

Молекулярная биология, 2021, T. 55, № 1, стр. 164-176
Гликолевые и фосфатные депо-формы 4- и/или 5-модифицированных нуклеозидов, проявляющих противобактериальную активность
С. Д. Негря a, М. В. Ясько a, Д. А. Макаров a, d, П. Н. Сольев a, И. Л. Карпенко a, О. В. Шевченко a, О. В. Чехов a, c, А. А. Глухова b, Б. Ф. Васильева b, Т. А. Ефименко b, И. Г. Сумарукова b, О. В. Ефременкова b, С. Н. Кочетков a, Л. А. Александрова a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Научно-исследовательский институт по изысканию новых антибиотиков им. Г.Ф. Гаузе
119021 Москва, Россия
c Московский физико-технический институт (государственный университет)
141700 Моcковcкая облаcть, Долгопpудный, Россия
d Российский химико-технологический университет им. Д.И. Менделеева, Высший химический колледж
Российской академии наук
125047 Москва, Россия
* E-mail: ala2004_07@mail.ru
Поступила в редакцию 19.05.2020
После доработки 24.06.2020
Принята к публикации 25.06.2020
Аннотация
Возникновение резистентности к большинству препаратов, применяемых при инфекционных заболеваниях, требует создания новых соединений, эффективных в отношении лекарственно-устойчивых штаммов патогенов. Недавно мы синтезировали несколько групп модифицированных нуклеозидов, показавших значительную противобактериальную активность in vitro, однако их дальнейшее изучение затрудняла низкая растворимость в водных средах. В связи с этим мы синтезировали соединения, хорошо растворимые в водно-органических средах, которые оказались более эффективными ингибиторами роста грамположительных бактерий и микобактерий. Мы предположили, что рассмотренные в настоящем сообщении растворимые формы модифицированных нуклеозидов представляют собой их депо-формы. Для подтверждения этого предположения изучена способность этих соединений гидролизоваться в различных средах, а также проведен молекулярный докинг соединений в активный центр предполагаемого белка-мишени – флавинзависимой тимидилатсинтазы Mycobacterium tuberculosis (ThyX). Результаты компьютерного моделирования показали, что водорастворимые производные не являются ингибиторами тимидилатсинтазы ThyX, что может подтвердить наше предположение о действии производных нуклеозидов как депо-форм. Соединения были устойчивы к химическому гидролизу, но гидролизовались и карбоксилэстеразой печени свиньи, и в сыворотке крови человека, а также при инкубации со Staphylococcus aureus 209P. Полученные данные позволяют со значительной долей уверенности утверждать, что изученные соединения являются депо-формами модифицированных нуклеозидов.
ВВЕДЕНИЕ
Одно из важнейших достижений медицины ХХ века – широкое использование противовирусных и противобактериальных средств, позволило значительно облегчить протекание инфекционных заболеваний и существенно снизить смертность. Однако в настоящее время большая часть патогенных бактерий и вирусов выработала резистентность к используемым в клинике лекарственным препаратам [1], в частности, появились новые штаммы возбудителя туберкулеза Mycobacterium tuberculosis с множественной лекарственной устойчивостью (MDR) и широкой лекарственной устойчивостью (XDR) [2].
Нуклеотиды и нуклеозиды – это не только основные структурные единицы ДНК и РНК, они также принимают участие в различных биохимических реакциях. В связи с этим даже небольшие модификации нуклеинового основания или углеводного фрагмента нуклеозида могут оказывать существенное влияние на узнавание и ингибирование соответствующих ферментов и/или рецепторов, и, как следствие, на его активность как антипатогена. На сегодняшний день аналоги и производные нуклеозидов используются в качестве противоопухолевых, противовирусных и, в значительно меньшей степени, противогрибковых препаратов [3–8]. В то же время обнаружена антибактериальная активность нуклеозидов, что привело к активному развитию этой области [9–13]. Только в начале XXI века появились сообщения о нескольких группах модифицированных нуклеозидов, обладающих заметным антимикобактериальным действием in vitro [13–18]. В частности, 5-модифицированные пиримидиновые нуклеозиды с протяженными 1-алкинильными, алкилоксиметильными и алкилтриазолилметильными заместителями обладают ингибирующей активностью in vitro в отношении ряда микобактерий (M. tuberculosis, M. avium и M. bovis) [19–27]. Несмотря на интенсивные исследования, биологические мишени и механизм действия соединений этой группы пока окончательно не выявлены. В работах бельгийских ученых показано, что 5'-монофосфаты 5-модифицированных 2'-дезоксиуридинов эффективно ингибируют флавин зависимую тимидилатсинтазу ThyX M. tuberculosis [КФ 2.1.1.148] (уникальный фермент микобактерий), практически не взаимодействуя с основным бактериальным ферментом ThyA (близким к эукариотическим тимидилатсинтазам) [28–31]. В связи с этим можно предположить, что одной из возможных мишеней 5‑модифицированных 2'-дезоксиуридинов может быть этот фермент [28–31]. С другой стороны, нами показано, что неспособные к фосфорилированию 5'-иодо-, азидо- и аминопроизводные 5-додецилоксиметил-2'-дезоксиуридина [24] и 5‑замещенные карбоциклические 2',3'-дидезокси-2',3'-дидегидро-5'-норуридины с протяженными 1-алкинильными [25], алкилоксиметильными и алкилтриазолилметильными [26, 27] заместителями обладают значительной ингибирующей активностью in vitro в отношении M. tuberculosis, связанной с разрушением клеточной стенки микобактерии [26, 27]. Таким образом, можно предположить несколько механизмов действия модифицированных уридинов в отношении M. tuberculosis.
Серьезную проблему при изучении биологических свойств модифицированных нуклеозидов, проявляющих противобактериальную и/или противовирусную активность, представляет их низкая растворимость в воде. Для решения этой задачи необходимо ввести в состав молекулы гидрофильные группировки, синтезируя так называемые депо-формы (“пролекарства”), с помощью которых можно модулировать фармакокинетику, фармакодинамику и токсичность препарата. Термин депо-форма означает биологически инертное или слабо активное соединение, содержащее исходное лекарственное средство, которое подвергается трансформации in vivo вследствие химического или ферментативного расщепления. Такой подход, с одной стороны, позволяет эффективно доставлять молекулу в клетки, а с другой, улучшать фармакокинетические характеристики препарата за счет более медленного высвобождения активного компонента [32–39].
Недавно мы синтезировали ряд водорастворимых форм 5-модифицированных пиримидиновых 2'-дезоксинуклеозидов. Они оказались как минимум на два порядка лучше растворимыми по сравнению с исходными формами. Полученные соединения были активны против ряда грамположительных бактерий (MIC 20–95 мкг/мл), включая лекарственно-устойчивые штаммы Staphylococcus aureus и M. smegmatis, обладали низкой цитотоксичностью в отношении клеточных линий человека (CD50$ \gg $ 100 мкг/мл) [40, 41].
Настоящая работа посвящена изучению химической и ферментативной стабильности синтезированных соединений с целью определения возможного механизма их действия для подтверждения нашей гипотезы о том, что они являются депо-формами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследуемые соединения синтезированы согласно разработанным нами ранее методам [31, 40, 41].
Идентификацию продуктов гидролиза проводили методом хромато-масс-спектрометрии. Масс-спектры высокого разрешения регистрировали на приборе Bruker Daltonics micrOTOF-Q II методом электрораспылительной ионизации (ESI). Измерения выполнены на положительных ионах в соответствии с применяемыми ранее условиями [42].
Количественный анализ смеси продуктов гидролиза проводили методом тонкослойной хроматографии (ТСХ) на пластинках Kieselgel 60 F254 (“Merck”, Германия) в системах хлороформ– этанол 9 : 1 (система A), диоксан–аммиак (25% водн.) 4 : 1 (система Б) или диоксан–аммиак (25% водн.)–вода 6 : 1 : 4 (система В). Анализ ТСХ проводили на приборе ChemiDoc Imaging System (“Bio-Rad”, США). Результаты обрабатывали с помощью программы Image Lab Software 6.1 (“Bio-Rad”). Средние значения и стандартные отклонения, приведенные на диаграммах, получали исходя из трех независимых экспериментов. Экспериментальные данные, полученные в программе Image Lab, анализировали с применением программного обеспечения Excel.
Растворимость соединений определяли путем перемешивания на магнитной мешалке 20 мг соединения в 3 мл воды в течение 24 ч. Осадок отделяли центрифугированием (10 мин, 14 000 об./мин). Концентрацию вещества определяли, измеряя УФ-поглощение полученного раствора.
Химический гидролиз соединений и гидролиз соединений в сыворотке крови человека проводили в соответствии с разработанными ранее условиями [43].
Ферментативный гидролиз соединений проводили карбоксилэстеразой из свиной печени [КФ 3.1.1.1] (“Sigma-Aldrich”, лиофилизат, ≥15 ед./мг).
Химический гидролиз соединений. К 100 мкл раствора нуклеозида (40 мМ) в ДМСО добавляли 1.9 мл растворов, содержащих 0.2 М глицин + 0.2 М HCl-буфер (рН 2.2), или 0.1 М калий-фосфатный буфер (рН 7.5), или 0.2 М глицин + 0.2 М NaOH-буфер (рН 9.0), и инкубировали в течение 24 ч при 37°С. Аликвоты (0, 0.5, 2, 4, 8, 12, 16, 24 ч) анализировали с помощью ТСХ (в системе A).
Гидролиз соединений в сыворотке крови человека. В сыворотку крови человека (100%, 196 мкл) добавляли 4 мкл раствора изучаемых соединений (20 мМ) в ДМСО. Смесь инкубировали в течение 24 ч при 37°С. Аликвоты (15 мкл) отбирали через определенные промежутки времени, добавляли этанол (60 мкл), смесь выдерживали в течение 20 мин при –20°C и центрифугировали (10 мин, 14 000 об./мин). Отбирали супернатант и выпаривали его до получения сухого остатка. Полученные остатки растворяли в этаноле (10 мкл) и анализировали методами хромато-масс-спектрометрии и ТСХ в системах A для производных нуклеозидов, Б и В для производных нуклеотидов.
Ферментативный гидролиз соединений. Карбоксилэстеразу (5 ед.) растворяли в 28 мкл буферной смеси №1 (25 мМ Трис-HCl, 50 мМ KCl, рН 8.0). Затем к полученному раствору добавляли 5 мкл раствора изучаемых соединений (20 мМ) в этаноле, 40 мкл буферной смеси № 2 (50 мМ Трис-HCl, 6 мМ CaCl2, 350 мМ NaCl, рН 7.6) и 127 мкл H2O. Полученную реакционную смесь инкубировали при 37°С в течение 3 ч. Аликвоты (15 мкл) отбирали через определенные промежутки времени, добавляли этанол (60 мкл), смесь выдерживали в течение 20 мин при –20°C и центрифугировали (10 мин, 14000 об./мин). Отбирали супернатант и выпаривали его до получения сухого остатка. Полученные остатки растворяли в этаноле (10 мкл) и анализировали методами хромато-масс-спектрометрии или ТСХ в системе А.
Гидролиз соединений 1c и 5c при инкубировании с Staphylococcus aureus FDA 209P. Соединения 1c и 5c (3.3 мг) растворяли в смеси MeOH : H2O 1 : 1 (5 мл) и 180 мкл смеси добавляли к 2820 мкл модифицированной питательной среды № 2 Гаузе, инфицированной S. aureus FDA 209P (106 клеток/мл). Пробы инкубировали при 37°С, отбирая аликвоты (300 мкл) в 0, 1, 2, 4 и 8 ч, после чего выдерживали во льду в течение 5 мин и отделяли супернатант центрифугированием (13 000 об./мин, 5 мин, Eppendorf). К отобранному супернатанту (300 мкл) добавляли 1 мл этанола и выдерживали в течение 20 мин при –20°С. Далее полученную суспензию центрифугировали в тех же условиях, отбирали супернатанты и выпаривали их до получения сухого остатка, который растворяли в этаноле (20 мкл) и анализировали с помощью хромато-масс-спектрометрии или ТСХ. К осадкам, содержащим бактериальные клетки, добавляли по 100 мкл водного раствора лизоцима с концентрацией 10 мкг/мкл, встряхивали на вортексе в течение 1 мин и помещали в термостат с температурой 37°С на 30 мин. Полное разрушение клеток стафилококка было подтверждено после завершения инкубирования путем микроскопирования. В пробирки вносили по 800 мкл этанола, выдерживали в течение 20 мин при –20°С, отделяли супернатант центрифугированием в описанном режиме и анализировали по приведенной выше методике.
Компьютерное моделирование связывания 5'-O- и 3'-O-(триэтиленгликоль)карбонил-5-додецилоксиметил-2'-дезоксиуридина (5с и 1с) и dUMP с белком (ThyX). Связывание 5c и 1c с белком ThyX изучали на его структуре, полученной с разрешением 2.01 Å (идентификатор PDB – 2AF6 [44]). В качестве исходной брали геометрию комплекса ThyX с 5'-монофосфатом 5-додецилоксиметил-2'-дезоксиуридина, полученную в [31]. На основе этой же работы применили процедуру пошагового наращивания звеньев фрагмента каждого соединения – 5c или 1c. В результате получили структуры комплексов 5c и 1c с активным центром ThyX и определили их энергии. Далее определили структуры и энергии этих комплексов, когда 5c и 1c удалены более чем на 15 Å от ThyX. Разность этих энергий и энергий 5c и 1c в активном центре ThyX интерпретировали как искомые энергии связывания.
Использование системы параметров MMFF94x [45] и критериев окончания расчетов в программе Molecular Operating Environment (MOE) версии 2009.10 [46] подробно описаны в [31].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Отнесение противобактериальных модифицированных нуклеозидов к депо-формам требует, прежде всего, изучения их химической и ферментативной стабильности. С этой целью мы выбрали ряд модифицированных 2'-дезоксинуклеозидов, содержащих различные заместители как в пиримидиновом основании, так и в углеводном фрагменте молекулы (рис. 1).
Были исследованы 3'-O- и 5'-O-карбонилтри- и тетраэтиленгликолевые производные 5-алкилоксиметил-2'-дезоксиуридина (3': 1a–c, 2c и 5': 5a–c, 6с), 3'-O- и 5'-O-карбонилтри- и тетраэтиленгликолевые производные 5-(4-алкилтриазол-1-ил)метил-2'-дезоксиуридина (3': 3a–c и 4а и 5': 7a, c, 8a), 3'-O-карбонилтриэтиленгликолевые производные N4-алкил-2'-дезокси-5-метилцитидина (9а, с), 3'-O-(триэтиленгликоль)карбонил-4-тио-5-ундецилоксиметил-2'-дезоксиуридин (10b), 3'-(триэтиленгликоль)карбониламино-5-додецилоксиметил-2',3'-дидезоксиуридин (11с), 3'-O-(6-гидроксигексилоксикарбонил)-5-ундецилоксиметил-2'-дезоксиуридин (12b), 3'-O-(6-аминогексилоксикарбонил)-5-ундецилоксиметил-2'-дезоксиуридин (13b) и также 5'-монофосфаты 5-алкилоксиметил-2'-дезоксиуридина (14a–c) и 5-(4-алкилтриазол-1-ил)метил-2'-дезоксиуридина (15a, c).
При изучении стабильности гликолевых производных модифицированных нуклеозидов в качестве контролей использовали 3',5'-диацетат тимидина и 5'-монофосфат тимидина, а при исследовании стабильности 5'-монофосфатов модифицированных нуклеозидов применяли 5'-монофосфаты тимидина и 5-азидометил-2'-дезоксиуридина (16).
Как сказано во введении, растворимость депо-форм изученных модифицированных пиримидиновых 2'-дезоксинуклеозидов (рис. 2) в воде была лучше, чем у исходных соединений.
В табл. 1 в качестве примера приведены растворимости в воде исходных 5-модифицированных производных 2'-дезоксиуридина и их 5'-триэтиленгликолевых производных.
Таблица 1.
Значения растворимости в воде некоторых гликолевых производных 5-модифицированных 2'-дезоксиуридинов
| № | Заместитель при нуклеиновом основании | Заместитель при углеводном фрагменте | Раствори-мость, мг/мл | № | Заместитель при нуклеиновом основании | Заместитель при углеводном фрагменте | Раствори-мость, мг/мл |
|---|---|---|---|---|---|---|---|
| 17a | 5-CH2–OC10H21 | – | 0.028 | 18a | 5-CH2–TriC10H21 | – | 0.01 |
| 17b | 5-CH2–OC11H23 | – | 0.017 | 18c | 5-CH2–TriC12H25 | – | 0.005 |
| 17c | 5-CH2–OC12H25 | – | 0.009 | 3a | 5-CH2–TriC10H21*** | 3'-O–C(O)TEG | 1.25 |
| 1a | 5-CH2–OC10H21 | 3'-O–C(O)TEG* | 4.46 | 3c | 5-CH2–TriC12H25 | 3'-O–C(O)TEG | 0.78 |
| 1b | 5-CH2–OC11H23 | 3'-O–C(O)TEG | 3.61 | 7a | 5-CH2–TriC10H21 | 5'-O–C(O)TEG | 2.82 |
| 1c | 5-CH2–OC12H25 | 3'-O–C(O)TEG | 1.3 | 7c | 5-CH2–TriC12H25 | 5'-O–C(O)TEG | 1.18 |
| 5a | 5-CH2–OC10H21 | 5'-O–C(O)TEG | 5.5 | 4а | 5-CH2–TriC10H21 | 3'-O–C(O)TetEG | 2.14 |
| 5b | 5-CH2–OC11H23 | 5'-O–C(O)TEG | 2.26 | 8a | 5-CH2–TriC10H21 | 5'-O–C(O)TetEG | 4.36 |
| 5c | 5-CH2–OC12H25 | 5'-O–C(O)TEG | 0.85 |
Химическая и ферментативная стабильность
Стабильность соединений в буферных растворах и в сыворотке крови человека оценивали, используя метод [43]. Все изученные производные нуклеозидов были устойчивы в буферных растворах при двух значениях рН: 2.2 и 7.5 – и незначительно гидролизовались при рН 9.0. Это указывает на химическую стабильность исследованных соединений, и их гидролиз в сыворотке крови был практически полностью ферментативным.
Оптимальное время полугидролиза депо-форм составляет 5–12 ч, согласно [47]. Практически все полученные гликолевые производные, за исключением 3'-O-карбонилтриэтиленгликолевых производных N4-алкил-2'-дезокси-5-метилцитидина (9а, с) и 3'-O-(триэтиленгликоль)карбониламино-5-додецилоксиметил-2',3'-дидезоксиуридина (11с), имели оптимальное время полугидролиза 2–12 ч (табл. 2) и расщеплялись только до исходных 5-модифицированных нуклеозидов. Наличие более протяженного алкильного или гликолевого заместителя обычно увеличивало время гидролиза.
Таблица 2.
Значения времени полугидролиза соединений в сыворотке крови человека
| № | Заместитель при нуклеиновом основании | Заместитель при углеводном фрагменте | t1/2 (ч ± 0.5) |
№ | Заместитель при нуклеиновом основании | Заместитель при углеводном фрагменте | t1/2 (ч ± 0.5) |
|---|---|---|---|---|---|---|---|
| 1a | 5-CH2–OC10H21 | 3'-O–C(O)TEG* | 2 | 8a | 5-CH2–TriC10H21 | 5'-O–C(O)TetEG | 12 |
| 1b | 5-CH2–OC11H23 | 3'-O–C(O)TEG | 3 | 9a | 4-NHC10H21 | 3'-O–C(O)TEG | >24 |
| 1c | 5-CH2–OC12H25 | 3'-O–C(O)TEG | 4 | 9с | 4-NHC12H25 | 3'-O–C(O)TEG | >24 |
| 5a | 5-CH2–OC10H21 | 5'-O–C(O)TEG | 3 | 10b | 4-S 5-CH2–OC11H23 |
3'-O–C(O)TEG | 6 |
| 5b | 5-CH2–OC11H23 | 5'-O–C(O)TEG | 3 | 11c | 5-CH2–OC12H25 | 3'-NH–C(O)TEG | >24 |
| 5c | 5-CH2–OC12H25 | 5'-O–C(O)TEG | 5 | 12b | 5-CH2–OC11H23 | 3'-O–C(O)OC6H12OH | 14 |
| 2c | 5-CH2–OC12H25 | 3'-O–C(O)TetEG** | 7 | 13b | 5-CH2–OC11H23 | 3'-O–C(O)OC6H12NH2 | 16 |
| 6c | 5-CH2–OC12H25 | 5’-O–C(O)TetEG | 8 | 14a | 5-CH2–OC10H21 | 5'-O–P(O)(OH)2 | >12 |
| 3a | 5-CH2–TriC10H21*** | 3'-O–C(O)TEG | 6 | 14b | 5-CH2–OC11H23 | 5'-O–P(O)(OH)2 | >12 |
| 3c | 5-CH2–TriC12H25 | 3'-O–C(O)TEG | 8 | 14c | 5-CH2–OC12H25 | 5'-O–PO(OH)2 | >12 |
| 7a | 5-CH2–TriC10H21 | 5'-O–C(O)TEG | 7 | 15а | 5-CH2–TriC10H21 | 5'-O–P(O)(OH)2 | 9 |
| 7c | 5-CH2–TriC12H25 | 5'-O–C(O)TEG | 5 | 15c | 5-CH2–TriC12H25 | 5'-O–P(O)(OH)2 | 11 |
| 4а | 5-CH2–TriC10H21 | 3'-O–C(O)TetEG | 10 | 16 | 5-CH2N3 | 5'-O–P(O)(OH)2 | 1.5 |
| TMP | – | 5'-O–P(O)(OH)2 | 40 ± ± 10 мин. | Ac2T | – | 3'-OAc 5'-OAc |
3 |
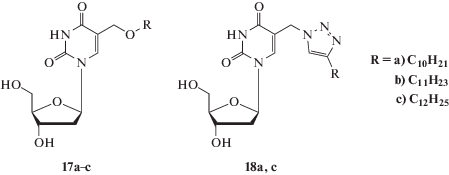
На рис. 3 в качестве примера представлена диаграмма гидролиза 3'-O-(триэтиленгликоль)карбонил-5-додецилоксиметил-2'-дезоксиуридина 1с в сыворотке крови человека, из которой следует, что время полугидролиза (t1/2) 1с с образованием “родительского” 5-додецилоксиметил-2'-дезоксиуридина 17c (Схема 1 ) составляет 4 ± 0.5 ч.
Рис. 3.
Гидролиз соединения 1с в сыворотке крови человека. По оси Х отложено время отбора проб, по оси Y – количество вещества в процентах. Гидролиз проводилия в условиях, приведенных в Экспериментальной части (см. Гидролиз соединений в сыворотке крови человека).
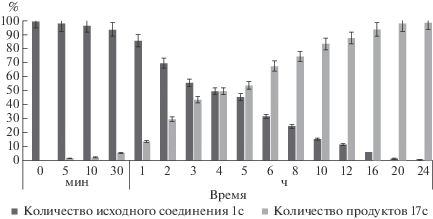
Время полугидролиза соединений 1с и 5с карбоксилэстеразой составило 60 ± 10 мин (рис. 4).
Гидролиз соединений 1с и 5с при инкубации с S. aureus 209P
Изучение продуктов метаболизма 1с и 5с показало, что родительский нуклеозид 5-додецилоксиметил-2'-дезоксиуридин 17с образуется при инкубации соединений 1с и 5с (в концентрациях, соответствующих ½ МПК) в инкубационной среде, содержащей S. aureus FDA 209P. Обнаружено, что после инкубации в течение 8 ч конверсия 1с и 5с в родительский нуклеозид 17с составляет около 30% как внутри бактерий, так и в инкубационной среде.
Компьютерное моделирование соединений 1с и 5с с активным центром ThyX
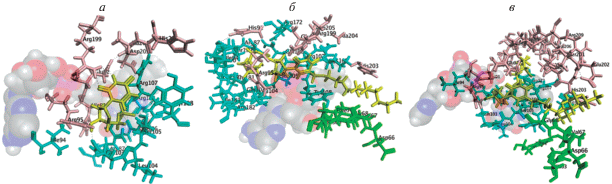
Как сказано выше, данные об эффективном ингибировании микобактериальной флавинзависимой тимидилатсинтазы ThyX 5'-монофосфатами 5-модифицированных 2'-дезоксиуридинов [28–31] позволяют рассматривать данный фермент в качестве одной из мишеней ингибирующего действия 5-модифицированных 2'-дезоксиуридинов. С целью выявления возможности связывания полученных нами водорастворимых производных с ThyX проведен молекулярный докинг 5'-O- и 3'-O-(триэтиленгликоль)карбонил-5-додецилоксиметил-2'-дезоксиуридина (5с и 1с, в качестве примера) с активным центром фермента и сравнение полученных энергий связывания ThyX c соединениями 5с, 1с и субстратом фермента – монофосфатом 2'-дезоксиуридина (dUMP). Результаты расчетов приведены в табл. 3, а геометрия полученных комплексов – на рис. 5.
Рис. 4.
Ферментативный гидролиз соединения 1с карбоксилэстеразой. По оси Х отложено время отбора проб, по оси Y – количество вещества в процентах. Гидролиз проводили в условиях, приведенных в Экспериментальной части (см. Ферментативный гидролиз соединений).
Таблица 3.
Энергия связывания соединений с активным центром флавинзависимой тимидилатсинтазы M. tuberculosis (белка ThyX)
| Соединение | Субстрат dUMP [31] | Аналог 5с | Аналог 1с |
|---|---|---|---|
| Энергия связывания, ккал/моль | –189.4 | –50.1 | –77.1 |
Рис. 5.
Расположение субстрата и его аналогов в активном центре ThyX. а – ThyX с субстратом, б – ThyX с аналогом 1c, в – ThyX с аналогом 5c. Желтым цветом показаны субстрат и, соответственно, его аналоги 1c и 5c, кремовым – цепь A ThyX, зеленым – цепь B ThyX, бирюзовым – цепь C ThyX. FAD располагается на переднем плане каждого рисунка и поэтому изображен полупрозрачным.
Максимальные значения энергии связывания исследуемых аналогов с ThyX 77.1 и 50.1 ккал/моль, соответственно, что хуже энергии связывания с субстратом. Это свидетельствует о том, что данные аналоги, скорее всего, не являются ингибиторами ThyX.
Попытки оптимизации геометрии веществ в активном центре для увеличения энергии связывания исследуемых аналогов с ThyX не привели к существенному изменению энергии.
Сравнение положений субстрата, аналога 1c и аналога 5c относительно FAD и цепей A, B, C белка ThyX позволяет сделать следующие заключения. Как показано ранее, наличие длинного алифатического заместителя по положению 5 урацила обеспечивает дополнительный контакт этого заместителя с цепью B и не меняет расположение дезоксиуридинового фрагмента относительно FAD. Это обеспечивает успешное ингибирование ThyX 5’-монофосфатами 5-модифицированных пиримидиновых нуклеозидов с протяженными 1‑алкинильными, алкилоксиметильными и алкилтриазолилметильными заместителями, отмечаемое в [28–31].
Появление объемного заместителя по положению 3'-дезоксиуридина приводит к существенному смещению всего аналога относительно FAD. Одновременно ослабляется контакт с цепью A и существенно возрастает контакт с цепью C.
Наконец, такой же заместитель в положении 5'-дезоксиуридина оставляет контакт FAD с урацильным остатком близким к контакту FAD с субстратом, но, одновременно, усиливает контакт аналога с цепью A.
Из этого можно заключить, что контакт 5'-заместителя с цепью A оказывается энергетически невыгодным и, несмотря на остальные относительно удачные контакты, приводит к практически нулевому связыванию аналога 5c в активном центре ThyX.
Взаимодействие 3'-заместителя с цепью C не является столь же невыгодным, как взаимодействие с цепью A, но сопровождающие его изменения в расположении аналога 1c относительно активного центра фермента (и FAD) все же приводят к уменьшению энергии связывания ThyX и 1c настолько, что он является плохим ингибитором этого фермента.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Кризис антибиотикотерапии, обусловленный резистентностью патогенов к одобренным антибактериальным препаратам, привел к необходимости создания принципиально новых малотоксичных веществ – эффективных ингибиторов лекарственно-устойчивых штаммов патогенов. Очевидно, что для получения лекарственных форм таких соединений желательно, чтобы они были растворимы в воде. Дизайн депо-формы представляет собой широко используемую стратегию молекулярной модификации, которая направлена на оптимизацию физико-химических и фармакологических свойств лекарств, для улучшения их растворимости и фармакокинетических свойств и, зачастую, снижения их токсичности [33, 37]. Как указывалось выше, предложен ряд способов получения депо-форм соединений [32, 35–43, 48, 49]. В качестве пероральных пролекарств хорошо зарекомендовали себя аминокислотные (в первую очередь валиновые) эфиры нуклеозидов [50–52].
Другой подход к повышению растворимости биологически активных нуклеозидов – синтез их 5'-фосфатных или 5'-фосфонатных производных, которые под действием клеточных ферментов гидролизуются с постепенным образованием активного нуклеозидного ингибитора. В качестве примера можно привести 5'-фосфонатные гомо- и гетеродимеры нуклеозидов с противовирусной и противобактериальной активностью [41, 53] или применяемый в терапии онкологических заболеваний 5'-фосфат 2-фторарааденозина (флударабин) [54], а также созданные в нашей лаборатории 5'-H-фосфонат 3'-азидо-3'-дезокситимидина (никавир), используемый в России для лечения инфицированных вирусом иммунодефицита человека [55], и 5'-H-фосфонат 2',3'-дидезокси-3'-тиацитидина [49].
Мы воспользовались этими подходами для синтеза растворимых производных полученных нами ранее нуклеозидов, проявляющих противобактериальную активность.
Известно, что оптимальное время полугидролиза депо-форм в сыворотке крови человека составляет от 3 до 12 ч [47]. Синтезированные нами 5'-монофосфаты 5-алкилоксиметил-2'-дезоксиуридина (14а-с) оказались довольно устойчивыми к ферментативному гидролизу (время полугидролиза в сыворотке крови человека >12 ч), в то время как 5'-монофосфаты 5-(4-алкилтриазол-1-ил)метил-2'-дезоксиуридина (15а,с) имели оптимальное время полугидролиза (t1/2: 3–11 ч). Для сравнения: t1/2 5'-монофосфатов тимидина и 5-азидометил-2'-дезоксиуридина (16) в этих условиях составляет 40 и 90 мин соответственно. Однако применение 5'-монофосфатов в большинстве случаев ограничено неспособностью заряженных соединений проникать через клеточную стенку бактерий [23, 56].
Более перспективным нам представляется применение в качестве депо-форм 3'-O- и 5'-O-карбонилтри- и тетраэтиленгликолевых производных нуклеозидов. Как сказано во введении и показано в нашей работе (табл. 1), растворимость депо-форм изученных модифицированных пиримидиновых 2'-дезоксинуклеозидов оказалась, как минимум, на два порядка лучше чем у исходных форм (для примера, 8.5 мг/мл у 3'-триэтиленгликолевого производного 5с и 13 мг/мл у 5'-производного по сравнению с 0.09 мг/мл у исходного 5-додецилоксиметил-2'-дезоксиуридина 17с, табл. 1) При этом растворимость 3'- и 5'-производных различалась незначительно, а замена триэтиленгиколевого фрагмента на тетраэтиленгликолевый приводила к двукратному улучшению растворимости, например, 2.14 у триэтиленгликолевого производного 4а и 4.36 у тетраэтиленгликолевого производного 8а.
Практически все полученные соединения, за исключением N4-алкил-5-метил-2'-дезоксицитидинов (9а, с) и 3'-(триэтиленгликоль)карбониламино-5-додецилоксиметил-2',3'-дидезоксиуридина (11с), имели время полугидролиза 2–12 ч (табл. 2) и расщеплялись только до исходных нуклеозидов, что соответствует поставленной нами цели. Стоит отметить, что соединения оказались стабильными в буферных растворах с незначительным гидролизом при pH 9.0 и эффективно гидролизовались карбоксилэстеразой (рис. 4), что указывает на ферментативную природу гидролиза в сыворотке крови.
Эти наблюдения также подтверждают, что карбонатные и карбаматные группы в депо-формах соединений отличаются по прочности связи. Оксикарбонильная связь в них не равноценна связи в природных сложных эфирах, однако ее гидролиз в сыворотке крови, вероятно, также осуществляется карбоксилэстеразами в соответствии со схемой 1 , но лишь в случае карбонатов, что объясняет сильно различающиеся времена полугидролиза соединений 11c и 1c в сыворотке крови человека.
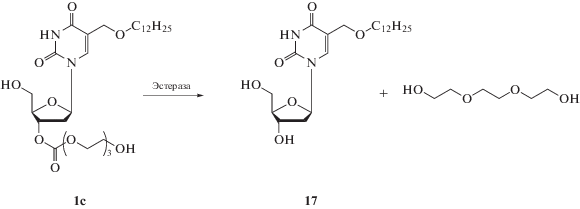
Схема 1 . Предполагаемый механизм гидролиза водорастворимых форм нуклеозидов (на примере 1с).
Из табл. 2 следует, что карбонилтри- и тетраэтиленгликолевые производные различаются по скорости гидролиза в сыворотке крови (тетраэтилегликолевые производные гидролизуются медленнее). С другой стороны, замена гликолевого фрагмента на гидроксигексильный или аминогексильный приводила к значительному увеличению времени гидролиза соединений 12b и 13b (t1/2 = 14 и 16 ч соответственно).
На устойчивость соединений к гидролизу влияет также модификация нуклеинового основания, например, замена атома кислорода в 4-м положении в соединении 1b на атом серы 10b приводила к двукратному увеличению времени гидролиза (t1/2 = = 3 и 6 ч соответственно). Наблюдается также зависимость стабильности гликолевых производных от размера заместителя в позиции 5 основания. Как правило, соединения с более протяженными углеводородными заместителями имеют большее время гидролиза. С другой стороны, нахождение гликолевых заместителей в 3'- или 5'-положении углеводного фрагмента не влияет существенно на устойчивость гликолевых производных.
Среди всех исследованных соединений, три- и тетраэтиленгликолевые производные 5-додецилоксиметил-2'-дезоксиуридина 1c, 2c, 5c и 6c представляются наиболее перспективными для использования в качестве депо-форм, поскольку обладают как оптимальным временем полугидролиза (4–8 ч), так и заметно увеличенной растворимостью в воде (8.5–13 мг/мл по сравнению с 0.09 мг/мл у исходного соединения).
Результаты теоретических расчетов связывания 5'-O- и 3'-O-(триэтиленгликоль)карбонил-5-додецилоксиметил-2'-дезоксиуридина (5с и 1с) с флавинзависимой тимидилатсинтазой M. tuberculosis (белком ThyX) в сравнении с природным субстратом dUMP позволяют предположить, что эти аналоги 5с и 1с не являются ингибиторами ThyX, поскольку значение энергии связывания с исследуемыми аналогами значительно хуже энергии связывания с субстратом. Можно предположить, что проникшие в клетку соединения должны подвергаться расщеплению эстеразами, а уже затем связываться с предполагаемой мишенью ThyX, как это, собственно, и следует из самой концепции депо-формы.
ЗАКЛЮЧЕНИЕ
Таким образом, полученные данные (результаты ферментативного гидролиза и теоретические расчеты) позволяют со значительной долей уверенности утверждать, что синтезированные нами хорошо растворимые в водно-органических средах карбонилтри- и тетраэтиленгликолевые производные и 5'-монофосфаты модифицированных нуклеозидов являются депо-формами нуклеозидов, обладающих противобактериальной активностью. Показана перспективность создания подобных производных нуклеозидов для получения эффективных ингибиторов роста микроорганизмов.
Авторы благодарны М.К. Кухановой (ИМБ РАН) за ценные советы и помощь в работе.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (гранты № 20-04-01085, 20-04-00094, 20-34-90056, 17-00-00393 и 18-29-08010) и Программы фундаментальных исследований государственных академий наук на 2013–2020 годы (№ 01201363818).
В настоящей работе не использовали людей и животных в качестве объектов исследования.
Авторы подтверждают отсутствие конфликта интересов.
Список литературы
Ventola C.L. (2015) The antibiotic resistance crisis: part 1: causes and threats. P&T. 40, 277–283.
Brown E.D., Wright G.D. (2016) Antibacterial drug discovery in the resistance era. Nature. 529, 336–343.
Jordheim L.P., Durantel D., Zoulim F., Dumontet C. (2013) Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug. Discov. 12, 447–464.
Matsuda A., Sasaki T. (2004) Antitumor activity of sugar-modified cytosine nucleosides. Cancer Sci. 95,105–111.
De Clercq E. (2012) Human viral diseases: what is next for antiviral drug discovery? Curr. Opin. Virol. 2, 572–579.
De Clercq E., Li G. (2016) Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 29. 695–747.
Winn M., Goss R.J.M., Kimura K., Timothy D.H., Bugg T.D.H. (2010) Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 27, 279–304.
Yssel A.E.J., Vanderleyden J., Steenackers H.P. (2017) Repurposing of nucleoside- and nucleobase-derivative drugs as antibiotics and biofilm inhibitors. J. Antimicrob. Chemother. 72, 2156–2170.
Cui Z., Wang X., Koppermann S., Thorson J.S., Ducho C., Van Lanen S.J. (2018) Antibacterial muraymycins from mutant strains of Streptomyces sp. NRRL 30471. J. Nat. Prod. 81. 942–948.
Abbas M., Elshahavi S.I., Wang X., Ponomareva L.V., Sajid I., Shaaban K.A., Thorson J.S. (2018) Puromycins B-E, naturally occurring amino-nucleosides produced by the Himalayan isolate Streptomyces sp. PU-14G. J. Nat. Prod. 81, 2560–2566.
Seydlova G., Pohl R., Zbornikova E., Ehn M., Simak O., Panova N., Kolar M., Bogdanova K., Vecerova R., Fi-ser R., Sanderova H., Vitovska D., Sudzinova P., Pospisil J., Benada O., Krizek T., Sedlak D., Bartunek P., Krasny L., Rejman D. (2017) Lipophosphonoxins II: design, synthesis, and properties of novel broad spectrum antibacterial agents. J. Med. Chem. 60, 6098 6118.
Bockman M.R., Engelhart C.A., Dawadi S., Larson P., Tiwari D., Ferguson D.M., Schnappinger D., Aldrich C.C. (2018) Avoiding antibiotic inactivation in Mycobacterium tuberculosis by Rv3406 through strategic nucleoside modification. ACS Infect. Dis. 4, 1102–1113.
Serpi M., Ferrari V., Pertusati F. (2016) Nucleoside derived antibiotics to fight microbial drug resistance: new utilities for an established class of drugs? J. Med. Chem. 59, 10343–10382.
Van Calenbergh S., Pochet S., Munier-Lehmann H. (2012) Drug design and identification of potent leads against Mycobacterium tuberculosis thymidine monophosphate kinase. Curr. Top. Med. Chem. 12, 694–705.
Duckworth B.P., Nelson K.M., Aldrich C.C. (2012) Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 12, 766–796.
Duckworth B.P., Wilson D.J., Nelson K.M., Boshoff H.I., Barry C.E. 3rd, Aldrich C.C. (2012) Development of a selective activity-based probe for adenylating enzymes: profiling MbtA involved in siderophore biosynthesis from Mycobacterium tuberculosis. ACS Chem. Biol. 7, 1653–1658.
Шмаленюк Э.Р., Кочетков С.Н., Александрова Л.А. (2013) Новые ингибиторы роста M. tuberculosis на основе модифицированных пиримидиновых нуклеозидов и их аналогов. Успехи химии. 82, 896–915.
Ferrari V., Serpi M. (2015) Nucleoside analogs and tuberculosis: new weapons against an old enemy. Future Med. Chem. 7, 291–314.
Rai D., Johar M., Manning T., Agrawal B., Kunimoto D.Y., Kumar R. (2005) Design and studies of novel 5-substituted alkynylpyrimidine nucleosides as potent inhibitors of mycobacteria. J. Med. Chem. 48, 7012–7017.
Johar M., Manning T., Tse C., Desroches N., Kunimoto D.Y., Agrawal B., Kumar R. (2007) Growth inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium in vitro: effect of 1-beta-D-2'-arabinofuranosyl and 1-(2'-deoxy-2'-fluoro-beta-D-2'-ribofuranosyl) pyrimidine nucleoside analogs. J. Med. Chem. 50, 3696–3705.
Srivastav N.C., Manning T., Kunimoto D.Y., Kumar R. (2007) Studies on acyclic pyrimidines as inhibitors of mycobacteria. Bioorg. Med. Chem. 15, 2045–2053.
Srivastav N.C., Manning T., Kunimoto D.Y., Kumar R. (2006) In vitro anti-mycobacterial activities of various 2'-deoxyuridine, 2'-arabinouridine and 2'-arabinofluoro-2'-deoxyuridine analogues: synthesis and biological studies. Med. Chem. 2, 287–293.
Александрова Л.А., Шмаленюк Э.Р., Кочетков С.Н., Ерохин В.В., Смирнова Т.Г., Андреевская С.Н., Черноусова Л.Н. (2010) Новые 5-модифицированные пиримидиновые нуклеозиды – ингибиторы роста микобактерий. Acta Naturae. 1, 115–118.
Shmalenyuk E.R., Chernousova L.N., Karpenko I.L., Kochetkov S.N., Smirnova T.G., Andreevskaya S.N., Efremenkova O.V., Alexandrova L.A. (2013) Inhibition of Mycobacterium tuberculosis strains H37Rv and MDR MS-115 by a new set of C5 modified pyrimidine nucleosides. Bioorg. Med. Chem. 21, 4874–4884.
Matyugina E., Khandazhinskaya A., Chernousova L., Andreevskaya S., Smirnova T., Chizhov A., Karpenko I., Kochetkov S., Alexandrova L. (2012) The synthesis and antituberculosis activity of 5'-nor carbocyclic uracil derivatives. Bioorg. Med. Chem. 20, 6680–6686.
Khandazhinskaya A.L., Alexandrova L.A., Matyugina E.S., Solyev P.N., Efremenkova O.V., Buckheit K.W., Wilkinson M., Buckheit Jr. R.W., Chernousova L.N., Smirnova T.G., Andreevskaya S.N., Kochetkov S.N., Seley-Radtke K.L. (2018) Novel 5'-norcarbocyclic pyrimidine derivatives as antibacterial agents. Molecules. 23, pii: E3069.
Khandazhinskaya A.L, Matyugina E.S., Alexandrova L.A., Kezin V.A., Chernousova L.N., Smirnova T.G., Andreevskaya S.N., Popenko V.I., Leonova O.G., Kochetkov S.N. (2020) Interaction of 5-substituted pyrimidine nucleoside analogues and M. tuberculosis: a view through an electron microscope. Biochimie. 171–172, 170–177.
Kögler M., Vanderhoydonck B., De Jonghe S., Rozenski J., Van Belle K., Herman J., Louat T., Parchina A., Sibley C., Lescrinier E., Herdewijn P. (2011) Synthesis and evaluation of 5-substituted 2'-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in Mycobacterium tuberculosis. J. Med. Chem. 54, 4847–4862.
Kögler M., Busson R., De Jonghe S., Rozenski J., Van Belle K., Louat T., Munier-Lehmann H., Herdewijn P.S. (2012) Synthesis and evaluation of 6-aza-2'-deoxyuridine monophosphate analogs as inhibitors of thymidylate synthases, and as substrates or inhibitors of thymidine monophosphate kinase in Mycobacterium tuberculosis. Chem. Biodiv. 9, 536–556.
Parchina A., Froeyen M., Margamuljana L., Rozenski J., De Jonghe S., Briers Y., Lavigne R., Herdewijn P., Eveline Lescrinier E. (2013) Discovery of an acyclic nucleoside phosphonate that inhibits Mycobacterium tuberculosis ThyX based on the binding mode of a 5‑alkynyl substrate analogue. ChemMedChem. 8, 1373–1383.
Alexandrova L.A., Chekhov V.O., Shmalenyuk E.R., Kochetkov S.N., Abu El-Asrar R., Herdewijn P. (2015) Synthesis and evaluation of C-5 modified 2'-deoxyuridine monophosphates as inhibitors of M. tuberculosis thymidylate synthase. Bioorg. Med. Chem. 23, 7131–7137.
Rautio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Järvinen T., Savolainen J. (2008) Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 7, 255–270.
Huttunen K., Raunio H., Rautio J. (2011) Prodrugs – from serendipity to rational design. Pharmacol. Rev. 63, 750–771.
Stella V.J., Nti-Addae K.W. (2007) Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 59, 677–694.
Beaumont K., Webster R., Gardner I., Dack K. (2003) Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: challenges to the discovery scientist. Curr. Drug Metab. 4, 461–485.
Testa B. (2009) Prodrugs: bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol. 13, 338–344.
Jornada D.H., Dos Santos Fernandes G.F., Chiba D.E., De Melo T.R.F., Dos Santos J.L., Chung M.C. (2016) The prodrug approach: a successful tool for improving drug solubility. Molecules. 21. https://doi.org/10.3390/molecules21010042
Mori G., Chiarelli L.R., Riccardi G., Pasca M.R. (2017) New prodrugs against tuberculosis. Drug Discov. Today. 22, 519–525.
Pochet S., Kansall V., Destouesse F., Sarfatil S.R. (1990) Alkylglycoside carbonates of 3’-azido-3’-deoxythymidine. Tetrahedron Lett. 31, 6021–6024.
Negrya S.D., Jasko M.V., Solyev P.N., Karpenko I.L., Efremenkova O.V., Vasilyeva B.F., Sumarukova I.G., Kochetkov S.N., Alexandrova L.A. (2020) Synthesis of water-soluble prodrugs of 5-modified 2'-deoxyuridines and their antibacterial activity. J. Antibiotics. 73, 236–246.
Alexandrova L. Zicari S., Matyugina E., Khandazhinskaya A., Smirnova T., Andreevskaya S., Chernousova L., Vanpouille C., Kochetkov S., Margolis L. (2017) Dual-targeted anti-TB/anti-HIV heterodimers. Antiviral Res. 145, 175–183.
Александрова Л.А., Ефременкова О.В., Андронова В.Л., Галегов Г.А., Сольев П.Н., Карпенко И.Л., Кочетков С.Н. (2016) 5-(4-алкил-1,2,3-триазол-1-ил)метильные производные 2'-дезоксиуридина – ингибиторы роста бактерий и вирусов. Биоорган. химия. 42, 746–754.
Khandazhinskaya A.L., Shirokova E.A., Karpenko I.L., Zakirova N.F., Tarussova N.B., Krayevsky A.A. (2000) P-(alkyl)-nucleoside 5ʹ-hydrogenphosphonates as depot forms of antiviral nucleotide analogues. Nucleosides Nucleotides Nucl. Acids. 19, 1795–1804.
Sampathkumar P., Turley S., Ulmer J.E., Rhie H.G., Sibley C.H., Hol W.G. (2005) Structure of the Mycobacterium tuberculosis flavin dependent thymidylate synthase (MtbThyX) at 2.0A resolution. J. Mol. Biol. 352, 1091–1104.
Halgren T.A. (1999) MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 20, 720–729.
Chemical Computing Group Inc. Molecular Operating Environment (MOE) (2012). 10.
De Clercq E. (2001) Antiviral drugs: current state of the art. J. Clin. Vir. 22, 73–89.
Solyev P.N., Shipitsin A.V., Karpenko I.L., Nosik D.N., Kalnina L.B., Kochetkov S.N., Kukhanova M.K., Jasko M.V. (2012) Synthesis and anti-HIV properties of new carbamate prodrugs of AZT. Chem. Biol. Drug Des. 80, 947–952.
Khandazhinskaya A.L., Jasko M.V., Karpenko I.L., Solyev P.N., Golubeva N.A., Kukhanova M.K. (2011) 5′-Phosphonated derivatives of 2′,3′-dideoxy-3′-thiacytidine as new anti-HIV prodrugs. Chem. Biol. Drug Des. 78, 50–56.
McGuigan C., Pathirana R.N., Migliore M., Adak R., Luoni G., Jones A.T., Díez-Torrubia A., Camarasa M.J., Velázquez S., Henson G., Verbeken E., Sienaert R., Naesens L., Snoeck R., Andrei G., Balzarini J. (2007) Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus. J. Antimicrob. Chemother. 60, 1316–1330.
McGuigan C., Balzarini J. (2009) FV100 as a new approach for the possible treatment of varicella-zoster virus infection. J. Antimicrob. Chemother. 64, 671–673.
Snoeck R., Andrei G., De Clercq E. (1994) Chemotherapy of varicella zoster virus infections. Int. J. Antimicrob. Agents. 4, 211–226.
Solyev P.N., Jasko M.V., Karpenko I.L., Sharkin Y.A., Shipitsyn A.V., Kukhanova M.K. (2014) New dinucleoside phosphonate derivatives as prodrugs of 3′-azido-3′-deoxythymidine and β-L-2′,3′-dideoxy-3′-thiacytidine: synthesis and anti-HIV properties. Nucleosides Nucleotides Nucl. Acids. 33, 64–79.
Jain N., O’Brien S. (2015) Initial treatment of CLL: integrating biology and functional status. Blood. 126, 463–470.
Хандажинская А.Л., Широкова Е.А. (2013) 5'-Фосфонаты AZT: достижения и перспективы в лечении и профилактике ВИЧ-инфекции. Acta Naturae. 5, 57–65.
McGuigan C., Derudas M., Gonczy B., Hinsinger K., Kandil S., Pertusati F., Serpi M., Snoeck R., Andrei G., Balzarini J., McHugh T.D., Maitra A., Akorli E., Evangelopoulos D., Bhakta S. (2014) ProTides of N-(3-(5-(2'-deoxyuridine))prop-2-ynyl)octanamide as potential anti-tubercular and anti-viral agents. Bioorg. Med. Chem. 22, 2816–2824.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология