Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 400, 284–288.
Belotserkovskaya R., Oh S., Bondarenko V.A., Orphanides G., Studitsky V.M., Reinberg D. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science. 301, 1090–1093.
Shi J., Whyte W.A., Zepeda-Mendoza C.J., Milazzo J.P., Shen C., Roe J.S., Minder J.L., Mercan F., Wang E., Eckersley-Maslin M.A., Campbell A.E., Kawaoka S., Shareef S., Zhu Z., Kendall J., Muhar M., Haslinger C., Yu M., Roeder R.G., Wigler M.H., Blobel G.A., Zuber J., Spector D.L., Young R.A., Vakoc C.R. (2013) Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662. https://doi.org/10.1101/gad.232710.113
Pulikkan J.A., Hegde M., Ahmad H.M., Belaghzal H., Illendula A., Yu J., O’Hagan K., Ou J., Muller-Tidow C., Wolfe S.A., Zhu L.J., Dekker J., Bushweller J.H., Castilla L.H. (2018) CBFβ-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell. 174, 172–186.e21. https://doi.org/10.1016/j.cell.2018.05.048
Lucena-Araujo A.R., de Oliveira F.M., Leite-Cueva S.D., dos Santos G.A., Falcao R.P., Rego E.M. (2011) High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leuk. Res. 35, 260–264. https://doi.org/10.1016/j.leukres.2010.07.034
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология