

Молекулярная биология, 2021, T. 55, № 2, стр. 181-193
Интерактом систем репарации оснований и нуклеотидов
Н. И. Речкунова a, *, Ю. С. Красикова a, О. И. Лаврик a, b
a Институт химической биологии и фундаментальной медицины Сибирского отделения
Российской академии наук
630090 Новосибирск, Россия
b Новосибирский государственный университет
630090 Новосибирск, Россия
* E-mail: nadyarec@niboch.nsc.ru
Поступила в редакцию 12.08.2020
После доработки 12.08.2020
Принята к публикации 24.08.2020
Аннотация
Системы эксцизионной репарации оснований и нуклеотидов (BER и NER) специфично удаляют повреждения ДНК определенного типа. BER удаляет модифицированные основания (окисленные, алкилированные, дезаминированные), а NER – объемные повреждения, вызванные УФ-облучением или химическими канцерогенами. Однако в ряде случаев процесс исправления проходит по более сложному сценарию, при котором пути репарации обмениваются белками и взаимодействуют друг с другом, формируя общий интерактом. В настоящем обзоре представлены основные сведения о механизмах BER и NER, рассмотрены данные, показывающие участие белков NER в восстановлении повреждений ДНК, вызванных окислительным стрессом, и белков BER в удалении объемных аддуктов ДНК. Обсуждается также роль поли(ADP-рибоза)полимеразы 1 в регуляции процессов BER и NER и их координации при исправлении сложных повреждений.
ВВЕДЕНИЕ
В геномной ДНК постоянно возникают повреждения, обусловленные как внутренней неустойчивостью, так и воздействием эндогенных факторов и экзогенных генотоксических агентов. Повреждения могут вызывать нарушения основных процессов метаболизма ДНК, таких как репликация и транскрипция, что приводит к возникновению мутаций, хромосомных аберраций, а также к остановке клеточного цикла и гибели клеток. Последствия повреждений ДНК могут быть причиной различных патологий человека, включая рак и нейродегенеративные заболевания. Для противостояния этим неблагоприятным последствиям в ходе эволюции возникло несколько механизмов репарации ДНК [1].
Эксцизионная репарация оснований (BER – base excision repair) – одна из основных систем репарации ДНК, действие которой направлено на исправление наиболее многочисленных повреждений ДНК – модифицированных оснований. Такие повреждения возникают в результате спонтанных реакций (дезаминирование, метилирование, окисление) или под действием продуктов метаболизма клетки (активные формы кислорода), а также таких экзогенных источников, как алкилирующие агенты (метилметансульфонат) и ионизирующее излучение [2].
Эксцизионная репарация нуклеотидов (NER – nucleotide excision repair) наиболее универсальный путь репарации, с помощью которого происходит удаление широкого спектра повреждений, дестабилизирующих дуплексную структуру ДНК. Субстратами NER служат индуцированные ультрафиолетовым (УФ) излучением фотопродукты – циклобутанпиримидиновые димеры (CPD) и пиримидин-пиримидон-(6-4)-фотопродукты (6-4PP), а также внутрицепочечные сшивки и объемные аддукты оснований ДНК с реакционноспособными метаболитами некоторых химических канцерогенов или химиотерапевтическими агентами [3].
В ходе многолетних исследований процессов BER и NER накопилось достаточно данных, показывающих, что эти системы репарации работают не как изолированные пути, а динамически взаимодействуют друг с другом, формируя общий интерактом, что позволяет осуществлять модуляцию и координацию функций этих процессов [4, 5]. С одной стороны, показано влияние белков NER на активность ключевых ферментов BER, ДНК-гликозилаз и апуриновой/апиримидиновой эндонуклеазы 1 (АРЕ1) [6–9], а также взаимодействие белков NER с окислительными повреждениями ДНК in vivo [10, 11]. С другой стороны, ключевой фермент BER – APE1, регулирует удаление аддуктов ДНК с производными платины в процессе NER [12, 13].
Некоторые генотоксические факторы вызывают образование в ДНК повреждений нескольких типов. Так, УФ-излучение приводит к образованию не только пиримидиновых димеров, но и окисленных оснований, а также к разрывам цепи ДНК. В результате интенсивного воздействия одного или нескольких факторов в ДНК могут возникать кластерные повреждения, включающие модифицированные основания, разрывы цепи и объемные аддукты, расположенные в пределах одного–двух витков спирали в одной или обеих цепях дуплекса. При репарации таких повреждений особенно необходимы согласованные действия разных систем репарации, их взаимодействие и взаимная регуляция активности, чтобы предотвратить образование наиболее токсичных для клетки двухцепочечных разрывов ДНК. Взаимное влияние белков-участников разных систем может осуществляться путем белок-белковых взаимодействий, в том числе опосредованных посттрансляционными модификациями белков одной или обеих систем репарации [4]. В настоящем обзоре представлены основные сведения о механизмах BER и NER, рассмотрены известные к настоящему времени данные, показывающие участие белков NER в восстановлении повреждений ДНК, вызванных окислительным стрессом, и белков BER в удалении объемных аддуктов ДНК. Обсуждается также роль поли(ADP-рибоза)полимеразы 1 (PARP1) в регуляции процессов BER и NER и их координации при исправлении сложных повреждений ДНК.
ЭКСЦИЗИОННАЯ РЕПАРАЦИЯ ОСНОВАНИЙ
Процесс BER инициируется ДНК-гликозилазами, узнающими поврежденное основание и катализирущими гидролиз N-гликозидной связи. ДНК-гликозилазы специфичны к определенным повреждениям оснований, в зависимости от выполняемых функций они делятся на две группы – моно- и бифункциональные. В результате действия монофункциональных ДНК-гликозилаз, из которых наиболее известна урацил-ДНК-гликозилаза, удаляющая из ДНК урацил, образовавшийся в результате спонтанного дезаминирования цитозина, в ДНК образуются апуриновые/апиримидиновые (AP) сайты. Бифункциональные ДНК-гликозилазы, действие которых направлено на окисленные основания, не только удаляют поврежденные основания, но и способны вносить разрыв с 3'-стороны от образовавшегося AP-сайта. В результате реакции в ДНК возникают разрывы, фланкированные 3'-α,β-4-гидроксипентен-2-алем (3'-PUA) и 5′-концевым фосфатом (β-элиминирование) или с 3′- и 5′-фосфатными группами (β,δ-элиминирование) [14]. Эффективность расщепления сахарофосфатного остова бифункциональными ДНК-гликозилазами значительно отличается, так, например, OGG1, удаляющая наиболее распространенное окисленное основание – 8-оксогуанин (8-oxoG), функционирует преимущественно как монофункциональный фермент [15].
AP-сайты могут возникать также за счет спонтанного гидролиза N-гликозидной связи в нуклеотидах, в основном пуриновых [16]. Основной фермент, гидролизующий АР-сайты в клетках высших эукариот, – апуриновая/апиримидиновая эндонуклеаза 1 (АРЕ1) [17]. В результате гидролиза АР-сайта образуется одноцепочечный разрыв, фланкированный гидроксильной группой на 3'-конце и остатком 2'-дезоксирибозо-5'-фосфата (dRP) на 5'‑конце. АРЕ1 необходима не только для гидролиза АР-сайтов, но и для удаления остатка 3′-PUA в разрыве, возникающем под действием бифункциональных ДНК-гликозилаз, катализирующих β-элиминирование, а также при спонтанном расщеплении АР-сайтов, при котором также образуется разрыв, содержащий 3'-PUA-группу [18].
При расщеплении АР-сайта бифункциональными ДНК-гликозилазами, функционирующими по механизму β,δ-элиминирования, продукт расщепления содержит 3'-фосфатную группу, которую также необходимо удалять с помощью фосфатазы. Основным ферментом, осуществляющим эту реакцию в клетках млекопитающих, является бифункциональная полинуклеотидкиназа-фосфатаза (PNKP), которая, кроме фосфорилирования 5'-концевой гидроксильной группы цепи ДНК, обладает способностью выщеплять 3'-концевую фосфатную группу, генерируя 3'-гидроксильную группу [19]. Недавно мы показали, что расщепление АР-сайта способен катализировать еще один фермент – тирозил-ДНК-фосфодиэстераза 1 (TDP1) [20], в результате действия которого также образуется разрыв с 3'-фосфатной группой [21], удаление которой осуществляет PNKP. Таким образом, последовательное действие бифункциональных ДНК-гликозилаз, расщепляющих АР-сайт по механизму β,δ-элиминирования, либо TDP1 и PNKP приводит концы цепи ДНК на месте расщепленного АР-сайта в состояние, пригодное для включения нуклеотидных звеньев ДНК-полимеразами и последующего лигирования разрыва. Такую последовательность событий можно рассматривать как АРЕ1-независимый путь BER [19, 21].
Следующая стадия процесса BER – включение нуклеотида в 3'-конец цепи ДНК. На этой стадии происходит разделение процесса BER на пути, называемые “короткозаплаточной” и “длиннозаплаточной” BER, которые осуществляются с участием разных ферментов. В короткозаплаточной BER ДНК-полимераза β (Polβ) включает один dNMP на место поврежденного звена. Остаток dRP удаляется из ДНК под действием 2'-дезоксирибозо-5'-фосфатлиаз (dRP-лиаз), которые выщепляют dRP по механизму β-элиминирования. В клетках высших эукариот эта функция осуществляется преимущественно за счет лиазной активности Polβ, от эффективности удаления 5′-dRP зависит выбор пути BER [22, 23]. Затем происходит лигирование, катализируемое в клетках высших эукариот ДНК-лигазой III в комплексе с белком XRCC1 [24].
Если по какой-то причине (например, модификации остатка сахара) Polβ не может удалить dRP-остаток, то происходит переключение BER на длиннозаплаточный путь, в котором после включения первого dNMP продолжается репаративный синтез с заменой 2–20 нуклеотидных звеньев и вытеснением цепи ДНК. В клетках высших эукариот этот синтез осуществляется “репликативными” ДНК-полимеразами δ и/или ε с привлечением вспомогательных факторов (PCNA, RFC). Вытесненный участок ДНК удаляется флэп-эндонуклеазой (FEN1), и одноцепочечный разрыв лигируется ДНК-лигазой I [25]. Потенциально репаративный синтез с вытеснением цепи может осуществляться по механизму трансляции бреши с помощью FEN1 и Polβ [26]. Кроме того, в нашей лаборатории показано, что Polβ в сочетании с АРE1 способна осуществлять синтез с вытеснением цепи с одновременной “коррекцией ошибок” с помощью 3' → 5'-экзонуклеазной активности АРЕ1 [27]. Дальнейшие исследования показали, что Polβ физически взаимодействует с АРE1 [28, 29].
Очевидно, что для эффективного восстановления поврежденной ДНК и предотвращения накопления токсичных для клетки разрывов ДНК требуется координация действий отдельных ферментов, катализирующих последовательные стадии процесса BER. Перенос ДНК-интермедиата от фермента, связанного с продуктом катализируемой им реакции, к следующему ферменту, аналогично передаче эстафетной палочки (passing the baton), рассматривается как основной механизм координации BER [30–32]. В то же время количественный анализ взаимодействий белков BER, выполненный в нашей лаборатории [29], показал, что многие белки образуют прочные комплексы друг с другом как в отсутствие, так и в присутствии ДНК-интермедиатов BER, и эти взаимодействия могут вносить вклад в координацию процесса BER (см. обзор [33]). Наиболее прочный комплекс образуют Polβ и XRCC1 [29], что согласуется с данными о ключевой роли взаимодействия этих белков в процессе BER [32, 34]. Интересно отметить, что связывание с XRCC1 защищает Polβ от убиквитинирования и последующей протеасомной деградации [35].
ЭКСЦИЗИОННАЯ РЕПАРАЦИЯ НУКЛЕОТИДОВ
Процесс NER был открыт в клетках бактерий еще в начале 60-х годов прошлого столетия как способность к вырезанию поврежденных УФ-светом участков ДНК с последующим ресинтезом ДНК в образовавшихся брешах [36]. В клетках эукариот существуют два пути NER: общегеномная репарация (global genome repair, GG-NER), удаляющая повреждения во всей геномной ДНК, и репарация, связанная с транскрипцией (transcription coupled repair, TC-NER), с помощью которой происходит удаление повреждений в транскрибируемой цепи ДНК. Эти пути отличаются способом узнавания повреждения и набором белков, участвующих на начальных этапах процесса. В TC-NER узнавание повреждения сопряжено с остановкой на нем РНК-полимеразы II (RNAP II) [37], что служит сигналом к сборке инициирующего репарацию комплекса, в состав которого входят белки CSA и CSB [38]. Мутации в генах этих белков вызывают синдром Кокейна (Cockayne syndrome, CS).
В случае GG-NER повреждение узнает фактор пигментной ксеродермы С (ХРС) в комплексе с белками RAD23B и центрин-2 (Cen2). Комплекс ХРС-RAD23B-Cen2 (далее комплекс XPC) определяет нарушение комплементарности в парах оснований и/или дестабилизацию ДНК-дуплекса, вызванную повреждением, а не конкретное повреждение [39, 40], и взаимодействует с неповрежденным участком противоположной цепи [41–44]. Этот механизм обеспечивает широкую субстратную специфичность процесса GG-NER, однако не позволяет узнавать УФ-индуцированные фотопродукты, особенно CPD, которые практически не дестабилизируют ДНК-дуплекс. Узнавание таких повреждений осуществляет специализированный белок – гетеродимер UV-DDB (UV-damaged DNA-binding protein), который специфически взаимодействует с CPD и 6-4PP и привлекает к ним XPC [45, 46]. Далее комплекс XPC (либо комплекс факторов TC-NER) взаимодействует с мультифункциональным 10-субъединичным белком – фактором транскрипции IIH (transcription factor IIH, TFIIH), который осуществляет частичное раскручивание ДНК-дуплекса за счет геликазной активности субъединицы XPD и одновременно проверку повреждения ДНК, останавливаясь на нем [47, 48]. На частично открытом дуплексе формируется так называемый “предрасщепляющий” комплекс, в состав которого входят репликативный белок А (RPA), фактор пигментной ксеродермы А (XPA), а также структуроспецифичные эндонуклеазы XPF-ERCC1 и XPG, расщепляющие поврежденную цепь ДНК с 5′- и 3′-стороны от повреждения соответственно [48–50]. Ресинтез удаленного участка осуществляет репликативный комплекс, лигирование ника – ДНК-лигаза I или комплекс ДНК-лигазы III с XRCC1 [51].
Таким образом, исправление повреждений системой NER – сложный многостадийный процесс, протекающий с образованием множества промежуточных комплексов, сборка и функционирование которых осуществляются за счет ДНК-белковых и белок-белковых взаимодействий, требующих четкой координации и регуляции. Как оказалось, помимо факторов NER в этот процесс вовлечены белки других процессов репарации, в том числе BER, а также регуляторные белки, в частности PARP1.
ВЗАИМОДЕЙСТВИЕ ПУТЕЙ BER И NER УВЕЛИЧИВАЕТ ЭФФЕКТИВНОСТЬ РЕПАРАЦИИ ДНК
Предположение о возможном участии белков NER в восстановлении окислительных повреждений ДНК впервые было высказано в работе, выполненной в лаборатории будущего нобелевского лауреата Aziz Sancar [52]. Оказалось, что в экстрактах линий клеток человека, дефектных по белкам NER (XP-A, XP-B, XP-C, XP-D, XP-F и XP-G), заметно снижена способность к репарации окисленных оснований, 8-oxoG и тимингликоля [52]. В реконструированной из очищенных белков (XPA, RPA, TFIIH (в состав которого входят XPB и XPD), XPC-RAD23B, XPG и ERCC1-XPF) системе NER наблюдалось выщепление фрагментов поврежденной ДНК, характерных для этой системы репарации, поэтому предположили, что NER может участвовать в исправлении повреждений, репарируемых по механизму BER, в качестве резервной системы, либо белки NER вовлечены в процесс BER.
Прямые доказательства участия факторов NER в исправлении окисленных оснований получены группой исследователей под руководством Eugenia Dogliotti. В экспериментах на клетках человека они обнаружили, что линии клеток с мутациями в гене, кодирующем XPC, чрезвычайно чувствительны к окислительным агентам, что свидетельствует о важной роли XPC в защите от окислительного стресса [53]. Кроме того, с использованием очищенных белков показана стимуляция активности ДНК-гликозилазы OGG1 белком XPC-RAD23B, а также непосредственное взаимодействие этих белков. В отличие от XPC, XPA в исследованных концентрациях не стимулировал OGG1. Таким образом показано, что XPC-RAD23B стимулирует OGG1-зависимый процесс BER. Роль XPC в процессинге 8-oxoG подтверждена в [54]. Следует заметить, что несколько ранее способность XPC-RAD23B стимулировать активность тимин-ДНК-гликозилазы, субстратом которой является дезаминированный 5-метилцитозин, была показана группой японских авторов [6]. В более позднем исследовании этой группы установлено, что XPC стимулирует не только активность TDG, но и гликозилазу SMUG1, удаляющую урацил в паре G/U, а также в оцДНК. Кроме того, стимулирующий эффект XPC обеспечивается за счет белок-белковых взаимодействий и сохраняется при сумоилировании гликозилаз [55].
Дальнейшие исследования роли факторов NER в репарации окисленных оснований были проведены в лаборатории Dogliotti с использованием эмбриональных фибробластов мыши c дефицитом белков NER (Csbm/m, Csa–/–, Xpa–/–, Xpc–/– и их комбинации) и/или OGG1 (Ogg1–/–) [56]. Анализ содержания 8-oxoG в клетках, обработанных окислительным агентом (бромат калия), выявил снижение скорости удаления 8-oxoG из ДНК в клетках с дефицитом белков NER по сравнению с клетками дикого типа, хотя и в меньшей степени, чем в клетках Ogg1–/–. Кроме того, в клетках с двойными мутациями Csb–/– Xpa–/– и Csb–/– Xpc–/– удаление 8-oxoG происходило медленнее, чем с одиночными мутациями, а снижение скорости было сопоставимо с клетками Ogg1–/–. В то же время эффективность репарации 8-oxoG в клетках с двойными мутациями Xpa–/– Xpc–/– практически не отличалась от эффективности в клетках с одиночными мутациями. На основании полученных результатов предположили, что повышенный уровень окислительных повреждений ДНК в клетках с комбинацией мутаций в CSB и XPA/XPC свидетельствует об участии этих белков в разных путях репарации. Данные, полученные на клетках мыши, подтверждены на первичных фибробластах человека XP-A, которые оказались более чувствительными к бромату калия по сравнению с нормальными фибробластами. Проблемы с исправлением окислительных повреждений в клетках с дефицитом факторов NER XPA и ХРС [57], а также в клетках XP-G/CS [58] обнаружены и другими исследователями. Участие XPA и CSB в удалении 8-oxoG связано преимущественно с TC-NER [59].
Привлечение факторов NER к местам окислительных повреждений показано также в живых клетках, экспрессирующих химерные флуоресцентные белки XPC-GFP и CSB-GFP [60]. После локального лазерного облучения клеточного ядра наблюдалось накопление флуоресцентных белков в определенных местах: CSB рекрутировался в ядрышко, а XPC накапливался в области гетерохроматина, что соответствует роли этих белков в TC-NER и GG-NER соответственно. Участие CSB в репарации 8-oxoG получило дальнейшее подтверждение в более позднем исследовании, проведенном той же группой [61]. Используя, как и в предыдущей работе, метод визуализации флуоресцентных белков в живых клетках, показано, что рекрутирование OGG1 к повреждению не зависит от CSB, тогда как привлечение XRCC1, белка-платформы BER, стимулируется CSB транскрипционно-зависимым способом. Предполагается, что в качестве белка, ремоделирующего хроматин, CSB помогает загрузке XRCC1 в труднодоступные участки транскрибируемых генов, тем самым обеспечивая доступ для последующих белков BER.
Недавно показали, что UV-DDB, один из ключевых факторов GG-NER, функционирующий как сенсор УФ-индуцированных повреждений и ремоделирующий хроматин фактор, увеличивает эффективность OGG1 и APE1, а также значительно стимулирует активность Polβ в заполнении бреши [62]. Визуализация на уровне одной молекулы в режиме реального времени показала, что UV-DDB образует временные комплексы с OGG1 или APE1, облегчая их диссоциацию из комплекса с ДНК. Кроме того, UV-DDB перемещается в клетке к местам репарации 8-oxoG, а истощение UV-DDB повышает чувствительность клеток к окислительному повреждению ДНК. Предполагается, что UV-DDB служит основным сенсором повреждений ДНК не только в NER, но и в BER, способствуя распознаванию повреждений в контексте хроматина.
Поврежденное основание 8-oxoG может подвергаться дальнейшему окислению, что приводит к образованию спироиминодигидантоина (Sp) и 5-гуанидиногидантоина (Gh), которые узнаются ДНК-гликозилазой NEIL1 [63–67], а также служат субстратами системы NER [68]. Недавно группа исследователей из университета Нью-Йорка, возглавляемая N.E. Geacintov, проанализировала репарацию ДНК, содержащих эти повреждения, в клетках человека (интактных и дефектных по BER (NEIL1−/−) и NER (XPA−/−)) и оценили относительный вклад BER и NER в их удаление [69]. Клетки трансфицировали ДНК, содержащими радиоактивную метку вблизи Gh или Sp, инкубировали в течение определенных промежутков времени, затем лизировали, выделяли низкомолекулярную ДНК и оценивали присутствие продуктов процессинга повреждений по пути BER и NER. Показано, что оба повреждения удаляются как по пути BER, так и с помощью NER, при этом количество образующихся продуктов прямо коррелировало с активностью системы. В клетках NEIL1−/− репарация проходила преимущественно по механизму NER, тогда как дефект в системе NER приводил к повышению количества продуктов BER. Полученные данные свидетельствуют о конкуренции между двумя путями репарации, которые узнают и связывают одни и те же повреждения, при этом вклад каждого из путей зависит от концентрации белков-участников процесса. Влияние факторов NER на различные стадии процесса BER схематично представлено на рис. 1.
Рис. 1.
Схема короткозаплаточного пути BER, инициируемого моно- и бифункциональными ДНК-гликозилазами. Белки NER, влияющие на активность ферментов BER, и катализируемые ими стадии процесса, приведены в рамках со стрелками.
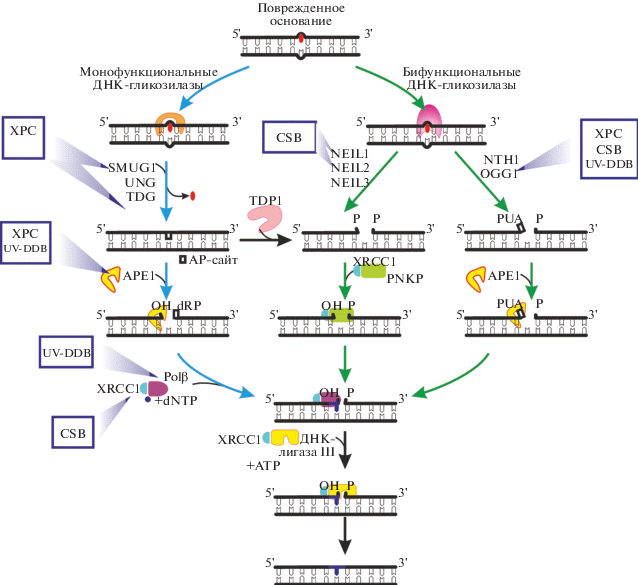
Система NER вносит основной вклад в репарацию аддуктов ДНК с платиной, возникающих под действием препаратов, используемых в химиотерапии (цисплатин, оксалиплатин и карбоплатин). В результате ковалентного связывания Pt с двумя нуклеотидными остатками в одной или в противоположных цепях ДНК-дуплекса образуются внутри- или межцепочечные сшивки (ICL) соответственно. Хотя эти повреждения удаляются главным образом в процессе NER, обнаружено, что удаление 1,2-(GpG)-аддуктов из ДНК зависит от уровня экспрессии ключевого фермента BER – APE1 [12, 13]. Подавление экспрессии APE1 с помощью РНК-интерференции приводило к ингибированию репарации аддуктов ДНК-Pt, а инфицирование клеток лентивирусом, несущим ген APE1, восстанавливало эффективность репарации [13]. Кроме того, изменение экспрессии APE1 влияло на уровни экспрессии белков NER, RPA и XPA, что свидетельствует о взаимодействии между двумя путями репарации.
Дальнейшее изучение репарации ICL, индуцированных воздействием цисплатина и оксиплатина, с применением технологии CRISPR/Cas9 подтвердило, что в исправление этих повреждений вовлечены белки не только NER, но и BER [70]. Аналогичные данные получены при изучении репарации повреждений, индуцированных производными псоралена, которые при облучении УФ-светом образуют моноаддукты как с ДНК, так и с ICL. Показано, что в репарацию таких повреждений вовлечена гликозилаза NEIL1, хотя данные, касающиеся вклада этого фермента в удаление каждого из повреждений, противоречивы. Сначала получили результаты, свидетельствующие об участии NEIL1 в репарации моноаддуктов [71], но не ICL, а позднее обнаружили способность NEIL1 удалять оба вида аддуктов [72, 73], тогда как другими исследователями показано, что в обработанных триоксаленом клетках NEIL1 привлекается только к ICL и не обнаруживается в местах образования моноаддуктов [74]. Таким образом, полученные результаты, несмотря на некоторую их противоречивость, показывают, что системы NER и BER взаимодействуют и дополняют друг друга, что обеспечивает более эффективную защиту ДНК при генотоксическом стрессе.
Значительный прогресс в понимании физических и функциональных взаимодействий путей NER и BER может обеспечить применение биоинформатических методов. База данных STRING содержит информацию о белок-белковых взаимодействиях, объединяющую известные данные о межбелковых ассоциациях у большого числа организмов [75]. В опубликованном недавно обзоре [76] представлены результаты анализа на основе STRING взаимодействий 32 белков, относящихся к NER, и 23 белков, ассоциированных с BER. Построенная сеть взаимодействий (интерактом) белков NER и BER выявляет значительную связь между этими путями репарации. Дальнейшие исследования могут выявить новые функции, общие для этих путей репарации, и их взаимодействия в поддержании стабильности генома.
ПОЛИ(ADP-РИБОЗА)ПОЛИМЕРАЗА 1 – РЕГУЛЯТОР ПРОЦЕССОВ BER И NER
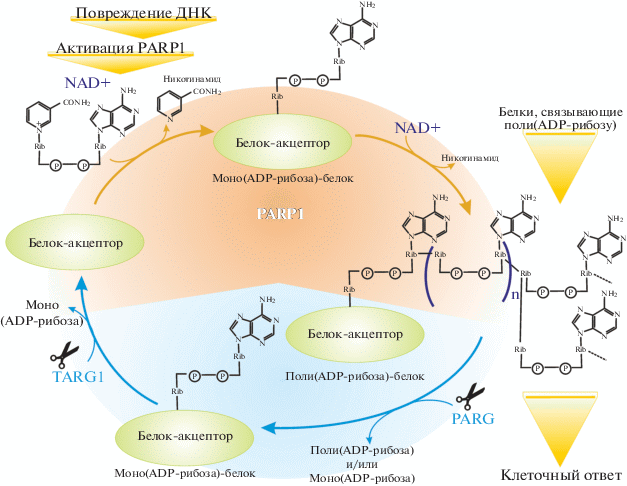
В ответ на повреждение ДНК в клетках высших эукариот происходит модификация ядерных белков с помощью полимера ADP-рибозы (PAR). Поли(ADP-рибозил)ирование (PAR-илирование) белков катализируется несколькими PARP, которые постоянно и в значительном количестве присутствуют в клетке. Белки PARP образуют суперсемейство, включающее 17 представителей, объединенных по характерному признаку – наличию консервативного домена, содержащего мотив “PARP signature” – высококонсервативную последовательность, которая участвует в формировании активного центра [77]. В качестве источника ADP-рибозы PARP использует NAD+ и переносит эту группу на белок-акцептор.
Из 17 белков семейства PARP в клеточном ответе на повреждение ДНК участвуют три – PARP1, PARP2 и PARP3 [78, 79]. Их мишенями преимущественно служат белки, участвующие в укладке хроматина и метаболизме ДНК, включая гистоны, белки репарации ДНК и транскрипционные факторы, а также сами PARP. Из трех ферментов PARP, активируемых поврежденной ДНК, наиболее хорошо изучен PARP1, на долю которого приходится до 90% синтезируемой в клетке PAR [79]. Реакция PAR-илирования обратима: PAR подвергается расщеплению с помощью фермента поли(ADP-рибоза)гликогидролазы, что обеспечивает дополнительную регуляцию уровня PAR-илирования белков и синтеза PAR в клетке. Остаток ADP-рибозы, присоединенный к акцепторному аминокислотному остатку белка, удаляется с помощью специальных ферментов, в частности терминальной (ADP-рибоза)гликогидролазы 1 [80], в результате чего PARP1 и другие белки-акцепторы могут участвовать в следующем раунде PAR-илирования (рис. 2).
Рис. 2.
Регуляция синтеза поли(ADP-рибозы). В ответ на повреждение ДНК PARP1 катализирует поли(ADP-рибозил)ирование белков (включая автомодификацию), используя в качестве субстрата NAD+. PAR-связывающие белки участвуют в клеточном ответе на повреждение ДНК. Поли(ADP-рибоза)гликогидролаза (PARG) расщепляет PAR, регулируя уровень PAR-илирования белков и синтеза PAR в клетке. Остаток ADP-рибозы, присоединенный к белку, удаляется с помощью терминальной (ADP-рибоза)гликогидролазы 1 (TARG1).
PARP1 считается одним из ключевых регуляторов репарации ДНК и других клеточных процессов [81–84]. Участие PARP1 в процессе BER изучено достаточно подробно (полученные результаты систематизированы в обзорах [85, 86]). Показано, что PARP1 взаимодействует с белками BER и модулирует их активность [27, 87], ряд белков BER подвергается PAR-илированию [88, 89]. В последнее время роль PARP1 и катализируемого им PAR-илирования в процессе NER активно изучается. Известные к настоящему времени данные обсуждены нами в обзорной статье [90]. Некоторые белки репарации способны связывать как свободный PAR, так и присоединенный к PARP1 и/или другим белкам-акцепторам (рис. 2), что рассматривается как один из механизмов вовлечения этих белков в соответствующие процессы и их регуляцию [78]. Взаимодействие осуществляется через PAR-связывающие домены, либо в нем участвуют РНК- или ДНК-связывающие домены белков. В этом случае возможна конкуренция между нуклеиновыми кислотами (PAR – полимер нуклеотидной природы, “третья нуклеиновая кислота”) за связывание с белком.
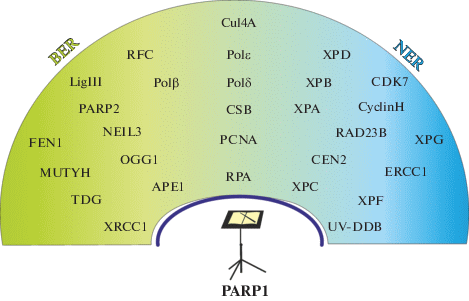
Развитие протеомных методов привело к обнаружению большого массива белков, подвергающихся PAR-илированию в ответ на генотоксический стресс и/или взаимодействующих с PAR, среди которых белки BER и NER [91, 92]. Известные к настоящему времени взаимодействия PARP1 с белками этих процессов репарации ДНК представлены на рис. 3.
И PARP1, и катализируемый этим ферментом синтез PAR могут участвовать в пространственно-временной регуляции процессов репарации при возникновении в ДНК множественных повреждений различной природы. Недавно опубликовали результаты масштабного исследования кинетики рекрутирования и диссоциации 70 белков репарации на индуцированные лазером сайты повреждения ДНК в клетках HeLa [93]. Эти белки в зависимости от времени их появления на поврежденном участке сгруппировали в семь групп. Белки-сенсоры повреждений NER (XPC, DDB2) и BER (NEIL3), а также PARP1, некоторые PAR-связывающие белки, в том числе PARG и XRCC1, факторы деконденсации хроматина вошли в первые две группы (полупериод рекрутирования 1.8–3.7 и 4.2–9.4 с соответственно). Анализ кинетики диссоциации выявил четыре группы белков, причем в первую группу (полупериод диссоциации с ДНК 24.7–110.5 с) вошли PARP1, PAR-связывающие и PAR-гидролизующие белки, что согласуется с предполагаемой ролью диссоциации PARP1 и гидролиза PAR в высвобождении белков репарации из комплексов с ДНК после исправления повреждения [94]. Изучено также изменение динамики ассоциации/диссоциации белков репарации при ингибировании PARP. Полученные данные свидетельствуют, что присутствие ингибитора PARP приводит к значительной перестройке порядка рекрутирования белков, что может изменить механизм и результат процесса репарации ДНК. Таким образом, исследование динамики белков репарации при множественных повреждениях ДНК выявляет многогранность взаимодействий и координацию путей репарации и может быть использовано для изучения нестабильности генома и влияния на эти процессы противоопухолевых препаратов – ингибиторов PARP.
В заключение хотелось бы подчеркнуть, что исследование интерактома репарации, взаимного влияния белков разных систем, выявление перекрывающихся или дополняющих функций этих белков имеет не только фундаментальное значение для понимания механизмов поддержания стабильности генома, но и актуальны для развития новых подходов к терапии онкологических заболеваний. Такие исследования необходимы в связи с известным фактом существования и формирования дополнительных путей репарации повреждений ДНК в раковых клетках, обеспечивающих лекарственную устойчивость. Особый интерес представляют взаимодействия белков BER и NER при исправлении множественных (кластерных) повреждений ДНК, возникающих при интенсивном генотоксическом стрессе, а также при радиотерапии. Регуляция активности процессов репарации при исправлении таких повреждений необходима для предотвращения образования двухцепочечных разрывов ДНК, возникновения мутаций и генетической нестабильности. С другой стороны, понимание механизмов регуляции процессов репарации ДНК может быть полезным для направленной дисрегуляции этих процессов в раковых клетках с целью усиления действия ДНК-повреждающих агентов.
Работа выполнена при поддержке Российского научного фонда (грант № 19-14-00107) и Программы фундаментальных научных исследований государственных академий наук на 2013–2020 гг. (№ АААА-А17-117020210022-4).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Friedberg E.C. (2003) DNA damage and repair. Nature. 421, 436–440. https://doi.org/10.1038/nature01408
Krokan H.E., Bjørås M. (2013) Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583. https://doi.org/10.1101/cshperspect.a012583
Gillet L.C., Scharer O.D. (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 106, 253–276. https://doi.org/10.1021/cr040483f.1
Limpose K.L., Corbett A.H., Doetsch P.W. (2017) BERing the burden of damage: pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair. 56, 51–64. https://doi.org/10.1016/j.dnarep.2017.06.007
Kumar N., Moreno N.C., Feltes B.C., Menck C.F., Van Houten B. (2020) Cooperation and interplay between base and nucleotide excision repair pathways: from DNA lesions to proteins. Genet. Mol. Biol. 43(1 suppl. 1), e20190104. https://doi.org/10.1590/1678-4685-GMB-2019-0104
Shimizu Y., Iwai S., Hanaoka F., Sugasawa K. (2003) Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 22, 164–173. https://doi.org/10.1093/emboj/cdg016
Muftuoglu M., de Souza-Pinto N.C., Dogan A., Aamann M., Stevnsner T., Rybanska I., Kirkali G., Dizdaroglu M., Bohr V.A. (2009) Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 284, 9270–9279. https://doi.org/10.1074/jbc.M807006200
Aamann M.D., Hvitby C., Popuri V., Muftuoglu M., Lemminger L., Skeby C.K., Keijzers G., Ahn B., Bjø-rås M., Bohr V.A., Stevnsner T. (2014) Cockayne syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 135, 1–14. https://doi.org/10.1016/j.mad.2013.12.008
Старостенко Л.В., Мальцева Е.А., Лебедева Н.А., Пестряков П.Е., Лаврик О.И., Речкунова Н.И. (2016) Взаимодействие фактора эксцизионной репарации нуклеотидов XPC–RAD23B с ДНК, содержащей кластерное повреждение – производное бенз[а]пирена и апуриновый/апиримидиновый сайт. Биохимия. 81, 350–360.
Menoni H., Hoeijmakers J.H., Vermeulen W. (2012) Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J. Cell. Biol. 199, 1037–1046. https://doi.org/10.1083/jcb.201205149
Menoni H., Wienholz F., Theil A.F., Janssens R.C., Lans H., Campalans A., Radicella J.P., Marteijn J.A., Vermeulen W. (2018) The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucl. Acids Res. 46, 7747–7756. https://doi.org/10.1093/nar/gky579
Kelley M.R., Jiang Y., Guo C., Reed A., Meng H., Vasko M.R. (2014) Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One. 9, e106485. https://doi.org/10.1371/journal.pone.0106485
Kim H.-S. Guo C., Thompson E.L., Jiang Y., Kelley M.R., Vasko M.R., Lee S.-H. (2015) APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat. Res. 779, 96–104. https://doi.org/10.1016/j.mrfmmm.2015.06.010
Fromme J.C., Verdine G.L. (2004) Base excision repair. Adv. Protein Chem. 69, 1–41. https://doi.org/10.1016/S0065-3233(04)69001-2
Boiteux S., Coste F., Castaing B. (2017) Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic. Biol. Med. 107, 179–201. https://doi.org/10.1016/j.freeradbiomed.2016.11.042
Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature. 362, 709–715. https://doi.org/10.1038/362709a0
Wilson 3rd D.M., Barsky D. (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 485, 283–307. https://doi.org/10.1016/s0921-8777(01)00063-5
Lhomme J., Constant J.F., Demeunynck M. (1999) Abasic DNA structure, reactivity, and recognition. Biopolymers. 52, 65–83. https://doi.org/10.1002/1097-0282
Wiederhold L., Leppard J.B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A.E., Izumi T., Prasad R., Wilson S.H., Mitra S., Hazra T.K. (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell. 15, 209–220. https://doi.org/10.1016/j.molcel.2004.06.003
Lebedeva N.A., Rechkunova N.I, Lavrik O.I. (2011) AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett. 585, 683–686. https://doi.org/10.1016/j.febslet.2011.01.032
Lebedeva N.A., Rechkunova N.I., Ishchenko A.A., Saparbaev M., Lavrik O.I. (2013) The mechanism of human tyrosyl-DNA phosphodiesterase 1 in the cleavage of AP site and its synthetic analogs. DNA Repair (Amst.). 12, 1037–1042. https://doi.org/10.1016/j.dnarep.2013.09.008
Srivastava D.K., Berg B.J., Prasad R., Molina J.T., Beard W.A., Tomkinson A.E., Wilson S.H. (1998) Mammalian abasic site base excision repair. identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 273, 21203–21209. https://doi.org/10.1074/jbc.273.33.21203
Horton J.K., Prasad R., Hou E., Wilson S.H. (2000) Protection against methylation-induced cytotoxicity by DNA polymerase beta-dependent long patch base excision repair. J. Biol. Chem. 275, 2211–2218. https://doi.org/10.1074/jbc.275.3.2211
Hoeijmakers J.H. (2001) Genome maintenance mechanisms for preventing cancer. Nature. 411, 366–374. https://doi.org/10.1038/35077232
Fortini P., Dogliotti E. (2007) Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair (Amst.). 6, 398–409. https://doi.org/10.1016/j.dnarep.2006.10.008
Liu Y., Beard W.A., Shock D.D., Prasad R., Hou E.W., Wilson S.H. (2005) DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J. Biol. Chem. 280, 3665–3674. https://doi.org/10.1074/jbc.M412922200
Sukhanova M.V., Khodyreva S.N., Lebedeva N.A., Prasad R., Wilson S.H., Lavrik O.I. (2005) Human base excision repair enzymes apurinic/apyrimidinic endonuclease 1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucl. Acids Res. 33, 1222–1229. https://doi.org/10.1093/nar/gki266
Prasad R., Williams J.G., Hou E.W., Wilson S.H. (2012) Polβ associated complex and base excision repair factors in mouse fibroblasts. Nucl. Acids Res. 40, 11571–11582.https://doi.org/10.1093/nar/gks898
Moor N.A., Vasil’eva I.A., Anarbaev R.O., Antson A.A., Lavrik O.I. (2015) Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucl. Acids Res. 43, 6009–6022. https://doi.org/10.1093/nar/gkv569
Prasad R., Shock D.D., Beard W.A., Wilson S.H. (2010) Substrate channeling in mammalian base excision repair pathways: passing the baton. J. Biol. Chem. 285, 40479–40488. https://doi.org/10.1074/jbc.M110.155267
Caldecott K.W. (2020) Mammalian DNA base excision repair: dancing in the moonlight. DNA Repair (Amst.). 93, 102921.https://doi.org/10.1016/j.dnarep.2020.102921
Saville K.M., Clark J., Wilk A., Rogers G.D., Andrews J.F., Koczor C.A., Sobol R.W. (2020) NAD+-mediated regulation of mammalian base excision repair. DNA Repair (Amst.). https://doi.org/10.1016/j.dnarep.2020.102930
Moor N.A., Lavrik O.I. (2019) Coordination of DNA base excision repair by protein-protein interactions. In: DNA Repair. An Update. Ed. Mognato M. p., 184–217. https://doi.org/10.5772/intechopen.82642
London R.E. (2015) The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst.). 30, 90–103. https://doi.org/10.1016/j.dnarep.2015.02.005
Fang Q., Inanc B., Schamus S., Wang X.H., Wei L., Brown A.R., Svilar D., Sugrue K.F., Goellner E.M., Zeng X., Yates N.A., Lan L., Vens C., Sobol R.W. (2014) HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat. Commun. 5, 5513. https://doi.org/10.1038/ncomms6513
Pettijohn D., Hanawalt P. (1964) Evidence for repair-replication of ultraviolet damaged DNA in bacteria. J. Mol. Biol. 9, 395–410. https://doi.org/10.1016/s0022-2836(64)80216-3
Tornaletti S., Hanawalt P.C. (1999) Effect of DNA lesions on transcription elongation. Biochimie. 81, 139–146. https://doi.org/10.1016/s0300-9084(99)80046-7
Fousteri M., Mullenders L.H. (2008) Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 18, 73–84. https://doi.org/10.1038/cr.2008.6
Sugasawa K., Shimizu Y., Iwai S., Hanaoka F. (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair (Amst.). 1, 95–107.
Maillard O., Camenisch U., Clement F.C., Blagoev K.B., Naegeli H. (2007) DNA repair triggered by sensors of helical dynamics. Trends Biochem. Sci. 32, 494–499. https://doi.org/10.1016/j.tibs.2007.08.008
Min J.H., Pavletich N.P. (2007) Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 449, 570–575. https://doi.org/10.1038/nature06155
Maltseva E.A., Rechkunova N.I., Petruseva I.O., Vermeulen W., Scharer O.D., Lavrik, O.I. (2008) Crosslinking of nucleotide excision repair proteins with DNA containing photoreactive damages. Bioorg. Chem. 36, 77–84. https://doi.org/10.1016/j.bioorg.2007.11.004
Rechkunova N.I., Lavrik O.I. (2010) Nucleotide excision repair in higher eukaryotes: mechanism of primary damage recognition in global genome repair. Subcell. Biochem. 50, 251–277. https://doi.org/10.1007/978-90-481-3471-7_13
Krasikova Y.S., Rechkunova N.I., Maltseva E.A., Pestryakov P.E., Petruseva I.O., Sugasawa K., Chen X., Min J.H., Lavrik O.I. (2013) Comparative analysis of interaction of human and yeast DNA damage recognition complexes with damaged DNA in nucleotide excision repair. J. Biol. Chem. 288, 10936–10947. https://doi.org/10.1074/jbc.M112.444026
Fitch M.E., Nakajima S., Yasui A., Ford J.M. (2003) In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem. 278, 46906–46910. https://doi.org/10.1074/jbc.M307254200
Moser J., Volker M., Kool H., Alekseev S., Vrieling H., Yasui A., van Zeeland A.A., Mullenders L.H. (2005) The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst.). 4, 571–582. https://doi.org/10.1016/j.dnarep.2005.01.001
Houten B.V., Kuper J., Kisker C. (2016) Role of XPD in cellular functions: to TFIIH and beyond. DNA Repair (Amst.). 44, 136–142. https://doi.org/10.1016/j.dnarep.2016.05.019
Evans E., Moggs J.G., Hwang J.R., Egly J.M., Wood R.D. (1997) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 16, 6559–6573. https://doi.org/10.1093/emboj/16.21.6559
Staresincic L., Fagbemi A.F., Enzlin J.H., Gourdin A.M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S.G., Vermeulen W., Scharer O.D. (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 28, 1111–1120. https://doi.org/10.1038/emboj.2009.49
Krasikova Y.S., Rechkunova N.I., Maltseva E.A., Petruseva I.O., Lavrik O.I. (2010) Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucl. Acids Res. 38, 8083–8094. https://doi.org/10.1093/nar/gkq649
Kemp M.G., Gaddameedhi S., Choi J.H., Hu J., Sancar A. (2014) DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J. Biol. Chem. 289, 26574–26583. https://doi.org/10.1074/jbc.M114.597088
Reardon J.T., Bessho T., Kung H.C., Bolton P.H., Sancar A. (1997) In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA. 94, 9463–9468. https://doi.org/10.1073/pnas.94.17.9463
D’Érrico M., Parlani E., Teson M., Jesus B.M.B., Degan P., Calcagnile A., Jaruga P., Bjøras M., Crescenzi B., Pedrini A.M., Egly J.-M., Zambruno G., Stefanini M., Dizdaroglu M., Dogliotti E. (2006) New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 25, 4305–4315.
Kassam S.N., Rainbow A.J. (2007) Deficient base excision repair of oxidative DNA damage induced by methylene blue plus visible light in xeroderma pigmentosum group C fibroblasts. Biochem. Biophys. Res. Commun. 359, 1004–1009. https://doi.org/10.1016/j.bbrc.2007.06.005
Shimizu Y., Uchimura Y., Dohmae N., Saitoh H., Hanaoka F., Sugasawa K. (2010) Stimulation of DNA glycosylase activities by XPC protein complex: roles of protein-protein interactions. J. Nucl. Acids. 2010, 805698. https://doi.org/10.4061/2010/805698
Parlanti E., D’Errico M., Degan P., Calcagnile A., Zijno A., van der Pluijm I., van der Horst G.T., Biard D.S., Dogliotti E. (2012) The cross talk between pathways in the repair of 8-oxo-7,8-dihydroguanine in mouse and human cells. Free Radic. Biol. Med. 53, 2171–2177. https://doi.org/10.1016/j.freeradbiomed.2012.08.593
Berra C.M., Oliveira C.S., Garcia C.C.M., Rocha C.R., Lerner L.K., Lima L.C., Baptista M.S., Menck C.F.M. (2013) Nucleotide excision repair activity on DNA damage induced by photoactivated methylene blue. Free Radic. Biol. Med. 61, 343–356. https://doi.org/10.1016/j.freeradbiomed.2013.03.026
Soltys D.T., Rocha C.R., Lerner L.K., Souza T.A., Munford V., Cabral F., Nardo T., Stefanini M., Sarasin A., Cabral-Neto J.B., Menck C.F. (2013) Novel XPG (ERCC5) mutations affect DNA repair and cell survival after ultraviolet but not oxidative stress. Hum. Mutat. 34, 481–489. https://doi.org/10.1002/humu.22259
Guo J., Hanawalt P.C., Spivak G. (2013) Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucl. Acids Res. 41, 7700–7712. https://doi.org/10.1093/nar/gkt524
Menoni H., Hoeijmakers J.H., Vermeulen W. (2012) Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J. Cell. Biol. 199, 1037–1046. https://doi.org/10.1083/jcb.201205149
Menoni H., Wienholz F., Theil A.F., Janssens R.C., Lans H., Campalans A., Radicella J.P., Marteijn J.A., Vermeulen W. (2018) The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucl. Acids Res. 46, 7747–7756. https://doi.org/10.1093/nar/gky579
Jang S., Kumar N., Beckwitt E.C., Kong M., Fouquerel E., Rapić-Otrin V., Prasad R., Watkins S.C., Khuu C., Majumdar C., David S.S., Wilson S.H., Bruchez M.P., Opresko P.L., van Houten B. (2019) Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 26, 695–703. https://doi.org/10.1038/s41594-019-0261-7
Hailer M.K., Slade P.G., Martin B.D., Rosenquist T.A., Sugden K.D. (2005) Recognition of the oxidized base damage spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair. 4, 41–50. https://doi.org/10.1016/j.dnarep.2004.07.006
Krishnamurthy N., Zhao X., Burrows C.J., David S.S. (2008) Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 47, 7137–7146. https://doi.org/10.1021/bi800160s
Luo W., Muller J.G., Rachlin E.M., Burrows C.J. (2000) Characterization of spiroiminodihydantoin as a product of one electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett. 2, 613–616. https://doi.org/10.1021/ol9913643
Niles J.C., Wishnok J.S., Tannenbaum S.R. (2001) Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org. Lett. 3, 963–966.
Zhao X., Krishnamurthy N., Burrows C.J., David S.S. (2010) Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry. 49, 1658–1666. https://doi.org/10.1021/bi901852q
Shafirovich V., Kropachev K., Anderson T., Liu Z., Kolbanovskiy M., Martin B.D., Sugden K., Shim Y., Chen X., Min J.H., Geacintov N.E. (2016) Base and nucleotide excision repair of oxidatively generated guanine lesions in DNA. J. Biol. Chem. 291, 5309−5319. https://doi.org/10.1074/jbc.M115.693218
Shafirovich V., Kropachev K., Kolbanovskiy M., Geacintov N.E. (2019) Excision of oxidatively generated guanine lesions by competing base and nucleotide excision repair mechanisms in human cells. Chem. Res. Toxicol. 32, 753–761. https://doi.org/10.1021/acs.chemrestox.8b00411
Slyskova J., Sabatella M., Ribeiro-Silva C., Stok C., Theil A.F., Vermeulen W., Lans H. (2018) Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucl. Acids Res. 46, 9537–9549. https://doi.org/10.1093/nar/gky764
Couve-Privat S., Mace G., Rosselli F., Saparbaev M.K. (2007) Psoralen-induced DNA adducts are substrates for the base excision repair pathway in human cells. Nucl. Acids Res., 35, 5672–5682. https://doi.org/10.1093/nar/gkm592
Couve S., Mace-Aim G., Rosselli F., Saparbaev M.K. (2009) The human oxidative DNA glycosylase NEIL1 excises psoralen-induced interstrand DNA cross-links in a three-stranded DNA structure. J. Biol. Chem. 284, 11963–11970. https://doi.org/10.1074/jbc.M900746200
Martin P.R., Couve S., Zutterling C., Albelazi M.S., Groisman R., Matkarimov B.T., Parsons J.L., Elder R.H., Saparbaev M.K. (2017) The human DNA glycosylases NEIL1 and NEIL3 excise psoralen-induced DNA-DNA cross-links in a four stranded DNA structure. Sci. Rep. 7, 17438. https://doi.org/10.1038/s41598-017-17693-4
McNeill D.R., Paramasivam M., Baldwin J., Huan J., Vyjayanti V.N., Seidman M.M., Wilson D.M. 3rd (2013) NEIL1 responds and binds to psoralen-induced DNA interstrand crosslinks. J. Biol. Chem. 288, 12426–12436. https://doi.org/10.1074/jbc.M113.456087
Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P., Jensen L.J., von Mering C. (2017) The STRING database in 2017: Quality-controlled proteinprotein association networks, made broadly accessible. Nucl. Acids Res. 45, D362–D368. https://doi.org/10.1093/nar/gkw937
Kumar N., Moreno N.C., Feltes B.C., Menck C.F.M., Van Houten B. (2020) Cooperation and interplay between base and nucleotide excision repair pathways: from DNA lesions to proteins. Genet. Mol. Biol. 43(1 suppl. 1), e20190104. https://doi.org/10.1590/1678-4685-GMB-2019-0104
Ame J.C., Spenlehauer C., de Murcia G. (2004) The PARP superfamily. Bioessays. 26, 882–893. https://doi.org/10.1002/bies.20085
Schreiber V., Dantzer F., Ame J.C., de Murcia G. (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 7, 517–528. https://doi.org/10.1038/nrm1963
Shieh W.M., Ame J.C., Wilson M.V., Wang Z.Q., Koh D.W., Jacobson M.K., Jacobson E.L. (1998) Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 273, 30069–30072. https://doi.org/10.1074/jbc.273.46.30069
Sharifi R., Morra R., Appel C.D., Tallis M., Chioza B., Jankevicius G., Simpson M.A., Matic I., Ozkan E., Golia B., et al. (2013) Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 32, 1225–1237. https://doi.org/10.1038/emboj.2013.51
Burkle A., Virag L. (2013) Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 34, 1046–1065. https://doi.org/10.1016/j.mam.2012.12.010
Kraus W.L., Hottiger M.O. (2013) PARP-1 and gene regulation: progress and puzzles, Mol. Aspects Med. 34, 1109–1123. https://doi.org/10.1016/j.mam.2013.01.005
Bock F.J., Todorova T.T., Chang P. (2015) RNA regulation by poly(ADP-ribose) polymerases. Mol. Cell. 58, 959–969. https://doi.org/10.1016/j.molcel.2015.01.037
Liu C., Vyas A., Kassab M.A., Singh A.K., Yu X. (2017) The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucl. Acids Res. 45, 8129–8141. https://doi.org/10.1093/nar/gkx565
Ходырева С.Н., Лаврик О.И. (2016) Поли(ADP-рибоза)полимераза 1 – ключевой регулятор репарации ДНК. Молекуляр. биология. 50, 655–673.
Lavrik O.I. (2020) PARPs’ impact on base excision DNA repair. DNA Repair (Amst.). https://doi.org/10.1016/j.dnarep.2020.102911
Суханова М.В., Ходырева С.Н., Лаврик О.И. (2004) Поли(ADP-рибоза)полимераза 1 ингибирует синтез ДНК с вытеснением цепи, катализируемый ДНК-полимеразой β. Биохимия. 69, 686–698.
Schreiber V., Ame J.C., Dolle P., Schultz I., Rinaldi B., Fraulob V., Menissier-de Murcia J., de Murcia G. (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 277, 23028–23036. https://doi.org/10.1074/jbc.M202390200
Moor N.A., Vasil’eva I.A., Kuznetsov N.A., Lavrik O.I. (2020) Human apurinic/apyrimidinic endonuclease 1 is modified in vitro by poly(ADP-ribose) polymerase 1 under control of the structure of damaged DNA. Biochimie. 168, 144–155. https://doi.org/10.1016/j.biochi.2019.10.011
Речкунова Н.И., Мальцева Е.А., Лаврик О.И. (2019) Посттрансляционные модификации белков эксцизионной репарации нуклеотидов и их роль в регуляции процесса. Биохимия. 84, 1244–1258.
Gagne J.P., Isabell M., Lo K.S., Bourassa S., Hendzel M.J., Dawson V.L., Dawson T.M., Poirier G.G. (2008) Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucl. Acids Res. 36, 6959–6976, https://doi.org/10.1093/nar/gkn771
Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M.O., Nielsen M.L. (2013) Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol. Cell. 52, 272–285. https://doi.org/10.1016/j.molcel.2013.08.026
Aleksandrov R., Dotchev A., Poser I., Krastev D., Georgiev G., Panova G., Babukov Y., Danovski G., Dyankova T., Hubatsch L., Ivanova A., Atemin A., Nedelcheva-Veleva M.N., Hasse S., Sarov M., Buchholz F., Hyman A.A., Grill S.W., Stoynov S.S. (2018) Protein dynamics in complex DNA lesions. Mol. Cell. 69, 1046–1061.e5. https://doi.org/10.1016/j.molcel.2018.02.016
Gibson B.A., Kraus W.L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424. https://doi.org/10.1038/nrm3376
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология