

Молекулярная биология, 2021, T. 55, № 2, стр. 210-222
Клеточный ответ на повреждения ДНК, возникающие в рибосомных генах
А. К. Величко a, b, c, С. В. Разин a, О. Л. Кантидзе a, *
a Институт биологии гена Российской академии наук
119334 Москва, Россия
b Первый Московский государственный медицинский университет им. И.М. Сеченова
Министерства здравоохранения Российской Федерации
119146 Москва, Россия
c Центр высокоточного редактирования и генетических технологий для биомедицины,
Институт биологии гена Российской академии наук
119334 Москва, Россия
* E-mail: kantidze@gmail.com
Поступила в редакцию 05.08.2020
После доработки 05.08.2020
Принята к публикации 12.08.2020
Аннотация
Ядрышки в клетках эукариот представляют собой места биосинтеза рибосом. Они формируются вокруг тандемно повторяющихся рибосомных генов, наиболее активно транскрибируемых в клетках человека. Исследования последних лет показывают, что для поддержания геномной стабильности рибосомных повторов существуют особые механизмы клеточного ответа на повреждение ДНК. В обзоре охарактеризована структура и функции рибосомных повторов, их организация в ядрышковом компартменте. Рассмотрены методические аспекты изучения повреждения ДНК в ядрышке, проанализированы уникальные особенности ответа на повреждения рибосомной ДНК, включая транскрипционную репрессию и ядрышковую реорганизацию, а также особенности механизмов репарации повреждений в рибосомных генах.
КЛЕТОЧНЫЙ ОТВЕТ НА ПОВРЕЖДЕНИЕ ДНК
Появление молекулы ДНК и переход к ней функций основного носителя генетической информации стали не только важнейшим скачком в процессе молекулярной эволюции, но и обеспечили предпосылки к формированию и развитию современных форм жизни. Этот переход был обусловлен большей химической стабильностью и прочностью молекулы ДНК по сравнению с РНК, склонной к спонтанному гидролизу и, в силу этого, неспособной формировать цепи большой длины. Тем не менее, молекула ДНК, несмотря на ее очень высокую стабильность, не является неуязвимой. Многообразные агрессивные факторы внешней среды, такие как ионизирующее и ультрафиолетовое излучение и ксенобиотики, способны индуцировать различные повреждения ДНК, называемые экзогенными. В масштабе же клетки или целого организма более вероятны эндогенные повреждения, спонтанно возникающие в процессе нормального клеточного метаболизма, например при коллапсе репликативных вилок [1], при столкновении между транскрипционной и репликативной машинами [2], неполном разделении сестринских хроматид во время митоза [3] и др. В результате совокупного действия экзо- и эндогенных факторов в геномной ДНК образуются десятки и даже сотни тысяч повреждений в сутки [4]. По приблизительным оценкам в число этих повреждений входят лишь от десяти до пятидесяти двухцепочечных разрывов ДНК (ДЦР) [5, 6]. ДЦР, несмотря на их сравнительно небольшое количество, считаются одним из самых токсичных для клетки видов повреждений ДНК, имеющих бесспорную значимость в процессе эволюционной изменчивости. В то же время связанная с ДЦР геномная нестабильность является причиной различных патологий, о чем свидетельствует существование прямой корреляции между дефектами систем репарации ДЦР и частотой развития иммунных и неврологических заболеваний [7], а также злокачественной трансформации клеток [8].
Для обеспечения целостности и стабильности ДНК эволюция наделила клетки целым арсеналом разнообразных молекулярных механизмов, которые в совокупности называются ответом на повреждения ДНК (DDR, DNA Damage Response). Ответ на повреждение ДНК отнюдь не ограничивается только репарацией. Он включает, в первую очередь, системы детекции повреждений, амплификацию сигналов о повреждении и привлечение в сайт разрыва необходимых репарационных белков, собственно репарацию, а также регуляцию клеточного цикла и механизмы запуска программируемой клеточной гибели при невозможности осуществления репарации, например при очень масштабных повреждениях [9]. В клетках высших эукариот используются два основных способа восстановления ДЦР. Во-первых, это гомологичная рекомбинационная репарация (HRR, от англ. “Homologous Recombination Repair”), обеспечивающая восстановление как физической целостности молекулы ДНК, так и ее нуклеотидной последовательности в сайте повреждения. Во-вторых, клетки могут использовать менее точный механизм соединения концов ДНК, который восстанавливает ее целостность, но не обязательно нуклеотидную последовательность. Эти механизмы включают классическое негомологичное соединение концов (c-NHEJ, от англ. “classic Non-Homologous End-Joining”), а также альтернативное соединение концов (alt-NHEJ). В отличие от системы NHEJ, активной на протяжении всего клеточного цикла, HRR работает предпочтительно в S/G2-фазах, поскольку в качестве донора гомологии ей требуется сестринская хроматида [10]. Однако выбор пути репарации не всегда зависит лишь от фазы клеточного цикла. Иногда он определяется типом повреждения ДНК [11], а также эпигенетическим контекстом и транскрипционным статусом региона [12]. Таким образом, ответ на повреждение отнюдь не одинаков для всей ядерной ДНК, и в некоторых геномных областях требуется определенная специализация. Это особенно актуально для тех регионов, в которых повреждения возникают значительно чаще, чем в других – так называемых горячих точках. К ним относятся различные участки генома, богатые повторяющимися последовательностями, такие как теломеры и центромеры [13], ломкие сайты, в частности поздно реплицирующиеся локусы [14], а также активно транскрибирующиеся области [15–17]. С этой точки зрения одним из главных источников нестабильности генома являются рибосомные гены (рДНК) [18, 19], что обусловлено несколькими причинами. Во-первых, это самые активно транскрибирующиеся в клеточном ядре гены, а высокая плотность РНК-полимеразы I в рДНК многократно увеличивает риск столкновения между транскрипционной и репликативной машинами. Еще одна связанная с транскрипцией уязвимость, типичная для рДНК – образование R‑петель. Синтезируемая РНК может гибридизоваться с ДНК, которая кодирует ее в цис-положении, образуя при этом гибридную структуру РНК : ДНК, известную как R-петля [20]. Вытесненная в результате образования R-петли оцДНК служит предпочтительной мишенью для таких мутагенных процессов, как дезаминирование и депуринизация [21]. Будучи достаточно стабильными, R-петли создают препятствия для продвижения РНК- и ДНК-полимераз, что в конечном итоге приводит к ДЦР [22]. Во-вторых, геномные области с высоким содержанием GC или простых повторов, такие как рДНК, сложны для репликации [23]. Если репликативный стресс сохраняется в течение длительного времени, то специфичные к структуре эндонуклеазы будут генерировать одно- и двухцепочечные разрывы в попытке перезапустить остановленные вилки репликации [24]. В-третьих, регионы с высоким содержанием GC склонны к образованию неканонических форм ДНК, состоящих из четырех цепей. Эти структуры известны как G-квадруплексы. По мере движения РНК-полимеразы I нематричная цепь ДНК может формировать G-квадруплексы, которые также представляют собой препятствие для последующего движения вилок репликации [25].
Наконец, рибосомные гены формируют длинные тандемные повторы, обладающие повышенной рекомбиногенностью, что может приводить к грубым перестройкам при повреждении ДНК. Именно поэтому нестабильность рДНК часто сопровождает многие онкологические [26] и нейродегенеративные заболевания [27], а также фенотип преждевременного старения [28].
За последнее время появляется все больше работ, которые показывают уникальные особенности ответа на повреждения ДНК рибосомных генов [29–32]. Именно этому посвящен наш обзор. Мы начнем с характеристики структуры и функционирования рибосомных повторов и их организации в ядрышковом компартменте. Затем рассмотрим методические аспекты изучения повреждения ДНК в ядрышке, проанализируем уникальные особенности ответа на повреждения рДНК, включая транскрипционную репрессию и ядрышковую реорганизацию, а закончим механизмами репарации повреждений рДНК и их особенностями.
СТРУКТУРНО-ФУНКЦИОНАЛЬНАЯ ОРГАНИЗАЦИЯ ЯДРЫШКА
Ядрышко – это большой безмембранный субъядерный компартмент, присутствующий в эукариотических клетках всех типов. Основная функция ядрышка состоит в транскрипции рибосомных генов, процессинге рРНК и сборке субъединиц рибосом. Рибосомные гены организованы в тандемные повторы, называемые ядрышковым организатором (NOR, от англ. “Nucleolar Organization Regions”). Гаплоидный геном человека содержит в среднем 300 копий таких повторов длиной 43 т.п.н., кластеры которых локализованы на коротких плечах всех акроцентрических хромосом (хромосомы 13, 14, 15, 21 и 22) (рис. 1) [33, 34]. Длина кластера рибосомных генов характеризуется поразительной изменчивостью – она различается не только у разных индивидов, но и в клетках одного человека, и может варьировать от 50 тысяч до нескольких миллионов пар нуклеотидов. [35]. Подобная изменчивость определяет уникальный кариотип рДНК каждого человека. Установленный максимум количества повторов составляет около 400 копий на геном человека, при этом минимальное зарегистрированное их количество – всего 14 [36]. Большинство повторов рДНК расположены в направлении “голова к хвосту” и являются каноническими. Однако около трети повторов организованы в виде палиндромных, или неканонических повторов. Каждый из повторов длиной 43 т.п.н. включает в себя кодирующий рРНК сегмент размером 13 т.п.н., а также межгенный спейсер длиной 30 т.п.н. (IGS, от англ. “InterGenic Spacer”). Этот спейсер содержит различные регуляторные элементы, такие как промотор, энхансерные и терминаторные последовательности [37] (рис. 1).
Рис. 1.
Схема организации рибосомного повтора. Сокращения: 18S, 5.8S, 28S – кодирующие последовательности соответствующей РНК, AsiSI – сайт узнавания эндонуклеазы рестрикции AsiSI, CORE – минимальный (кор-) промотор, ETS – внешний транскрибируемый спейсер, IGS – межгенный спейсер, I-PpoI – сайт узнавания эндонуклеазы рестрикции I-PpoI, ORI – участок начала (ориджин) репликации, Pol1 – промотор РНК-полимеразы I, Pol2 – промотор РНК-полимеразы II, T0 – терминатор-подобный элемент, T1–T10 – терминаторы транскрипции, UCE – 5′-концевой регуляторный элемент.
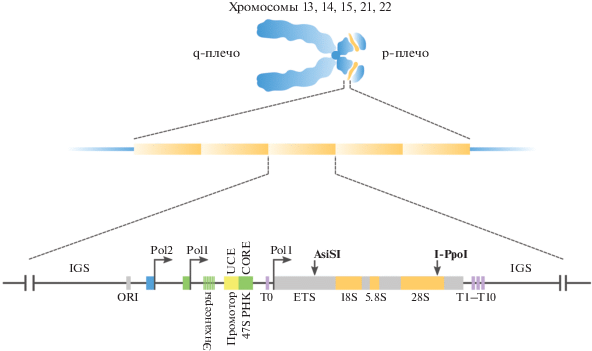
Повторы рДНК существуют в двух формах: активно транскрибирующиеся повторы, представляющие собой открытый эухроматин, и неактивные, организованные в гетерохроматин. И те, и другие при этом локализуются в ядрышке. Таким образом, интенсивность синтеза рРНК может регулироваться не только за счет изменения суммарного количества копий рибосомных повторов, но и эпигенетически – путем изменения баланса между количеством активных и репрессированных копий повтора [38].
Транскрипция генов рРНК требует образования на промоторе рДНК преинициаторного комплекса, состоящего из РНК-полимеразы I, фактора активации транскрипции UBF и фактора селективности промотора SL1. Когда РНК-полимераза I начинает процесс синтеза рРНК, UBF и SL1 остаются связанными с промоторной областью, что позволяет непрерывно рекрутировать молекулы РНК-полимеразы I [38, 39]. Таким образом, каждый ген рРНК может транскрибироваться одновременно несколько раз. Это порождает структуру, напоминающую рождественскую елку, которую можно визуализировать с помощью электронной микроскопии [40]. Исходный 47S транскрипт-предшественник рРНК процессируется с образованием зрелых 18S, 5.8S и 28S рРНК, которые затем посттранскрипционно модифицируются при участии малых ядрышковых РНП и дополнительных факторов процессинга. Наконец, процессированные и модифицированные рРНК связываются с рибосомными белками и экспортируются в цитоплазму [41].
В ядрышках высших эукариот рибосомные повторы организованы в три внутриядрышковых компартмента: фибриллярный центр (FC, от англ. “Fibrillar Centre”), плотный фибриллярный компонент (DFC, от англ. “Dense Fibrillar Component”) и гранулярный компонент (GC, от англ. “Granular Component”). Каждый компонент можно легко идентифицировать по специфической морфологии с помощью электронной и световой микроскопии [41]. Ядрышко может иметь множество фибриллярных центров, каждый из которых организован вокруг одного–трех рибосомных генов [42, 43]. Фибриллярные центры содержат избыточный запас компонентов аппарата транскрипции – РНК-полимеразу I и транскрипционные факторы, такие как UBF, Treacle, SL1 и др. Тем не менее, транскрипция происходит не в самих фибриллярных центрах, а на границе между ними и плотным фибриллярным компонентом [43, 44]. Плотный фибриллярный компонент окружает каждый фибриллярный центр и содержит в основном факторы раннего процессинга рРНК. Плотный фибриллярный компонент, в свою очередь, окружен плотным гранулярным компонентом, куда перемещается транскрибированная рРНК для дальнейшего созревания и сборки рибосом. Таким образом, организация и структура этих внутриядрышковых компартментов тесно связана с последовательными шагами биогенеза рибосом и направленным транспортом рРНК.
Многие немембранные субъядерные компартменты, такие как центры сплайсинга (nuclear speckles) или тельца Кахаля, проявляют свойства жидких капель. Принцип, на котором основано образование таких органелл, получил название жидкость-жидкостное фазовое разделение (LLPS, от англ. “Liquid-Liquid Phase Separation”), и в настоящее время идея о том, что именно этот принцип лежит в основе внутренней организации клетки в целом, приобретает все большую популярность [45, 46]. Ядрышки также проявляют жидкостные свойства [47]. Показано, что неупорядоченные домены фибрилларина, нуклеофозмина и РНК-полимеразы I (ключевых структур плотного гранулярного компонента, гранулярного компонента и фибриллярного соответственно) необходимы для образования капель, а их мотивы связывания с РНК необходимы для поддержания LLPS [48, 49]. Именно за счет жидкостных свойств ядрышко обладает весьма динамической структурой. Так, например, ингибирование транскрипции рибосомных генов актиномицином D (AСD), который при низкой концентрации ингибирует преимущественно РНК-полимеразу I, приводит к крупномасштабной реорганизации ядрышка и образованию так называемых “ядрышковых кепов” [50]. Формирование кепов сопровождается транслокацией белков фибриллярного центра и плотного фибриллярного компонента вместе с рДНК на периферию ядрышка, при этом фибриллярный центр обращен к нуклеоплазме, а плотный фибриллярный компонент к внутренней части ядрышка. Одновременно с этим происходит перемещение многих рибосомных белков из ядрышка в нуклеоплазму. Высвобожденные рибосомные белки секвестрируют убиквитинлигазу MDM2 в нуклеоплазме и таким образом способствуют повышению уровня р53, что приводит к остановке клеточного цикла, старению или апоптозу [51]. Такой сценарий ядрышковой реорганизации реализуется не только при прямом ингибировании транскрипции рибосомных генов, но и при многих других типах стресса, в частности тепловом [52, 53], окислительном, осмотическом [31], и при повреждении ДНК, возникшем как непосредственно в рДНК [29, 30], так и в остальной части генома [32, 54]. Таким образом, изменение уровня транскрипции рибосомных генов в общем, как и их репрессия в частности, представляет собой универсальную реакцию клеток на стресс, а ядрышко считается сенсором и центром координации этой реакции [55].
СИСТЕМЫ ИНДУКЦИИ ПОВРЕЖДЕНИЙ В рДНК
Системы репарации повреждений ядрышковой ДНК экспериментально изучают с использованием различных способов внесения таких повреждений. Каждый способ обладает рядом преимуществ и недостатков. Тем не менее, выбор того или иного способа зависит в первую очередь от исследовательских задач. В первых работах, посвященных изучению ответа на повреждение рДНК, клетки подвергали действию γ-излучения или ультрафиолетового излучения [54, 56, 57]. Однако эти типы воздействия не являются специфичными и повреждают не только рибосомные гены, но ДНК всего генома в целом. Лазерное микрооблучение и ионные микропучки позволяют получить более локальные повреждения, поскольку их можно сфокусировать непосредственно на ядрышке [54, 58]. Однако они также имеют свои ограничения и недостатки. Во-первых, эти подходы по-прежнему не являются сайт-специфичными. Во-вторых, лазерные и ионные пучки индуцируют не только ДЦР, но и множество других типов повреждений ДНК (так называемые кластерные повреждения), что значительно усложняет интерпретацию результатов относительно систем распознавания и репарации таких повреждений. В подходах, лишенных обозначенных недостатков, используются специфичные эндонуклеазы, позволяющие вносить повреждения известного типа в известный участок рДНК. Эктопическая экспрессия в клетках хоминг-эндонуклеазы I-PpoI из Physarum polycephalum – система, которая регулярно используется для индукции ДЦР в рДНК при изучении ответа на повреждение ДНК в рибосомных генах [29–31, 59–61]. В кодирующем 28S РНК регионе рибосомного повтора находится отсутствующий в остальной части генома 13–15-нуклеотидный участок узнавания ДНК-нуклеазой I-PpoI (рис. 1), который на 100% идентичен у всех эукариот [62]. Экспрессируемая в клетках человека I-PpoI индуцирует повреждения приблизительно в 10% своих сайтов-мишеней рДНК, что составляет около 30 ДЦР на все рибосомные гены [63]. I-PpoI также атакует еще примерно 13 сайтов в геноме, большинство из которых, однако, представлены псевдогенами 28S рРНК [64].
Еще одна система индукции ДЦР в рДНК – эктопическая экспрессия эндонуклеазы AsiSI из Arthrobacter [29, 65], которая имеет 8-нуклеотидный сайт в кодирующем регионе 47S РНК (рис. 1). Однако AsiSI индуцирует повреждения не только в рибосомном повторе, но и еще в 174 сайтах генома человека [66]; поэтому для изучения повреждений именно рДНК AsiSI применяется значительно реже, чем I-PpoI. Кроме того, отсутствует даже приблизительный количественный анализ индуцированных AsiSI повреждений в рДНК. Стоит также отметить, что AsiSI не может расщеплять участки узнавания, содержащие метилированные CpG-динуклеотиды, а так как некоторая часть повторов рДНК инактивирована именно за счет метилирования [38], то AsiSI расщепляет, вероятно, только активные, деметилированные повторы.
Наконец, технология CRISPR/Cas9 также весьма эффективна для адресного внесения ДЦР в рДНК. С помощью этой технологии можно индуцировать ДЦР практически в любой части рибосомного повтора, конечно, при наличии подходящего PAM-сайта (мотив, смежный с протоспейсером). В исследованиях ответа на повреждение рДНК систему CRISPR/Cas9 успешно использовали для индукции ДЦР как в транскрибирующемся регионе рибосомного повтора, так и в его нетранскрибируемом межгенном спейсере [29, 60]. Тем не менее, надо иметь в виду, что длительная экспрессия I-PpoI, AsiSI или Cas9 в клетках приводит к повторным циклам разрезания-восстановления таргентных участков ДНК до тех пор, пока ошибки репарации не приведут к изменению целевой последовательности [63]. Это ограничение метода не относится к значительным, однако его стоит учитывать в долгосрочных экспериментах.
ПОВРЕЖДЕНИЯ РИБОСОМНОЙ ДНК ИНДУЦИРУЮТ PIK-ЗАВИСИМУЮ РЕПРЕССИЮ ЯДРЫШКОВОЙ ТРАНСКРИПЦИИ
Самые ранние результаты изучения ответа на повреждение ДНК в ядрышке получены в лаборатории R. Casellas в 2007 году. Kruhlak и соавт. показали, что при генотоксическом стрессе, индуцированном γ-излучением, лазерным микрооблучением или этопозидом (ингибитор ДНК-топоизомеразы II), в эмбриональных фибробластах мыши происходит масштабная репрессия транскрипции рибосомных генов [54]. Эта репрессия сопровождается реорганизацией ядрышка, в том числе формированием “ядрышковых кепов”, идентичных тем, которые образуются при ингибировании рибосомной транскрипции ACD [54]. В дальнейшем, изучая механизмы наблюдаемого эффекта, обнаружили поразительный факт: транскрипция в ядрышке была не просто пассивно заблокирована за счет повреждений ДНК. Наоборот, транскрипционная репрессия была регулируемым процессом и зависела от активности киназы ATM – одного из основных белков группы фосфатидилинозитол-3-киназа-подобных киназ (PIK-киназ), ответственных за распознавание и репарацию ДЦР [67, 68]. Впоследствии роль киназы ATM в индуцированной ДЦР репрессии ядрышковой транскрипции многократно подтвердили с использованием эндонуклеаз I-PpoI [29, 31, 59–61], AsiSI [65] и CRISPR/Cas9 [29, 60]. C помощью CRISPR/Cas9-индуцированных разрывов обнаружено, что ATM-зависимая репрессия транскрипции происходит вне зависимости от того, в кодирующей или некодирующей части рибосомного повтора возникают повреждения [60]. Это окончательно исключило возможность нерегулируемой физической остановки РНК-полимеразы I разрывами в матричной цепи ДНК.
Как выяснилось в дальнейшем, ATM – не единственный представитель PIK-киназ, регулирующий транскрипцию рДНК в ответ на повреждения. Оказалось, что PIK-киназа ATR также принимает участие в этом процессе. Несмотря на то, что ATR в меньшей степени вовлечена в репарацию ДЦР и, в основном, ответственна за поддержание стабильности репликации ДНК, в ряде работ показано, что ATR в равной степени с ATM обеспечивает репрессию ядрышковой транскрипции при индукции ДЦР различными агентами [29, 30]. В нашей лаборатории обнаружено, что киназа ATR совместно с ATM обеспечивает репрессию ядрышковой транскрипции в ответ на индуцированные гипоосмотическим стрессом котранскрипционные R-петли [31]. Однако осталось неясным, обусловлена ли активация ATR/ATM-зависимого сигнального пути конверсией R-петель в ДЦР, либо же сама структура R-петель является причиной его запуска.
Еще один представитель группы PIK-киназ – DNA-PKcs, которая, как и ATM, участвует в первую очередь в распознавании и репарации ДЦР. Известны всего две работы, показывающие возможность DNA-PKcs-зависимой репрессии РНК-полимеразы I. Первая работа выполнена с использованием in vitro систем [69], вторая выявляет участие DNA-PKcs в транскрипционной репрессии рДНК в ответ на повреждения, индуцируемые цисплатином и ультрафиолетовым излучением [70]. Тем не менее, в большинстве последующих работ с использованием CRISPR/Cas9, I-PpoI или ионизирующего излучения участие DNA-PKcs в репрессии транскрипции рДНК не подтвердилось [31, 54, 59]. Поэтому на сегодня главенствует мнение, что роль DNA-PKсs в ответе на повреждение рДНК ограничена только репарацией. Однако не исключено, что ATM, ATR, и DNA-PKcs, каждая из которых служит эффектором разного типа повреждений, в конечном итоге могут запускать одни и те же механизмы репрессии транскрипции.
МЕХАНИЗМЫ ТРАНСКРИПЦИОННОЙ РЕПРЕССИИ РИБОСОМНОЙ ДНК
В условиях воздействия ионизирующего излучения репарационные белки Nbs1 и MDC1, а также ATM необходимы для эффективного ингибирования ядрышковой транскрипции, в то время как некоторые факторы репарации, включая Ku80, BRCA1, 53BP1 и гистон H2AX, необязательны. Используя методы FRAP-микроскопии, удалось показать, что ATM-зависимая репрессия ядрышковой транскрипции обусловлена подавлением инициации транскрипции РНК-полимеразой I, а также ее удалением с хроматина в момент элонгации [54]. Однако конкретный механизм индуцированной ДЦР репрессии рДНК до сих пор остается предметом исследований. Ключом к пониманию этого механизма может быть поиск и идентификация мишеней АТМ в ядрышке. В ходе фосфопротеомных исследований обнаружено несколько таких мишеней [71, 72], среди которых субъединица РНК-полимеразы I RPA34, компонент SL1-инициаторного комплекса TAF1C, фактор терминации транскрипции 1 (TTF1), а также фактор транскрипции UBF и взаимодействующий с ним белок Treacle. Впоследствии прямую роль в индуцированной ДЦР репрессии рибосомных генов удалось доказать только для Treacle (рис. 2).
Рис. 2.
Обобщенная схема механизмов репрессии РНК-полимераза I-зависимой транскрипции, индуцированной повреждением ДНК в рибосомных повторах. Повреждение рДНК индуцирует метилирование гистона H3 и фосфорилирование гистона H2B, что обеспечивает компактизацию хроматина и его транскрипционный сайленсинг. Кроме того, повреждение рДНК сопровождается ATM/ATR-зависимым фосфорилированием Treacle, что стимулирует его взаимодействие с комплексом MRN и белком TOPBP1. Это взаимодействие также является необходимым условием транскрипционной репрессии рибосомных генов.
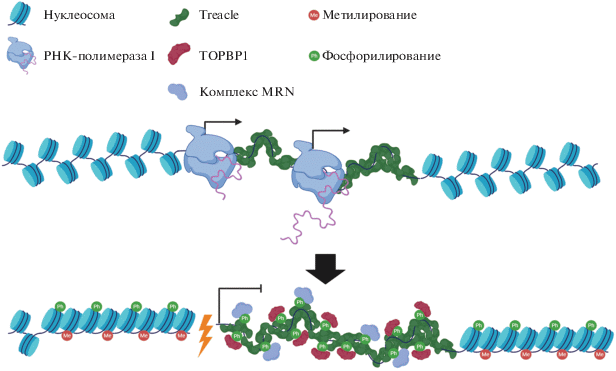
Treacle (также известный как TCOF1) – ядрышковый фосфопротеин, ген которого мутирован при синдроме Тричера–Коллинза, врожденном заболевании, характеризующемся черепно-лицевыми деформациями [73]. Treacle содержит 17 S/T-Q-мотивов – специфичных для ATM и других PIK-киназ сайтов фосфорилирования. В работах S.J. Elledge, M. Stucki и D.H. Larsen показано, что при индукции ДЦР в рибосомных генах с помощью CRISPR/Cas9 или ионизирующей радиации ATM-зависимое фосфорилирование Ser-1199 в одном из S/T-Q-мотивов Treacle необходимо для его взаимодействия с белком Nbs1. В свою очередь, это взаимодействие необходимо для транскрипционной репрессии рДНК в ответ на повреждение [29, 32, 54, 74]. Кроме того, Nbs1 взаимодействует с Treacle в составе комплекса Mre11-Nbs1-Rad50 (MRN). Это взаимодействие может происходить и без участия Mre11, однако отсутствие этого компонента компрометирует транскрипционную репрессию, индуцированную повреждением рДНК (рис. 2) [29].
Позже две исследовательские группы независимо показали, что активатор ATR-киназы – TOPBP1, также связывается c Treacle в ответ на повреждение рДНК [30, 31]. В лаборатории М. Stuki обнаружили, что при индукции повреждений с помощью I-PpoI для репрессии рибосомной транскрипции требуется взаимодействие Treacle и TOPBP1, причем такое взаимодействие опосредуется активностями обеих киназ – ATR и ATM, а также Nbs1 [30]. Поскольку Nbs1 взаимодействует с N-концевым доменом Treacle, а TOPBP1 с C-концевым доменом, то эти взаимодействия не интерферируют друг с другом и могут быть реализованы одновременно. При этом связывание Treacle с TOPBP1 по всей видимости также контролируется ATM/ATR-зависимым фосфорилированием S/T-Q-мотивов в Treacle. В работе нашей лаборатории установлено, что стабилизация R-петель при гипоосмотическом стрессе также приводит к ATR-зависимому взаимодействию Treacle и TOPBP1, необходимому, в свою очередь, для активации ATM и репрессии ядрышковой транскрипции [31]. Кроме того, в работе еще одного коллектива показано, что сверхэкспрессии TOPBP1 достаточно для ATR-зависимой репрессии рибосомных генов без какого-либо их повреждения [75]. Таким образом, можно предположить, что ATM/ATR-зависимая модификация Treacle и его прямое взаимодействие с репарационными белками TOPBP1 и Nbs1 может приводить к временной утрате им транскрипционной функции, что и обеспечивает репрессию рибосомных генов (рис. 2).
Несмотря на то, что рибосомные гены имеют низкую плотность нуклеосом, модификации гистонов также регулируют транскрипцию рДНК при ответе на повреждение. При повреждениях рДНК, индуцированных ионизирующим излучением или I-PpoI, происходит фосфорилирование гистона H2B по Ser-14 (H2BS14p), что является репрессирующей модификацией. Фосфорилирование H2B в данном случае осуществляется киназой MST2, которую при участии адапторного белка RASSF1A активирует киназа ATM. Отсутствие любого из компонентов цепи ATM–ASSF1A–MST2 приводит к потере H2BS14p и препятствует индуцируемой ДЦР репрессии рибосомных генов [76]. В исследовании, представленном лабораторией G. Legube [65], обнаружено, что AsiSI-индуцируемое повреждение рДНК приводит к увеличению уровня гистона H3, триметилированного по Lys-9 (H3K9Me3), что также является фактором формирования неактивного хроматина. Метилирование при этом обеспечивается активностями репрессорного комплекса HUSH и гистон-метилтрансферазs SUV39H1. Нокдаун любого из этих белков приводит к снижению уровня ДЦР-индуцируемой репрессии и предотвращает формирование ядрышковых кепов [65]. Таким образом, репрессия транскрипции рибосомных генов, индуцируемая повреждением ДНК, может управляться совокупностью механизмов, включающих не только модификацию компонентов самой транскрипционной машины, но и изменение эпигенетического статуса ядрышкового хроматина.
ДАЛЬНОДЕЙСТВИЕ ТРАНСКРИПЦИОННОЙ РЕПРЕССИИ рДНК
В последние годы активно изучается влияние повреждений ДНК на транскрипцию не только тех генов, которые непосредственно содержат ДЦР, но и на активность смежных с ними неповрежденных генов. Показано, что индукция ДЦР в генах, активно транскрибируемых РНК-полимеразой II, приводит к их ATM/DNA-PK-зависимой репрессии [77, 78]. Обнаружено снижение транскрипции или накопление репрессивных хроматиновых меток в генах, лежащих на расстоянии до нескольких тысяч пар нуклеотидов от специфического сайта разрыва [77, 79]. Отмечено отсутствие включения рибонуклеотидов и активной формы РНК-полимеразы II в регионах, маркированных γH2AX, при индукции ДЦР с помощью ионизирующего или лазерного излучения [80]. Таким образом, расстояние, на которое распространяется эффект, репрессирующий транскрипцию, скорее всего, ограничено размером репарационного компартмента. Однако реализуются ли подобные дистанционные эффекты при повреждении рДНК? Этот вопрос недостаточно изучен. Как далеко от сайта повреждения в рДНК могут распространяться репрессирующие транскрипцию сигнальные каскады? Действуют ли они только в пределах того рибосомного повтора, в котором индуцировано повреждение, или же транскрипционная репрессия действует также in cis на соседние неповрежденные повторы?
Для индукции ДЦР в единичном ядрышке Kruhlak и соавт. [54] использовали лазерное микрооблучение, мощность которого обеспечивала внесение одного ДЦР на 1 млн.п.н. ДНК. С помощью этого подхода установлено, во-первых, что транскрипция репрессируется исключительно в том ядрышке, которое подвергается действию облучения, и не распространяется на остальные в пределах клеточного ядра. Во-вторых, транскрипционная репрессия ядрышка, его реорганизация и формирование “кепов” могут происходить при очень низкой плотности ДЦР – приблизительно один ДЦР на 23 рибосомных повтора. Тем не менее, количественная оценка частоты ДЦР, представленная в [54], является приблизительной и, скорее всего, занижена, поскольку основана не на прямом измерении количества разрывов, а на определении интенсивности фосфорилирования H2AX в ядрышке, для которого характерна низкая плотность нуклеосом [81].
Транскрипционную репрессию во всем ядрышке провоцирует также эндонуклеаза I-PpoI [59], хотя по некоторым оценкам I-PpoI индуцирует ДЦР всего лишь в 10% рибосомных повторов [63]. Однако эта величина также может быть заниженной, поскольку использованный метод оценки не учитывает ни эффективность трансфекции клеток I-PpoI, ни особенности распределения ДЦР между активными и неактивными повторами.
Более точный анализ количества ДЦР в рДНК выполнен в группе D. Larsen. ДЦР в межгенном спейсере рибосомного гена индуцировали с помощью CRISPR/Cas9, что приводило к транскрипционной репрессии и формированию кепов. Используя количественный ПЦР-анализ, обнаружили, что поврежденными оказываются около 30% всех повторов рДНК, что соответствует одному ДЦР на три повтора или одному ДЦР на один фибриллярный центр [29]. Принимая во внимание, что CRISPR/Cas9 индуцирует разрывы преимущественно в активно транскрибирующихся регионах [82], можно предполагать, что репрессирующий потенциал одного ДЦР может быть ограничен одним фибриллярным центром.
Показано также, что повреждения, индуцируемые лишь в части рибосомных повторов, провоцируют скорее локальную репрессию в поврежденной области, чем глобальную репрессию целого ядрышка. Так, с помощью эндонуклеазы AsiSI, которая индуцирует разрывы в меньшем количестве рибосомных повторов, чем I-PpoI или CRISPR/Cas9, установили, что лишь некоторая доля рибосомных повторов одного ядрышка оказывается репрессированной и перемещается в кeпы, в то время как оставшаяся часть неповрежденных повторов продолжает транскрибироваться внутри ядрышка [65]. Недавно была предпринята попытка изучения реакции ядрышка на локальные повреждения рДНК, индуцированные микропучками ионов углерода [58]. Этот подход позволяет сфокусировать излучение в точке диаметром не более 1 мкм и облучать не все ядрышко, а лишь небольшую внутриядрышковую зону. Варьируя количество ионов в пучке, можно индуцировать от 0.2 до 20 ДЦР/мкм. С помощью этого подхода показано, что независимо от количества ДЦР транскрипционная репрессия происходит исключительно в зоне облучения и полностью совпадает с фосфорилированной формой H2AX, являющейся маркером ДЦР. При этом не обнаружено крупномасштабной транскрипционной репрессии и ядрышковой реорганизации [58].
Вероятность глобальной транскрипционной репрессии ядрышка коррелирует, по всей видимости, с количеством индуцированных в нем повреждений. Не исключено и распространение репрессии in cis от одного ДЦР на несколько соседних рибосомных повторов. Тем не менее, этот эффект пространственно ограничен, предположительно одним фибриллярным центром, что не позволяет локальному повреждению спровоцировать лавинообразную транскрипционную репрессию всего массива рДНК.
ТРАНСКРИПЦИОННАЯ РЕПРЕССИЯ И ФОРМИРОВАНИЕ ЯДРЫШКОВЫХ КЕПОВ
Ингибирование ядрышковой транскрипции обычно коррелирует с реорганизацией ядрышек с образованием ядрышковых кепов, которые формируются как при прямом ингибировании РНК-полимеразы I ACD, так и при ATM/ATR-зависимой репрессии в условиях повреждения рДНК. При этом активные формы ATM и ATR локализуются непосредственно в кепах, а ингибирование киназ полностью блокирует их формирование с сохранением количества индуцированных в рДНК повреждений [29–31, 59]. Механизм формирования кепов вызывает много вопросов. Существуют две гипотезы, объясняющие этот механизм. Первая из них предполагает пассивный процесс, основанный на изменении свойств жидкой фазы ядрышек в условиях репрессии транскрипции. В активно транскрибирующем ядрышке фибриллярный центр и плотный фибриллярный компонент представлены в виде капель, слипание которых между собой ограничено за счет активной транскрипции и присутствия РНК. Остановка транскрипции приводит к локальному истощению компонента РНК, в результате чего капельные свойства компонентов фибриллярного центра начинают возобладать, что и приводит к их сливанию, агрегации и пассивной транслокации к периферии ядрышка [49, 83]. В соответствии с этой идеей формирование кепов обусловлено ингибированием транскрипции. Вторая гипотеза предполагает активное перемещение рДНК при участии моторных механизмов клетки. При индукции повреждений рДНК эндонуклеазой AsiSI транслокация части поврежденных повторов в кепы осуществляется за счет комплекса LINK (комплекс, связывающий ядерную ламину и цитоскелет) и актиновых филаментов [65], т.е. тех же факторов, которые, согласно ряду данных, обеспечивают перемещение и кластеризацию ДЦР в других геномных локусах [84–86]. Однако такая LINK/актинзависимая транслокация происходила только в S- и G2-фазах клеточного цикла, что указывало на зависимость этого процесса от специфики репарации разрывов, индуцированных AsiSI [65].
Таким образом, образование кепов может быть обусловлено сразу несколькими факторами. Например, при действии ACD, ионизирующей радиации, I-PpoI или CRISPR/Cas9, когда репрессии подвергается большинство рибосомных повторов, формирование кепов является прямым следствием репрессии транскрипции и обусловлено лишь силами LLPS. Если же поврежденной оказывается лишь небольшая доля повторов (как в случае эндонуклеазы AsiSI), и сил LLPS недостаточно, то транслокацию могут обеспечить LINK и актин. Однако в этом случае эффективность транслокации будет определяться типом повреждений и, как следствие, типом и скоростью их репарации.
Считается, что образование ядрышковых кепов решает две основные задачи. Во-первых, обеспечивает пространственное разделение рДНК, происходящей из разных хромосом, для предотвращения межхромосомной аберрантной рекомбинации. Во-вторых, обеспечивает собственно репарацию рДНК. Некоторые сигналы и факторы репарации (включая факторы HRR) при разрывах рДНК не обнаруживаются до тех пор, пока не образуются ядрышковые кeпы [87]. К таким факторам относятся, например, белки MDC1, 53BP1 и BRCA1 [29, 87]. В настоящее время точная причина этого не установлена. Предполагается, что у этих белков могут отсутствовать необходимые домены, обеспечивающие их совместимость с жидкой фазой внутри ядрышка [49]. В то же время некоторые из этих белков привлекаются в сайт повреждений за счет модификаций гистонов, в частности фосфорилирования H2AX [88]. Как известно, рибосомные гены имеют низкую плотность нуклеосом, поэтому отсутствие перечисленных белков репарации внутри ядрышка также может отражать слабый уровень ответа на повреждение ДНК. С этой точки зрения формирование кепов необходимо не столько для обеспечения репарации, сколько для амплификации репарационного сигнального каскада.
РЕПАРАЦИЯ ПОВРЕЖДЕНИЙ ДНК В ЯДРЫШКЕ
Репарация ДЦР в рибосомных генах, как и в остальных областях генома, происходит с помощью двух основных путей: NHEJ и HRR. Репарация по типу негомологичного соединения концов начинается с распознавания ДЦР гетеродимером Ku70-Ku80. Он выступает в качестве платформы для остальных факторов репарации, включая нуклеазы, полимеразы и лигазы. Комплекс Ku70-Ku80 связывается с киназой DNA-PKcs, что приводит к изменению ее конформации и активации. В дальнейшем к месту разрыва привлекается комплекс XLF/XRCC4/ДНК-лигаза IV, который обеспечивает лигирование концов ДНК. При индукции повреждений с помощью I-PpoI гетеродимер Ku70-80 связывается с рДНК еще внутри ядрышка задолго до формирования кепов [89]. Химическое ингибирование или нокдаун DNA-PK, а также нокдаун некоторых факторов NHEJ значительно повышают количество ДЦР, индуцированных в рДНК эндонуклеазами I-PpoI и AsiSI. Кроме того, отсутствие факторов NHEJ усугубляет индуцируемую I-PpoI и AsiSI репрессию транскрипции [59, 65]. Это явно указывает на функциональную зависимость репарации рДНК от NHEJ.
В ядрышковых кепах отсутствуют факторы NHEJ (за исключением 53BP1) [87]. При этом большинство факторов HRR, таких как RPA, MDC1, Rad51, BRCA1, наоборот, рекрутируются преимущественно в ядрышковые кепы. Именно поэтому считается, что HRR-репарация наиболее активна в кепах. Репарацию этим способом можно разделить на три основных этапа. На первом этапе с разрывом связывается комплекс MRE11–RAD50–NBS1 (MRN), нуклеазы Exo1, Dna2, CtIP и хеликаза BLM. Эти белки необходимы для резекции концов ДНК и формирования одноцепочечных участков, которые немедленно покрывает белок RPA. На втором этапе RPA замещается рекомбиназой RAD51 с образованием нуклеопротеинового филамента. Известно, что RPA обладает высокой аффинностью к оцДНК, поэтому для его замещения на RAD51 требуется участие специальных белков-медиаторов, один из которых BRCA1. Далее происходит поиск гомологии и инвазия репарируемой цепи ДНК в гомологичную цепь с образованием замещающей петли и структуры Холлидея. На третьем этапе синтезируется поврежденный участок ДНК [88].
Известно, что HRR реализуется преимущественно в S- и G2-фазах клеточного цикла, что обусловлено наличием сестринской хроматиды. Действительно, при индукции повреждений рДНК с помощью нуклеаз I-PpoI и AsiSI факторы HRR локализуются в ядрышковых кепах в основном в клетках в фазах S/G2 [30, 65]. Удивительно, но при репарации повреждений, индуцированных CRISPR/Cas9, факторы HRR локализуются в кепах, даже если клетки находятся в G1-фазе, где еще нет сестринских хроматид. Предполагается, что в этом случае неповрежденные повторы рДНК могут служить матрицей для гомологичной рекомбинации in cis. Таким образом выбор HRR-репарации в рДНК может быть обусловлен не столько фазой клеточного цикла, сколько спецификой повреждения. I-PpoI и AsiSI вносят простые разрывы, которые быстро репарируются, и относительно небольшое их количество может быть восстановлено NHEJ непосредственно внутри ядрышка до момента транскрипционной репрессии и формирования кепов. Разрывы, индуцируемые CRISPR/Cas9, более сложные, они обладают относительно медленной кинетикой восстановления (до 10 ч) [90]. В этом случае NHEJ оказывается недостаточно, и рДНК перемещается в ядрышковые кепы, где репарация продолжается уже с помощью HRR [87]. Таким образом, выбор между путями репарации в рДНК может быть обусловлен, в первую очередь, типом и сложностью повреждения.
ЗАКЛЮЧЕНИЕ
Основная функция ядрышка – крупнейшего субъядерного компартмента – биосинтез рибосом, который является одним из наиболее энергетически затратных метаболических процессов. Своевременная регуляция транскрипции в ядрышке необходима, во-первых, для поддержания стабильности рДНК при ее повреждении, а, во-вторых, для перераспределения энергетических ресурсов, обеспечивающих клеточный гомеостаз в условиях стресса. Полученные нами и другими исследовательскими группами результаты подтверждают, что ядрышко и РНК-полимераза I – это многофункциональные сенсоры клеточного стресса. Уровень транскрипции генов РНК-полимеразой I намного выше уровня транскрипции РНК-полимеразой II, что делает ядрышко более чувствительным к разным типам стрессовых факторов. Ядрышко-специфичный ответ на повреждение ДНК может запускать различные пути клеточного ответа на стресс. Активация ATR и ATM может приводить к активации p53 и киназ, регулирующих клеточный цикл, в результате чего индуцируется арест пролиферации либо программы клеточной смерти. Интересно, что остановка транскрипции в рДНК сама по себе может активировать p53. Так, остановка транскрипции в ядрышке приводит к высвобождению рибосомных белков, которые подавляют MDM2, что приводит к активации p53. Структурные изменения ядрышка вместе с ингибированием РНК-полимеразы I могут индуцировать перераспределение белков ядрышка (B23, фибрилларин, нуклеолин), которые участвуют в клеточном ответе на стресс. Тем не менее, несмотря на долгую историю исследований ядрышка, мы все еще далеки от полного понимания его функциональной роли в качестве сенсора клеточного стресса.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 17-00-00098) и Российского научного фонда (грант № 19-74-10009).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Pfeiffer P., Goedecke W., Obe G. (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 15, 289–302.
García-Muse T., Aguilera A. (2016) Transcription-replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell. Biol. 17, 553–563.
Ganem N.J., Pellman D. (2012) Linking abnormal mitosis to the acquisition of DNA damage. J. Cell. Biol. 199, 871–881.
Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature. 362, 709–715.
Sonoda E., Morrison C., Yamashita Y.M., Takata M., Takeda S. (2001) Reverse genetic studies of homologous DNA recombination using the chicken B-lymphocyte line, DT40. Philos. Trans. R Soc. Lond. B Biol. Sci. 356, 111–117.
Vilenchik M.M., Knudson A.G. (2003) Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA. 100, 12871–12876.
Jackson S.P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature. 461, 1071–1078.
Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., Ørntoft T., Lukas J., Bartek J. (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870.
Harper J.W., Elledge S.J. (2007) The DNA damage response: ten years after. Mol. Cell. 28, 739–745.
Ceccaldi R., Rondinelli B., D’Andrea A.D. (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64.
Mladenov E., Magin S., Soni A., Iliakis G. (2016) DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 37–38, 51–64.
Clouaire T., Legube G. (2015) DNA double strand break repair pathway choice: a chromatin based decision? Nucleus. 6, 107–113.
Bzymek M., Lovett S.T. (2001) Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. USA. 98, 8319–8325.
Durkin S.G., Glover T.W. (2007) Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192.
Crosetto N., Mitra A., Silva M.J., Bienko M., Dojer N., Wang Q., Karaca E., Chiarle R., Skrzypczak M., Ginalski K., Pasero P., Rowicka M., Dikic I. (2013) Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods. 10, 361–365.
Lensing S.V., Marsico G., Hänsel-Hertsch R., Lam E.Y., Tannahill D., Balasubramanian S. (2016) DSB capture: in situ capture and sequencing of DNA breaks. Nat. Methods. 13, 855–857.
Mourad R., Ginalski K., Legube G., Cuvier O. (2018) Predicting double-strand DNA breaks using epigenome marks or DNA at kilobase resolution. Genome Biol. 19, 34.
Tchurikov N.A., Yudkin D.V., Gorbacheva M.A., Kulemzina A.I., Grischenko I.V., Fedoseeva D.M., Sosin D.V., Kravatsky Y.V., Kretova O.V. (2016) Hot spots of DNA double-strand breaks in human rDNA units are produced in vivo. Sci. Rep. 6, 25866.
Lindström M.S., Jurada D., Bursac S., Orsolic I., Bartek J., Volarevic S. (2018) Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene. 37, 2351–2366.
Santos-Pereira J.M., Aguilera A. (2015) R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597.
Aguilera A., García-Muse T. (2012) R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 46, 115–124.
Sollier J., Cimprich K.A. (2015) Breaking bad: R-loops and genome integrity. Trends Cell. Biol. 25, 514–522.
Chastain M., Zhou Q., Shiva O., Fadri-Moskwik M., Whitmore L., Jia P., Dai X., Huang C., Ye P., Chai W. (2016) Human CST facilitates genome-wide RAD51 recruitment to GC-rich repetitive sequences in response to replication stress. Cell Rep. 16, 1300–1314.
Dehé P.-M., Gaillard P.-H.L. (2017) Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell. Biol. 18, 315–330.
Rhodes D., Lipps H.J. (2015) G-quadruplexes and their regulatory roles in biology. Nucl. Acids Res. 43, 8627–8637.
Stults D.M., Killen M.W., Williamson E.P., Hourigan J.S., Vargas H.D., Arnold S.M., Moscow J.A., Pierce A.J. (2009) Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104.
Hallgren J., Pietrzak M., Rempala G., Nelson P.T., Hetman M. (2014) Neurodegeneration-associated instability of ribosomal DNA. Biochim. Biophys. Acta. 1842, 860–868.
Diesch J., Hannan R.D., Sanij E. (2014) Perturbations at the ribosomal genes loci are at the centre of cellular dysfunction and human disease. Cell. Biosci. 4, 43.
Korsholm L.M., Gál Z., Lin L., Quevedo O., Ahmad D.A., Dulina E., Luo Y., Bartek J., Larsen D.H. (2019) Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucl. Acids Res. 47, 8019–8035.
Mooser C., Symeonidou I.E., Leimbacher P.A., Ribeiro A., Shorrocks A.K., Jungmichel S., Larsen S.C., Knechtle K., Jasrotia A., Zurbriggen D., Jeanrenaud A., Leikauf C., Fink D., Nielsen M.L., Blackford A.N., Stucki M. (2020) Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat. Commun. 11, 123.
Velichko A.K., Petrova N.V., Luzhin A.V., Strelkova O.S., Ovsyannikova N., Kireev I.I., Petrova N.V., Razin S.V., Kantidze O.L. (2019) Hypoosmotic stress induces R loop formation in nucleoli and ATR/ATM-dependent silencing of nucleolar transcription. Nucl. Acids Res. 47, 6811–6825.
Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B., Grøfte M., Lukas C., Nielsen M.L., Smerdon S.J., Lukas J., Stucki M. (2014) The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell. Biol. 16, 792–803.
Sakai K., Ohta T., Minoshima S., Kudoh J., Wang Y., de Jong P.J., Shimizu N. (1995) Human ribosomal RNA gene cluster: identification of the proximal end containing a novel tandem repeat sequence. Genomics. 26, 521–526.
Gonzalez I.L., Sylvester J.E. (1995) Complete sequence of the 43-kb human ribosomal DNA repeat: analysis of the intergenic spacer. Genomics. 27, 320–328.
Stults D.M., Killen M.W., Pierce H.H., Pierce A.J. (2008) Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18.
Gibbons J.G., Branco A.T., Godinho S.A., Yu S., Lemos B. (2015) Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. USA. 112, 2485–2490.
McStay B., Grummt I. (2008) The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell. Dev. Biol. 24, 131–157.
Grummt I., Pikaard C.S. (2003) Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell. Biol. 4, 641–649.
Russell J., Zomerdijk J.C. (2005) RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 30, 87–96.
Miller O.L., Jr., Beatty B.R. (1969) Visualization of nucleolar genes. Science. 164, 955–957.
Olson M.O., Dundr M. (2005) The moving parts of the nucleolus. Histochem. Cell. Biol. 123, 203–216.
Maiser A., Dillinger S., Längst G., Schermelleh L., Leonhardt H., Németh A. (2020) Super-resolution in situ analysis of active ribosomal DNA chromatin organization in the nucleolus. Sci. Rep. 10, 7462.
Yao R.W., Xu G., Wang Y., Shan L., Luan P.F., Wang Y., Wu M., Yang L.Z., Xing Y.H., Yang L., Chen L.L. (2019) Nascent pre-rRNA sorting via phase separation drives the assembly of dense fibrillar components in the human nucleolus. Mol. Cell. 76, 767–783 e711.
Raska I., Shaw P.J., Cmarko D. (2006) Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell. Biol. 18, 325–334.
Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell. Biol. 18, 285–298.
Разин С.В., Гаврилов А.А. (2020) Роль разделения жидких фаз в компартментализации клеточного ядра и пространственной организации генома. Биохимия. 85, 755–762.
Mangan H., Gailín M., McStay B. (2017) Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 284, 3977–3985.
Brangwynne C.P., Mitchison T.J., Hyman A.A. (2011) Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA. 108, 4334–4339.
Feric M., Vaidya N., Harmon T.S., Mitrea D.M., Zhu L., Richardson T.M., Kriwacki R.W., Pappu R.V., Brang-wynne C.P. (2016) Coexisting liquid phases underlie nucleolar subcompartments. Cell. 165, 1686–1697.
Reynolds R.C., Montgomery P.O., Hughes B. (1964) Nucleolar “caps” produced by actynomycin D. Cancer Res. 24, 1269–1277.
Zhang Y., Lu H. (2009) Signaling to p53: ribosomal proteins find their way. Cancer Cell. 16, 369–377.
Velichko A.K., Petrova N.V., Razin S.V., Kantidze O.L. (2015) Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucl. Acids Res. 43, 6309–6320.
Zhao Z., Dammert M.A., Hoppe S., Bierhoff H., Grummt I. (2016) Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucl. Acids Res. 44, 8144–8152.
Kruhlak M., Crouch E.E., Orlov M., Montaño C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R. (2007) The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 447, 730–734.
Iarovaia O.V., Minina E.P., Sheval E.V., Onichtchouk D., Dokudovskaya S., Razin S.V., Vassetzky Y.S. (2019) Nucleolus: a central hub for nuclear functions. Trends Cell. Biol. 29, 647–659.
Boulon S., Westman B.J., Hutten S., Boisvert F.M., Lamond A.I. (2010) The nucleolus under stress. Mol. Cell. 40, 216–227.
Moore H.M., Bai B., Boisvert F.M., Latonen L., Rantanen V., Simpson J.C., Pepperkok R., Lamond A.I., Laiho M. (2011) Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell. Proteomics. 10, M111 009241.
Siebenwirth C., Greubel C., Drexler G.A., Reindl J., Walsh D.W.M., Schwarz B., Sammer M., Baur I., Pospiech H., Schmid T.E., Dollinger G., Friedl A.A. (2019) Local inhibition of rRNA transcription without nucleolar segregation after targeted ion irradiation of the nucleolus. J. Cell Sci. 132, jcs232181.
Harding S.M., Boiarsky J.A., Greenberg R.A. (2015) ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell. Rep. 13, 251–259.
van Sluis M., McStay B. (2015) A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 29, 1151–1163.
Warmerdam D.O., van den Berg J., Medema R.H. (2016) Breaks in the 45S rDNA lead to recombination-mediated loss of repeats. Cell Rep. 14, 2519–2527.
Ellison E.L., Vogt V.M. (1993) Interaction of the intron-encoded mobility endonuclease I-PpoI with its target site. Mol. Cell. Biol. 13, 7531–7539.
Monnat R.J., Jr., Hackmann A.F., Cantrell M.A. (1999) Generation of highly site-specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochem. Biophys. Res. Commun. 255, 88–93.
Berkovich E., Monnat R.J., Jr., Kastan M.B. (2007) Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell. Biol. 9, 683–690.
Marnef A., Finoux A.L., Arnould C., Guillou E., Daburon V., Rocher V., Mangeat T., Mangeot P.E., Ricci E.P., Legube G. (2019) A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 33, 1175–1190.
Clouaire T., Rocher V., Lashgari A., Arnould C., Aguirrebengoa M., Biernacka A., Skrzypczak M., Aymard F., Fongang B., Dojer N., Iacovoni J.S., Rowicka M., Ginalski K., Côté J., Legube G. (2018) Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell. 72, 250–262 e256.
Kantidze O.L., Velichko A.K., Luzhin A.V., Petrova N.V., Razin S.V. (2018) Synthetically lethal interactions of ATM, ATR, and DNA-PKcs. Trends Cancer. 4, 755–768.
Shiloh Y., Ziv Y. (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol. 14, 197–210.
Kuhn A., Gottlieb T.M., Jackson S.P., Grummt I. (1995) DNA-dependent protein kinase: a potent inhibitor of transcription by RNA polymerase I. Genes Dev. 9, 193–203.
Calkins A.S., Iglehart J.D., Lazaro J.-B. (2013) DNA damage-induced inhibition of rRNA synthesis by DNA-PK and PARP-1. Nucl. Acids Res. 41, 7378–7386.
Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S.P., Elledge S.J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 316, 1160–1166.
Larsen D.H., Stucki M. (2016) Nucleolar responses to DNA double-strand breaks. Nucl. Acids Res. 44, 538–544.
Trainor P.A., Dixon J., Dixon M.J. (2009) Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur. J. Hum. Genet. 17, 275–283.
Ciccia A., Huang J.-W., Izhar L., Sowa M.E., Harper J.W., Elledge S.J. (2014) Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA. 111, 18631–18636.
Sokka M., Rilla K., Miinalainen I., Pospiech H., Syväoja J.E. (2015) High levels of TopBP1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation. Nucl. Acids Res. 43, 4975–4989.
Pefani D.E., Tognoli M.L., Pirincci Ercan D., Gorgoulis V., O’Neill E. (2018) MST2 kinase suppresses rDNA transcription in response to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 37, e98760.
Iannelli F., Galbiati A., Capozzo I., Nguyen Q., Magnuson B., Michelini F., D’Alessandro G., Cabrini M., Roncador M., Francia S., Crosetto N., Ljungman M., Carninci P., d’Adda di Fagagna F. (2017) A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 8, 15656.
Pankotai T., Bonhomme C., Chen D., Soutoglou E. (2012) DNA-PKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 19, 276–282.
Ayrapetov M.K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B.D. (2014) DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA. 111, 9169–9174.
Gong F., Clouaire T., Aguirrebengoa M., Legube G., Miller K.M. (2017) Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell. Biol. 216, 1959–1974.
Kruhlak M.J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W.G., McNally J.G., Bazett-Jones D.P., Nussenzweig A. (2006) Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell. Biol. 172, 823–834.
Verkuijl S.A., Rots M.G. (2019) The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr. Opin. Biotechnol. 55, 68–73.
Falahati H., Pelham-Webb B., Blythe S., Wieschaus E. (2016) Nucleation by rRNA dictates the precision of nucleolus assembly. Curr. Biol. 26, 277–285.
Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374.
Caridi C.P., D’Agostino C., Ryu T., Zapotoczny G., Delabaere L., Li X., Khodaverdian V.Y., Amaral N., Lin E., Rau A.R., Chiolo I. (2018) Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature. 559, 54–60.
Lottersberger F., Karssemeijer R.A., Dimitrova N., de Lange T. (2015) 53BP1 and the LIN–C complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 163, 880–893.
van Sluis M., McStay B. (2019) Nucleolar DNA double-strand break responses underpinning rDNA genomic stability. Trends Genet. 35, 743–753.
Blackford A.N., Jackson S.P. (2017) ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell. 66, 801–817.
Britton S., Coates J., Jackson S.P. (2013) A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell. Biol. 202, 579–595.
Brinkman E.K., Chen T., de Haas M., Holland H.A., Akhtar W., van Steensel B. (2018) Kinetics and fidelity of the repair of Cas9-induced double-strand DNA breaks. Mol. Cell. 70, 801–813 e806.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология