

Молекулярная биология, 2021, T. 55, № 4, стр. 697-704
Влияние адипонектина на продукцию аполипопротеинов А-1 и Е макрофагами человека
Д. А. Танянский a, *, А. С. Трулев a, Е. В. Агеева a, А. А. Никитин a, В. С. Шавва a, С. В. Орлов a, b
a Институт экспериментальной медицины
197376 Санкт-Петербург, Россия
b Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
* E-mail: dmitry.athero@gmail.com
Поступила в редакцию 09.12.2020
После доработки 15.02.2021
Принята к публикации 16.02.2021
Аннотация
Адипонектин – гормон жировой ткани, влияющий на энергетический обмен, метаболизм липопротеинов и модулирующий воспалительные реакции. Однако роль данного адипокина в атерогенезе остается малоизученной. В настоящей работе изучено влияние адипонектина на продукцию аполипопротеинов (апо) А-1 и Е макрофагоподобными клетками линии THP-1 двух сроков дифференцировки (3 и 5 дней). Адипонектин (10 мкг/мл) стимулировал экспрессию гена apoА-1 на уровне мРНК в 5-, но не в 3-дневных макрофагах. При этом уровень мРНК apoЕ в макрофагах под действием адипонектина не изменялся. Адипонектин подавлял экспрессию гена TNF в макрофагах и повышал экспрессию гена IL-10 в 5-дневных макрофагах. Адипонектин не изменял уровни апоА-1 и апоЕ в культуральных средах макрофагов обоих сроков дифференцировки и снижал уровень поверхностного апоА-1 в 5-дневных макрофагах. Инкубация 5-дневных макрофагов с антагонистом ядерного рецептора PPARα MK-886 либо с агонистом ядерных рецепторов LXR TO-901317 приводила к отмене стимулирующего эффекта адипонектина на экспрессию гена аpоА-1. Полученные данные свидетельствуют о том, что адипонектин не только влияет на воспалительные свойства макрофагов, но и модулирует продукцию апоА-1 макрофагами, что, вероятно, можно рассматривать как один из механизмов его влияния на атерогенез.
ВВЕДЕНИЕ
Адипонектин – секретируемый клетками жировой ткани белок (адипокин), продукция которого снижена у пациентов с ожирением и метаболическим синдромом [1–3]. Концентрация данного адипокина в плазме связана с риском развития атеросклероза, его клиническими проявлениями и осложнениями [4, 5]. Адипонектин замедляет развитие экспериментального атеросклероза у животных, вероятно, как путем воздействия на формирование метаболических факторов риска, так и на процессы в сосудистой стенке [6, 7]. Так, адипонектин повышает чувствительность клеток к инсулину [8–10] и уменьшает концентрацию триглицеридов в плазме [6, 11]. Последнее может быть обусловлено влиянием данного адипокина на метаболизм жирных кислот [6], катаболизм липопротеинов [11], а также на продукцию аполипопротеинов (апо) А-1 и В гепатоцитами [11–13]. По данным in vivo и in vitro адипонектин подавляет адгезию моноцитов на сосудистом эндотелии [14, 15], модулирует дифференцировку макрофагов (МФ) и продукцию ими цитокинов [16–19], подавляет захват МФ модифицированных липопротеинов низкой плотности (ЛПНП) [20–22] и усиливает отток из них холестерина [22, 23]. Адипонектин реализует свои эффекты посредством взаимодействия со своими рецепторами типа 1 и 2 (AdipoR1/2), которые далее передают сигнал на киназу АМРК (АМР-активируемая протеинкиназа) и ядерные рецепторы активаторов пролиферации пероксисом (PPARα) и LXR (печеночные Х-рецепторы) [24, 25].
Ранее было показано, что МФ человека синтезируют и секретируют апоЕ [26] и апоА-1 [27, 28]. Сверхэкспрессия генов этих апопротеинов в МФ мышей с атеросклерозом приводила к уменьшению площади атеросклеротических поражений [29, 30]. Влияние апоЕ и апоА-1 на атерогенез связывают с их участием в обратном транспорте холестерина из МФ в печень [27, 31] и c иммуномодулирующим действием [27, 32, 33]. В частности, оба аполипопротеина подавляют стимулированную липополисахаридом выработку провоспалительных цитокинов in vitro [27, 32, 33]. Синтез обоих апопротеинов в МФ индуцируется накоплением в клетках холестерина и его окисленных форм посредством активации LXR [26, 28]. Экспрессия генов apoA-1 и apoE в МФ регулируется также активностью рецепторов PPARα и PPARγ [26, 28, 34]. Кроме того, продукция апоА-1 и апоЕ МФ стимулируется провоспалительным цитокином TNF (фактор некроза опухоли) [27, 28, 35].
В данном исследовании мы проверили гипотезу о влиянии адипонектина на продукцию апоА-1 и апоЕ МФ. Выяснение этого вопроса позволит расширить представления о механизмах влияния адипонектина на процессы атерогенеза.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Культивирование макрофагов. Клетки моноцитарно-макрофагальной линии человека THP-1 (ЦКП “Коллекция клеточных культур позвоночных” Института цитологии РАН) культивировали в СО2-инкубаторе на полной среде RPMI-1640 с добавлением 4 мМ глутамина, 0.1 мг/мл гентамицина (“Биолот”, Россия) и 10% фетальной сыворотки крупного рогатого скота (FCS; “HyClone”, США). Клетки высевали на 96-луночные культуральные планшеты (“Sarstedt”, Германия) по 2 × 105 клеток на лунку. Дифференцировку клеток в макрофагоподобные клетки (МФ THP-1) вызывали, добавляя к клеткам 50 нг/мл форбол-12-миристат-13-ацетата (PMA, “Sigma”, США). Через 1 сут клетки отмывали от PMA и далее (спустя одни либо трое суток) инкубировали в течение 24 ч на среде без FCS c 10 мкг/мл адипонектина (“Biovendor”, Чехия; RD172023100-C) либо c 10 мкг/мл бычьего сывороточного альбумина (БСА; “Sigma”, 4503), либо c добавлением такого же объема фосфатно-солевого буфера (PBS, “Биолот”). Таким образом, общее время дифференцировки МФ, включая этапы добавления PMA, выдерживания клеток без PMA и инкубации с адипонектином и другими агентами, составляло 3 или 5 сут. В некоторых экспериментах за 1 ч до добавления адипонектина и в течение всего срока инкубации к клеткам дополнительно добавляли: 10 мкМ MK-886 (антагонист PPARα; “Sigma”, C7081) либо 10 мкМ TO-901317 (агонист LXR; “Sigma”, T2320), либо растворитель указанных агентов диметилсульфоксид (DMSO; “Биолот”) в тех же концентрациях (≤0.5%). После окончания срока инкубации клетки собирали для выделения РНК либо для проведения проточной цитофлуориметрии, супернатанты отбирали для проведения иммуноферментного анализа (ИФА).
Мононуклеары периферической крови человека выделяли из цельной донорской крови центрифугированием в градиенте фиколла (плотность 1.077, “Биолот”). Клетки дважды промывали раствором Хенкса (“Биолот”), затем ресуспендировали в полной среде RPMI-1640 и высевали на 96-луночные планшеты (1.5 × 105 клеток на лунку) [27]. Через 2 ч мононуклеары дважды отмывали от неадгезированных клеток раствором Хенкса, после чего клетки переводили в свежую культуральную среду с 10% FCS. Среду обновляли каждые 2–3 дня и на 7-е сутки дифференцировки к клеткам добавляли в условиях 2% FCS 10 мкг/мл адипонектина, либо БСА (10 мкг/мл). Клетки инкубировали в течение 1 сут и снимали для проведения проточной цитофлуориметрии реактивом “Accutase” (“ThermoFisher”, США).
Выделение РНК, обратная транскрипция и ПЦР в реальном времени (ОТ-ПЦР). Суммарную РНК выделяли из клеток при помощи реагента “Евроген” (Россия) согласно инструкции производителя. Концентрацию и степень очистки суммарной РНК в полученных препаратах определяли спектрофотометрически, целостность РНК определяли с помощью электрофореза в агарозном геле. Обратную транскрипцию проводили с использованием 1 мкг суммарной РНК, олиго-dT-праймеров, обратных праймеров к гену apoА-1 (“Синтол”, Россия) и набора реактивов M-MLV (“Promega”, США) по протоколу производителя. ПЦР в реальном времени проводили при помощи амплификатора CFX-96 (“Bio-Rad”, США) с использованием интеркалирующего красителя SYBR Green и наборов фирмы “Синтол” [27]. Праймеры подбирали с помощью программы Primer3 [36]. Последовательности праймеров к генам apoА-1, apoЕ, TNF, IL-10 и к референсным генам RPLP0 и PPIA описаны ранее [27, 37–39]. Последовательности праймеров к гену AdipoR1: 5′-CCTGGAAAATTTGACATATGGTTC-3′ и 5′-AGGCTCAGAGAAGGGTGTCA-3′ и к гену AdipoR2: 5′-CGGGGAGTAAGAGCAGGAG-3′ и 5′-GGGCAGCTCCTGTGATGTAG-3′. Экспрессию искомых генов нормиро-вали на среднее геометрическое значение экспрессии генов домашнего хозяйства RPLP0 и PPIA. Относительные значения количества мРНК искомого гена (в процентах от значений в контрольном образце) рассчитывали методом ΔΔСt, где Сt – пороговый цикл кривой флуоресценции.
Иммуноферментный анализ (ИФА). Концентрацию апоА-1 в культуральных средах МФ определяли при помощи сэндвич-варианта ИФА с использованием нижних поликлональных козьих антител к апоА-1 человека (“R&D”, США; AF-3664) в разведении 1 : 100 и конъюгатов антител к апоА-1 с пероксидазой хрена (“ИМТЕК”, Россия; P-GAH Laa) в разведении 1 : 250. Концентрацию апоЕ в культуральных средах определяли при помощи прямого варианта ИФА с использованием моноклональных антител мыши к апоЕ (“ИМТЕК”; MGH Lee) в разведении 1 : 100 и конъюгатов козьих антител к Ig мыши с пероксидазой хрена (“Abcam”, Великобритания; ab6006) в разведении 1 : 500. Зависимость значений А450 от lg концентраций определяемых веществ имела линейный характер, коэффициент линейной корреляции Пирсона составлял 0.992–0.999.
Проточная цитофлуориметрия. Поверхностный уровень апоА-1 макрофагов оценивали методом проточной цитофлуориметрии [40]. Использовали антитела мыши к апоА-1 (“Bio-Rad”, 0650-0050) в разведении 1 : 200 и вторичные козьи антитела против Ig мыши, меченные Alexa-647, (“Cell Signaling”, США; 4410) в разведении 1 : 500. Для контроля специфичности окраски клетки обрабатывали вторичными антителами без предварительной обработки антителами к апоА-1 (изотип–контроль). Анализ клеточных популяций проводили на проточном цитофлуориметре “Beckman Coulter’s Navios Flow” (США). Результаты анализировали с использованием программного обеспечения Kaluza Analysis (“Beckman Coulter”, США).
Статистический анализ. Результаты представляли как среднее значение ± ошибка среднего (SEM). Каждый опыт повторяли 3–4 раза. Cтатистическую значимость различий оценивали с помощью непарного t-критерия Стьюдента либо согласно критерию Даннета. Различия считали значимыми при p < 0.05. Статистический анализ выполняли с использованием программы Statistica 6.0 (StatSoft, США).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Экспрессия генов аполипопротеинов, цитокинов и рецепторов адипонектина в макрофагах в зависимости от времени дифференцировки
В ходе дифференцировки МФ экспрессия генов apoА-1 и apoЕ возрастала (рис. 1а), что сопровождалось увеличением секреции данных апобелков (рис. 2в, г). Как и ожидалось, дифференцировка моноцитов в МФ в течение 3 дней приводила к повышению содержания мРНК цитокинов TNF и IL-10. Однако более длительная дифференцировка клеток приводила к уменьшению экспрессии генов этих цитокинов (рис. 1а) и секреции TNF (10.4 ± 0.5 и 1.7 ± 0.2 пг/мкг суммарной РНК 5- и 3-дневных МФ соответственно; p < 0.0001). При этом экспрессия генов рецепторов к адипонектину AdipoR1 и AdipoR2 в МФ разных сроков дифференцировки не изменялась (рис. 1б).
Рис. 1.
Экспрессия генов apoА-1, apoЕ, TNF, IL-10 (а), AdipoR1 и AdipoR2 (б) в макрофагоподобных клетках линии THP-1 3 и 5 дней дифференцировки. Результаты количественной ОТ-ПЦР. Приведены средние значения ± SEM (n = 10–12). *p < 0.05, **p < 0.001 – статистически значимые различия между группами 3- и 5-дневных МФ согласно непарному t-критерию Стъюдента.
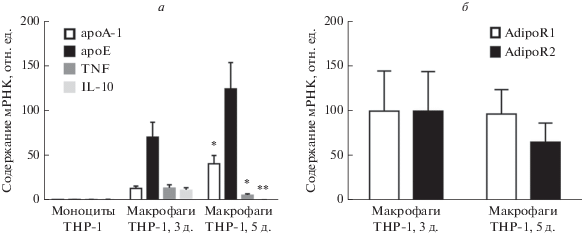
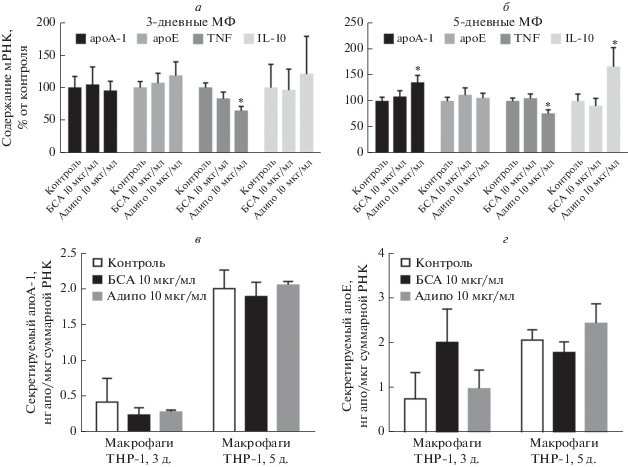
Рис. 2.
Влияние адипонектина на экспрессию генов apoА-1, apoЕ, TNF и IL-10 (а, б) в макрофагоподобных клетках линии THP-1, и содержание белков апо А-1 (в) и апо Е (г) в культуральных средах этих макрофагов. Приведены средние ± SEM (а, б – n = 16; в, г – n = 8–10). *p < 0.05 – статистически значимые отличия от контроля согласно непарному t-критерию Стъюдента. Адипо – адипонектин.
Влияние адипонектина на продукцию апоА-1 и апоЕ макрофагами
Добавление к клеткам 10 мкг/мл адипонектина, но не БСА в той же концентрации, приводило к повышению экспрессии гена apoА-1 на уровне мРНК в 5-дневных МФ, но не в МФ, дифференцированных в течение 3 дней (рис. 2а, б). При этом адипонектин не влиял на уровень экспрессии гена другого апобелка – apoЕ в МФ. Кроме того, адипонектин модулировал экспрессию генов цитокинов – подавлял экспрессию гена TNF в МФ обоих сроков дифференцировки и повышал экспрессию гена IL-10 в 5-дневных МФ (рис. 2а, б). Содержание апоА-1 и апоЕ в культуральных средах МФ под действием адипонектина не изменялось (рис. 2в, г).
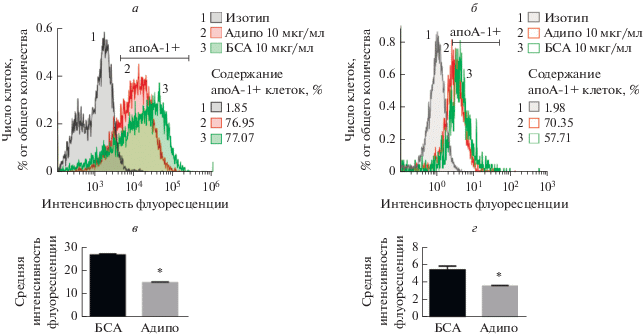
По данным проточной цитофлуориметрии адипонектин уменьшал уровень белка апоА-1 на поверхности 5-суточных МФ (рис. 3а). Эти данные подтверждены на первичных МФ, дифференцированных из мононуклеаров крови человека (рис. 3б). Таким образом, адипонектин модулирует продукцию апоА-1 МФ: повышает экспрессию гена apoА-1 на уровне РНК, но при этом уменьшает количество его белкового продукта на поверхности клеток.
Рис. 3.
Влияние адипонектина на уровень поверхностного апоА-1 в макрофагоподобных клетках линии THP-1, дифференцированных в течение 5 дней (а), и в макрофагах, дифференцированных из мононуклеаров периферической крови человека в течение 7 дней (б). Представлены диаграммы распределения уровня поверхностного апоА-1, средние значения ± SEM и % апоА-1-содержащих клеток. *p < 0.0001 – статистически значимые отличия от контроля (БСА) согласно непарному t-критерию Стъюдента.
Роль ядерных рецепторов PPARα и LXR в модуляции экспрессии гена apoА-1 под действием адипонектина
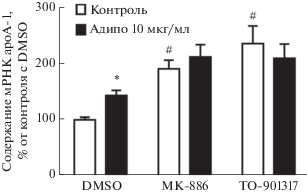
Экспрессия гена apoА-1 в МФ находится под контролем ядерных рецепторов: PPARα (лигандзависимый репрессор) и LXR (лигандзависимый активатор) [28]. В связи с этим для выяснения механизма влияния адипонектина на экспрессию гена apoА-1 в МФ мы прединкубировали клетки с MK-886 – антагонистом ядерного рецептора PPARα, и TO-901317 – агонистом ядерных рецепторов LXR. Как и ожидалось, добавление к клеткам данных агентов приводило к стимуляции экспрессии гена apoА-1 (рис. 4). Прединкубация клеток с MK-886 приводила к значительному снижению стимуляции экспрессии гена apoА-1 адипонектином, в то время как TO-901317 полностью отменял эффект адипонектина (рис. 4). Полученные данные свидетельствуют о том, что адипонектин повышает синтез мРНК apoA-1 при участии PPARα и LXR.
Рис. 4.
Участие ядерных рецепторов PPARα и LXR в стимуляции экспрессии гена apoА-1 адипонектином в макрофагоподобных клетках линии THP-1, дифференцированных в течение 5 дней. За 1 ч до начала инкубации с адипонектином к клеткам добавляли либо 10 мкМ MK-886 (ингибитор PPARα), либо 10 мкМ TO-901317 (активатор LXR), либо растворитель данных агентов DMSO. Приведены средние значения ± SEM (n = 12). *p < 0.01, #p < 0.0001– статистически значимые отличия от контроля с DMSO согласно критерию Даннета.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Макрофаги – ключевые клетки, участвующие в атерогенезе. Атеросклероз представляет собой хронический воспалительный процесс, вызванный очаговым накоплением ЛПНП в интиме крупных артерий [41]. Адипонектин не только влияет на привлечение мононуклеарных лейкоцитов в очаг воспаления [14, 15], но и воздействует на функцию МФ: модулирует их дифференцировку и продукцию провоспалительных цитокинов [16–19], подавляет захват модифицированных ЛПНП [20–22], усиливает отток холестерина из МФ [22, 23]. В настоящей работе впервые показано, что адипонектин модулирует экспрессию гена apoА-1 в МФ, не влияя при этом на экспрессию гена apoЕ в этих клетках. Интересно, что адипонектин уменьшает экспрессию гена TNF в МФ обоих сроков дифференцировки и в то же время повышает экспрессию генов apoА-1 и IL-10 только в МФ, дифференцированных в течение 5 сут. Обусловлены ли указанные различия в действии адипонектина разной степенью дифференцированности МФ или разницей во времени покоя МФ после стимуляции PMA, остается невыясненным. По данным Starr и соавт. [42], МФ THP-1, дифференцированные в течение 5 дней, обладают более высокой экспрессией макрофагальных маркеров CD11b и CD14 на клеточной мембране, чем 2-дневные МФ, но при этом 5-дневные МФ остаются менее активированными, ввиду длительного периода покоя без PMA [43]. Последнее может объяснять снижение выработки цитокинов 5-суточными МФ по сравнению с МФ, дифференцированными в течение 3 дней (рис. 1а).
Стимуляция экспрессии гена apoA-1 адипонектином не приводила к увеличению уровня белка апоА-1 в культуральной среде МФ, что может быть обусловлено тем, что основная часть продуцируемого МФ апоА-1 остается на поверхности мембраны, будучи связанной с кассетным транспортером ABCA-1 и липидными рафтами [27]. Снижение уровня апоА-1 на поверхности МФ под действием адипонектина свидетельствует, вероятно, об активации сигналов апоА-1, что приводит к ускорению деградации апоА-1. Показано, что, помимо участия в обратном транспорте холестерина, апоА-1 модулирует продукцию цитокинов в МФ, активируя сигнальный механизм ABCA-1/JAK2/STAT3 [32]. Ранее было установлено, что адипонектин повышает экспрессию гена ABCA-1 в МФ и активирует обратный транспорт холестерина [22, 23]. В какой мере продукция апоА-1 МФ опосредует влияние адипонектина на обратный транспорт холестерина и участие МФ в воспалительных реакциях, покажут будущие исследования.
Экспрессия гена apoА-1 в МФ, как и в гепатоцитах, зависит от взаимодействия ядерных рецепторов PPARα и LXR с регуляторной областью гена apoА-1 [28, 37]. При этом в гепатоцитах PPARα действует как лигандзависимый активатор транскрипции гена apoА-1, а LXR вызывает репрессию этого гена [37]. В МФ наблюдается зеркально противоположная картина: PPARα действует как лигандзависимый репрессор, а LXR – как активатор транскрипции гена apoА-1 [28]. Нами показано участие обоих ядерных рецепторов в регуляции транскрипции гена apoА-1 адипонектином (рис. 4). Адипонектин передает сигнал на указанные факторы транскрипции благодаря взаимодействию с рецепторами AdipoR1/2 и путем активации AMPK-киназы [24, 25].
Таким образом, показано, что адипонектин стимулирует экспрессию гена apoА-1 в МФ на уровне мРНК, но при этом уменьшает содержание белка апоА-1 на мембране этих клеток. На продукцию другого аполипопротеина, апоЕ, в МФ адипонектин не влияет. В активации транскрипции гена apoА-1 под действием адипонектина принимают участие ядерные рецепторы PPARα и LXR. Влияние адипонектина на продукцию МФ апоА-1 можно рассматривать как один из путей его воздействия на атерогенез.
Авторы приносят благодарность И.В. Кудрявцеву и М.К. Серебряковой (Отдел иммунологии ИЭМ) за помощь в проведении проточной цитофлуориметрии и А.Д. Денисенко (Отдел биохимии ИЭМ) за ценные критические замечания.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 15-04-07918).
В настоящей работе не использовали людей или животных в качестве объектов исследования.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Arita Y., Kihara S., Ouchi N., Takahashi M., Maeda K., Miyagawa J., Hotta K., Shimomura I., Nakamura T., Miyaoka K., Kuriyama H., Nishida M., Yamashita S., Okubo K., Matsubara K., Muraguchi M., Ohmoto Y., Funahashi T., Matsuzawa Y. (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 257, 79–83.
Танянский Д.А., Фирова Э.М., Шатилина Л.В., Денисенко А.Д. (2008) Адипонектин: снижение содержания при метаболическом синдроме и независимая связь с гипертриглицеридемией. Кардиология. 12, 20–25.
Разгильдина Н.Д., Бровин Д.Л., Побожева И.А., Пантелеева А.А., Мирошникова В.В., Беляева О.Д., Баранова Е.И., Полякова Е.А., Беркович О.А., Пчелина С.Н. (2018) Экспрессия гена ADIPOQ в подкожной и интраабдоминальной жировой ткани у женщин с различной степенью ожирения. Цитология. 60, 531–535.
Wang Y., Zheng A., Yan Y., Song F., Kong Q., Qin S., Zhang D. (2014) Association between HMW adiponectin, HMW-total adiponectin ratio and early-onset coronary artery disease in Chinese population. Atherosclerosis. 235, 392–397.
Wu Z.J., Cheng Y.J., Gu W.J., Aung L.H. (2014) Adiponectin is associated with increased mortality in patients with already established cardiovascular disease: a systematic review and meta-analysis. Metabolism. 63, 1157–1166.
Yamauchi T., Kamon J., Waki H., Imai Y., Shimozawa N., Hioki K., Uchida S., Ito Y., Takakuwa K., Matsui J., Takata M., Eto K., Terauchi Y., Komeda K., Tsunoda M., Murakami K., Ohnishi Y., Naitoh T., Yamamura K., Ueyama Y., Froguel P., Kimura S., Nagai R., Kadowaki T. (2003) Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J. Biol. Chem. 278, 2461–2468.
Wang X., Chen Q., Pu H., Wei Q., Duan M., Zhang C., Jiang T., Shou X., Zhang J., Yang Y. (2016) Adiponectin improves NF-κB-mediated inflammation and abates atherosclerosis progression in apolipoprotein E‑deficient mice. Lipids Health Dis. 15, 33.
Bruce C.R., Mertz V.A., Heigenhauser G.J., Dyck D.J. (2005) The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes. 54, 3154–3160.
Fu Y., Luo N., Klein R.L., Garvey W.T. (2005) Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J. Lipid Res. 46, 1369–1379.
Miller R.A., Chu Q., Le Lay J., Scherer P.E., Ahima R.S., Kaestner K.H., Foretz M., Viollet B., Birnbaum M.J. (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J. Clin. Invest. 121, 2518–2528.
Qiao L., Zou C., van der Westhuyzen D.R., Shao J. (2008) Adiponectin reduces plasma triglyceride by increasing VLDL triglyceride catabolism. Diabetes. 57, 1824–1833.
Matsuura F., Oku H., Koseki M., Sandoval J. C., Yuasa-Kawase M., Tsubakio-Yamamoto K., Masuda D., Maeda N., Tsujii K., Ishigami M., Nishida M., Hirano K., Kihara S., Hori M., Shimomura I., Yamashita S. (2007) Adiponectin accelerates reverse cholesterol transport by increasing high density lipoprotein assembly in the liver. Biochem. Biophys. Res. Commun. 358, 1091–1095.
Neumeier M., Sigruener A., Eggenhofer E., Weigert J., Weiss T.S., Schaeffler A., Schlitt H.J., Aslanidis C., Piso P., Langmann T., Schmitz G., Schölmerich J., Buechler C. (2007) High molecular weight adiponectin reduces apolipoprotein B and E release in human hepatocytes. Biochem. Biophys. Res. Commun. 352, 543–548.
Ouchi N., Kihara S., Arita Y., Maeda K., Kuriyama H., Okamoto Y., Hotta K., Nishida M., Takahashi M., Nakamura T., Yamashita S., Funahashi T., Matsuzawa Y. (1999) Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 100, 2473–2476.
Wang Y., Wang X., Lau W. B., Yuan Y., Booth D., Li J.J., Scalia R., Preston K., Gao E., Koch W., Ma X.L. (2014) Adiponectin inhibits tumor necrosis factor-α-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation Circ. Res. 114, 792–805.
Folco E.J., Rocha V.Z., López-Ilasaca M., Libby P. (2009) Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. J. Biol. Chem. 284, 25569–25575.
Ohashi K., Parker J.L., Ouchi N., Higuchi A., Vita J.A., Gokce N., Pedersen A.A., Kalthoff C., Tullin S., Sams A., Summer R., Walsh K. (2010) Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 285, 6153–6160.
Cheng X., Folco E.J., Shimizu K., Libby P. (2012) Adiponectin induces pro-inflammatory programs in human macrophages and CD4+ T cells. J. Biol. Chem. 287, 36896–36904.
van Stijn C.M., Kim J., Lusis A.J., Barish G.D., Tangirala R.K. (2015) Macrophage polarization phenotype regulates adiponectin receptor expression and adiponectin anti-inflammatory response. FASEB J. 29, 636–649.
Ouchi N., Kihara S., Arita Y., Nishida M., Matsuyama A., Okamoto Y., Ishigami M., Kuriyama H., Kishida K., Nishizawa H., Hotta K., Muraguchi M., Ohmoto Y., Yamashita S., Funahashi T., Matsuzawa Y. (2001) Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 103, 1057–1063.
Tian L., Luo N., Klein R.L., Chung B.H., Garvey W.T., Fu Y. (2009) Adiponectin reduces lipid accumulation in macrophage foam cells. Atherosclerosis. 202, 152–161.
Wang M., Wang D., Zhang Y., Wang X., Liu Y., Xia M. (2013) Adiponectin increases macrophages cholesterol efflux and suppresses foam cell formation in patients with type 2 diabetes mellitus. Atherosclerosis. 229, 62–70.
Tsubakio-Yamamoto K., Matsuura F., Koseki M., Oku H., Sandoval J.C., Inagaki M., Nakatani K., Nakaoka H., Kawase R., Yuasa-Kawase M., Masuda D., Ohama T., Maeda N., Nakagawa-Toyama Y., Ishigami M., Nishida M., Kihara S., Shimomura I., Yamashita S. (2008) Adiponectin prevents atherosclerosis by increasing cholesterol efflux from macrophages. Biochem. Biophys. Res. Commun. 375, 390–394.
Yamauchi T., Kamon J., Ito Y., Tsuchida A., Yokomizo T., Kita S., Sugiyama T., Miyagishi M., Hara K., Tsunoda M., Murakami K., Ohteki T., Uchida S., Takekawa S., Waki H., Tsuno N.H., Shibata Y., Terauchi Y., Froguel P., Tobe K., Koyasu S., Taira K., Kitamura T., Shimizu T., Nagai R., Kadowaki T. (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 423, 762–769.
Kemmerer M., Wittig I., Richter F., Brüne B., Namgaladze D. (2016) AMPK activates LXRα and ABCA1 expression in human macrophages. Int. J. Biochem. Cell Biol. 78, 1–9.
Laffitte B.A., Repa J.J., Joseph S.B., Wilpitz D.C., Kast H.R., Mangelsdorf D.J., Tontonoz P. (2001) LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA. 98, 507–512.
Mogilenko D.A., Orlov S.V., Trulioff A.S., Ivanov A.V., Nagumanov V.K., Kudriavtsev I.V., Shavva V.S., Ta-nyanskiy D.A., Perevozchikov A.P. (2012) Endogenous apolipoprotein A-I stabilizes ATP-binding cassette transporter A1 and modulates Toll-like receptor 4 signaling in human macrophages. FASEB J. 26, 2019–2030.
Shavva V.S., Mogilenko D.A., Nekrasova E.V., Tru-lioff A.S., Kudriavtsev I.V., Larionova E.E., Babina A.V., Dizhe E.B., Missyul B.V., Orlov S.V. (2018) Tumor necrosis factor α stimulates endogenous apolipoprotein A-I expression and secretion by human monocytes and macrophages: role of MAP-kinases, NF-κB, and nuclear receptors PPARα and LXRs. Mol. Cell. Biochem. 448, 211–223.
Major A.S., Dove D.E., Ishiguro H., Su Y.R., Brown A.M., Liu L., Carter K.J., Linton M.F., Fazio S. (2001) Increased cholesterol efflux in apolipoprotein AI (ApoAI)-producing macrophages as a mechanism for reduced atherosclerosis in ApoAI–/– mice. Arterioscler. Thromb. Vasc. Biol. 21, 1790–1795.
Fazio S., Babaev V.R., Burleigh M.E., Major A.S., Hasty A.H., Linton M.F. (2002) Physiological expression of macrophage apoE in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J. Lipid Res. 43, 1602–1609.
Kockx M., Jessup W., Kritharides L. (2008) Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 28, 1060–1067.
Yin K., Deng X., Mo Z.C., Zhao G.J., Jiang J., Cui L.B., Tan C.Z., We G.B., Fu Y., Tang C.K. (2011) Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. J. Biol. Chem. 286, 13834–13845.
Zhu Y., Kodvawala A., Hui D.Y. (2010) Apolipoprotein E inhibits toll-like receptor (TLR)-3- and TLR-4-mediated macrophage activation through distinct mechanisms. Biochem. J. 428, 47–54.
Galetto R., Albajar M., Polanco J.I., Zakin M.M., Rodríguez-Rey J.C. (2001) Identification of a peroxisome-proliferator-activated-receptor response element in the apolipoprotein E gene control region. Biochemical J. 357, 521–527.
Duan H., Li Z., Mazzone T. (1995) Tumor necrosis factor-alpha modulates monocyte/macrophage apoprotein E gene expression. J. Clin. Invest. 96, 915–922.
Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G. (2012) Primer3 – new capabilities and interfaces. Nucl. Acids Res. 40, e115.
Mogilenko D.A., Dizhe E.B., Shavva V.S., Lapikov I.A., Orlov S.V., Perevozchikov A.P. (2009) Role of the nuclear receptors HNF4 alpha, PPAR alpha, and LXRs in the TNF alpha-mediated inhibition of human apolipoprotein A-I gene expression in HepG2 cells. Biochemistry. 48, 11950–11960.
Mogilenko D.A., Kudriavtsev I.V., Trulioff A.S., Shavva V.S., Dizhe E.B., Missyul B.V., Zhakhov A.V., Ischenko A.M., Perevozchikov A.P., Orlov S.V. (2012) Modified low density lipoprotein stimulates complement C3 expression and secretion via liver X receptor and Toll-like receptor 4 activation in human macrophages. J. Biol. Chem. 287, 5954–5968.
Shavva V.S., Bogomolova A.M., Nikitin A.A., Dizhe E.B., Tanyanskiy D.A., Efremov A.M., Oleinikova G.N., Perevozchikov A.P., Orlov S.V. (2017) Insulin-mediated downregulation of apolipoprotein A-I gene in human hepatoma cell line HepG2: the role of interaction between FOXO1 and LXRβ transcription factors. J. Cell. Biochem. 118, 382–396.
Некрасова Е.В., Данько Е.В., Шавва В.С., Диже Э.Б., Олейникова Г.Н., Орлов С.В. (2020) Действие инсулина на экспрессию гена аполипопротеина А-I в макрофагах человека. Мед. Акад. Журн. 20(1), 65–74.
Kasikara C., Doran A.C., Cai B., Tabas I. (2018) The role of non-resolving inflammation in atherosclerosis. J. Clin. Invest. 128, 2713–2723.
Starr T., Bauler T.J., Malik-Kale P., Steele-Mortimer O. (2018) The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella typhimurium. PLoS One. 13, e0193601.
Lund M.E., To J., O’Brien B.A., Donnelly S. (2016) The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus. J. Immunol. Meth. 430, 64–70.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология