

Молекулярная биология, 2022, T. 56, № 1, стр. 135-146
Изменение экспрессии селенопротеинов эндоплазматического ретикулума и маркеров апоптоза при воздействии селенита натрия и дитиотреитола на клетки линии MCF7 рака молочной железы
В. Н. Мальцева a, *, М. В. Голтяев a, С. В. Новоселов a, Е. Г. Варламова a
a Институт биофизики клетки Российской академии наук “Федеральный исследовательский
центр “Пущинский научный центр биологических исследований Российской академии наук”
142290 Пущино, Московская обл., Россия
* E-mail: mvn3@mail.ru
Поступила в редакцию 09.02.2021
После доработки 02.05.2021
Принята к публикации 30.05.2021
- EDN: BKCRNS
- DOI: 10.31857/S0026898422010062
Аннотация
Изучено влияние индукторов стресса эндоплазматического ретикулума (ЭПР) – дитиотреитола (ДТТ) и селенита натрия (СН) – на экспрессию селенопротеинов ЭПР и маркеров апоптоза в клетках линии MCF7 рака молочной железы. Показано, что ДТТ в концентрациях 1 и 5 мМ не влиял на выживаемость клеток MCF7. Исходя из данных ПЦР в реальном времени и уровня маркеров стресса ЭПР мы предполагаем, что ДТТ в использованных концентрациях вызывает в клетках MCF7 развитие ЭПР-стресса по адаптационному пути, преимущественно с участием факторов транскрипции IRE1 и ATF6, а также селенопротеинов SELS, SELK, SELT, SELM, SELN. Обработка клеток MCF7 0.01 мкМ СН приводила к снижению уровня мРНК всех исследуемых генов. При увеличении концентрации СН до 0.1 мкМ наблюдалось повышение уровня экспрессии ключевых генов ЭПР-стресса и маркеров апоптоза: CHOP, GADD34, PUMA, BIM, ATF4, sXBP, uXBP, AKT1, BAX, BAK. Согласно полученным результатам, 0.1 мкМ СН инициирует развитие процесса, известного как “ответ на неправильную укладку белка” (unfolded protein response, UPR) по проапоптическому пути с участием PERK, а также по альтернативному пути IRE1. СН в концентрации 1 мкМ также вызывает повышение уровня мРНК маркеров апоптоза, усиливает экспрессию сплайсированной формы XBP1, а также сопровождается значительным снижением выживаемости опухолевых клеток. Мы предполагаем, что СН (1 мкМ) инициирует гибель клеток MCF7 по пути апоптоза. Показано, что в зависимости от природы и концентрации индуктора ЭПР-стресса в клетках активируются как адаптивные, так и проапоптотические пути передачи сигналов UPR.
ВВЕДЕНИЕ
Рак молочный железы – наиболее частая форма рака у женщин и одна из основных причин смертности от онкологических заболеваний [1]. Однако эффективность препаратов, применяемых в химиотерапии рака молочной железы, ограничена развитием лекарственной резистентности раковых клеток [2, 3].
Один из возможных процессов, приводящих к апоптотической гибели клеток, – стресс эндоплазматического ретикулума (ЭПР-стресс) – процесс, в развитии которого огромное значение имеет нарушение фолдинга белков, приводящее к развитию гомеостатического механизма, известного как ответ на неправильную упаковку белка (UPR). UPR – это сложный комплекс взаимосвязанных сигнальных путей, имеющих как адаптационную, так и проапоптотическую направленность, но объединенных общим пусковым механизмом, который представлен триадой трансмембранных белков – PERK, IRE1 и ATF6. В физиологических условиях регуляторные домены этих триггерных белков находятся в просвете ЭПР и связаны с иммуноглобулинсвязывающим белком BiP (BiP/GRP78). С увеличением нагрузки на ЭПР содержание свободных шаперонов в просвете уменьшается, в результате чего происходит диссоциация BiP от этих белков, активация PERK, IRE1 и ATF6 и запуск каскада сигнальных путей UPR.
ЭПР-стресс может индуцироваться как физическими факторами и соединениями различной химической природы, так и нарушениями метаболизма клетки [5–7]. Среди хорошо известных индукторов ЭПР-стресса особое место занимают селенсодержащие соединения. Одно из распространенных соединений селена – селенит натрия (СН) – широко используется в качестве потенциального противоопухолевого cредства. СН оказывает цитотоксическое, антипролиферативное и проапоптотическое действие на раковые клетки разной этиологии [8–16]. Основными механизмами цитотоксического действия СН считаются продукция активных форм кислорода (АФК), дисфункция митохондрий с высвобождением цитохрома с, активация каспаз и, как следствие, окислительный стресс [17], а также образование дисульфидных мостиков, что приводит к модификациям структуры белков и изменению их конформации. Накопление неправильно свернутых белков в просвете ЭПР приводит к стрессу ЭПР с последующей активацией проапоптотического сигнального пути UPR [18–22]. Показано, что характер воздействия СН на клетки зависит от концентрации: при низких концентрациях индуцируется остановка клеточного цикла в S-фазе, в то время как высокие концентрации СН способствуют задержке клеточного цикла в фазе G0/G1 [20]. Описан антиапоптотический эффект СН, обусловленный ингибированием окислительного стресса, индуцированного токоферилсукцинатом (α-TOS), и стимуляцией механизма аутофагии [21].
Одним из типичных индукторов ЭПР-стресса является низкомолекулярный серусодержащий восстанавливающий агент дитиотреитол (ДТТ). Цитотоксическое действие ДТТ обусловлено наличием тиольной группы, при окислении которой образуется пероксид водорода. Повышенный уровень пероксида водорода и других АФК, образующихся в реакции Фентона, вызывает, в конечном итоге, окислительный стресс и гибель клетки [22, 23]. Кроме того, ДТТ приводит к гибели клеток путем активации не связанного с митохондриями пути апоптоза с участием каспазы 3. Это подтверждается отсутствием деполяризации митохондриальной мембраны и раннего высвобождения цитохрома с из митохондрий в цитозоль, а также незначительной ролью каспазы 9 в индуцированном ДТТ апоптозе [22].
Известно, что селенопротеины вовлечены в процессы канцерогенеза, их рассматривают в качестве маркеров онкологических заболеваний [24–28]. Роль этих белков в регуляции стресса ЭПР активно исследуется, при этом особый интерес представляют селенопротеины, локализующиеся преимущественно в ЭПР, такие как SELK, SELN, SELS, SELM, SELT, SEP15 и DIO2 [29–33]. Вклад этих селенопротеинов в гомеостатическую функцию ЭПР становится все более очевидным: они чувствительны к действию различных источников ЭПР-стресса, экспрессия мРНК и количественное содержание самих белков существенно зависят от природы индуктора и его концентрации. Подобные исследования проведены нами ранее на раковых клетках линий HT-1080 (фибросаркома) и DU 145 (карцинома простаты). Обнаружено, что уровни экспрессии семи селенопротеинов-резидентов ЭПР по-разному изменялись в зависимости от клеточной линии и концентрации индуктора ЭПР [34–36]. Однако характеристика некоторых белков in vivo ограничена, а их связь с метаболической дисфункцией только предполагается, что делает необходимым изучение функций этих белков, их роли в ЭПР-стрессе и патофизиологии метаболических заболеваний, в том числе в канцерогенезе. В настоящей работе изучены молекулярные механизмы развития ЭПР-стресса, роль селенопротеинов, ключевых белков UPR и апоптоза в индукции ЭПР-стресса посредством СН и ДТТ на примере аденокарциномы молочной железы человека.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реагенты. В работе использованы: набор для анализа пролиферации клеток МТТ (“Abcam”, Великобритания), антитела для Вестерн-блотинга, включая анти-Gapdh, анти-ATF4, анти-ATF6, анти-Xbp1 и вторичные антитела, конъюгированные с пероксидазой хрена (“Abcam”), СН (“Sigma Aldrich”, США), ДТТ (“Sigma Aldrich”). Cуммарную РНК выделяли с использованием реагента ExtractRNA (“Евроген”, Россия). Использовали набор для проведения обратной транскрипции MMLV RT kit (“Евроген”), 5-кратную реакционную смесь для ПЦР в режиме реального времени qPCRmix-HS SYBR (“Евроген”).
Культура клеток и реагенты. Работа проведена на клетках линии MCF7 (аденокарцинома молочной железы человека), приобретенных в АТСС (Американская коллекция типовых культур клеток, Manassas, США). Клетки культивировали в среде DMEM с добавлением 10%-ной фетальной сыворотки крупного рогатого скота.
Анализ пролиферации и жизнеспособности клеток. Количество жизнеспособных клеток MCF7 и скорость их пролиферации в каждом образце определяли с помощью МТТ-теста. Клетки высевали на 96-луночный планшет из расчета 5500 клеток/лунку и инкубировали в течение 24 ч с СН (0.01, 0.1 и 1 мкМ) или ДТТ (1, 5 мМ). Далее клетки инкубировали в течение 3 ч с реагентом МТТ (20 мкл, 5 г/л в фосфатном буфере, pH 7.4) при 37°C. После инкубации клетки обрабатывали растворителем МТТ в течение 15 мин при комнатной температуре. Оптическую плотность растворов измеряли при λ = 590 нм. Каждое измерение повторяли трижды.
Выделение РНК, обратная транскрипция и количественная ПЦР в режиме реального времени. Клетки MCF7 инкубировали в течение 24 ч с СН (0.01, 0.1 или 1 мкМ) или ДТТ (1 или 5 мМ), а затем выделяли суммарную РНК с использованием реагента ExtractRNA, содержащего раствор фенола и гуанидинизотиоцианата, в соответствии с инструкциями производителя (“Евроген”). Концентрацию РНК определяли спектрофотометрически (NanoDrop ND-1000, “NanoDrop Technologies”, США) при длине волны 260 нм. Качество РНК проверяли по сохранности 18S и 28S рРНК с помощью электрофореза в 1%-ном агарозном геле. Растворы суммарной РНК хранили при температуре –75°С.
кДНК первой цепи синтезировали из 2 мкг суммарной РНК, реакцию проводили в присутствии oligo(dT)-праймеров и обратной транскриптазы MMLV в соответствии с инструкциями производителя (“Евроген”). Полученную кДНК использовали в качестве матрицы для ПЦР в реальном времени. ПЦР в реальном времени проводили с помощью смеси qPCRmix-HS SYBR (“Евроген”) на амплификаторе “ДТлайт 5” (“ДНК-Технология”, Россия). Специфичность праймеров и чистоту продуктов ПЦР проверяли с помощью электрофореза в 2%-ном агарозном геле.
Ген GAPDH, кодирующий глицеральдегид-3-фосфат–дегидрогеназу, использовали в качестве референсного. Нуклеотидные последовательности праймеров к исследуемым генам и гену GAPDH синтезированы фирмой “Евроген” (табл. 1).
Таблица 1.
Нуклеотидные последовательности праймеров для ПЦР в реальном времени
| Ген | Прямой праймер 5' → 3' | Обратный праймер 5' → 3' |
|---|---|---|
| GAPDH | ACATCGCTCAGACACCATG | GCCAGTGAGCTTCCCGTT |
| SELT | TCTCCTAGTGGCGGCGTC | GTCTATATATTGGTTGAGGGAGG |
| SELM | AGCCTCCTGTTGCCTCCGC | AGGTCAGCGTGGTCCGAAG |
| SEP15 | TACGGTTGTTGTTGGCGAC | CAAATTGTGCTTCCTCCTGAC |
| SELK | TTTACATCTCGAACGGACAAG | CAGCCTTCCACTTCTTGATG |
| SELS | TGGGACAGCATGCAAGAAG | GCGTCCAGGTCTCCAGG |
| SELN | TGATCTGCCTGCCCAATG | TCAGGAACTGCATGTAGGTGG |
| DIO2 | AGCTTCCTCCTCGATGCC | AAAGGAGGTCAAGTGGCTG |
| CHOP | GCTCTGATTGACCGAATGG | TCTGGGAAAGGTGGGTAGTG |
| GADD34 | CTCCGAGAAGGTCACTGTCC | GACGAGCGGGAAGGTGTGG |
| PUMA | CAGATATGCGCCCAGAGAT | CCATTCGTGGGTGGTCTTC |
| BIM | GGACGACCTCAACGCACAGTACGAG | GTAAGGGCAGGAGTCCCA |
| ATF4 | GTGTTCTCTGTGGGTCTGCC | GACCCTTTTCTTCCCCCTTG |
| ATF6 | AACCCTAGTGTGAGCCCTGC | GTTCAGAGCACCCTGAAGA |
| XBPu | ACTCAGACTACGTGCACCTC | GTCAATACCGCCAGAATCC |
| XBPs | CTGAGTCCGCAGCGGTGCAGG | GGTCCAAGTTGTCCAGAATG |
| CAS3 | GCATTGAGACAGACAGTGGTG | AATAGAGTTCTTTTGTGAGCATG |
| CAS4 | CACGCCTGGCTCTCATCATA | TAGCAAATGCCCTCAGCG |
| MAP3K5 | AACACCTGAAGCTTAAGTCCC | TCAATGATAGCCTTCCACAGTG |
| MAPK-8 | AAAGGGAACACACAATAGAAGAG | GCTGCTGCTTCTAGACTG |
| BAX | GGGCTGGACATTGGACTTC | AACACAGTCCAAGGCAGCTG |
| BAK | GAGAGTGGCATCAATTGGGG | CAGCCACCCCTCTGTGCAATCCA |
Изменение уровня экспрессии мРНК исследуемых белков до и после обработки СН и ДТТ определяли по формуле ОУЭ = 2–ΔΔСt, где ΔΔСt – разница значений ΔСt для каждого гена до и после обработки клеток. Каждое значение ΔСt рассчитывали по формуле ΔСt = Сt (исследуемый ген) – Сt (референсный ген).
Вестерн-блотинг. Клетки MCF7 после инкубации СН или ДТТ трижды промывали ледяным фосфатно-солевым буфером, далее гомогенизировали с помощью лизирующего буфера, содержащего 100 мМ Трис-HCl pH 8.0, 0.15 мМ NaCl, 1 мМ EDTA, 1 мМ фенилметансульфонилфторид (PMSF). Лизаты очищали центрифугированием при 14000 g в течение 10 мин при 4°C. Белки разделяли методом электрофореза в 12%-ном полиакриламидном геле (ПААГ) и переносили на нитроцеллюлозную мембрану (“GE Healthcare”, Франция). Неспецифическое связывание антител предупреждали, блокируя мембраны 5%-ным обезжиренным молоком на шейкере в течение 1 ч при комнатной температуре. Затем мембраны инкубировали в течение ночи при 4°C с первичными антителами (все 1 : 200). После отмывки в TBST мембраны с образцами инкубировали в течение 1 ч с вторичными антителами, конъюгированными с пероксидазой хрена (1 : 5000). Связавшиеся антитела, конъюгированные с пероксидазой хрена, выявляли с использованием раствора DAB (0.05% DAB в TBS + 10 мкл 30% Н2О2). Денситометрический анализ проводили, используя программу Image J Software.
Статистический анализ. Для анализа данных, построения графиков и статистической обработки использовали пакет программ Microsoft Excel и GraphPadPrism 5. Данные нескольких (не менее трех) независимых экспериментов объединяли и анализировали с помощью t-теста Стьюдента. Значения p < 0.05 рассматривали как статистически значимые. Количество белка в образцах определяли по методу Лоури. Уровень экспрессии белка количественно оценивали с помощью программы ImageJ. На графиках приведены средние значения ± стандартное отклонение (SD), вычисленные по результатам трех независимых экспериментов.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Влияние СН и ДТТ на пролиферацию и жизнеспособность клеток MCF7
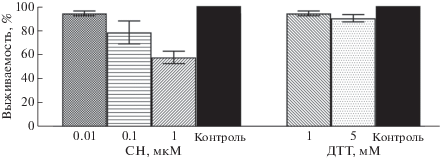
На первом этапе работы с помощью МТТ-анализа мы проверили цитотоксические свойства используемых нами индукторов стресса ЭПР на клетках MCF7. Показано, что обработка клеток ДТТ (1 и 5 мМ) и СН (0.01 мкМ) в течение 24 ч практически не повлияла на их пролиферацию и жизнеспособность. Оказалось, что СН постепенно снижает жизнеспособность клеток MCF7 с 94 до 62% (р < 0.05) по мере увеличения его концентрации с 0.01 до 1 мкМ соответственно (рис. 1).
Рис. 1.
Цитотоксический эффект СН и ДТТ на линию клеток MCF7. Клетки инкубировали с СН и ДТТ в разных концентрациях в течение 24 ч и определяли их жизнеспособность с помощью МТТ-теста. За 100% принимали значения, полученные для необработанных клеток. Стандартные отклонения рассчитывали по результатам не менее трех независимых экспериментов.
Влияние СН и ДТТ на экспрессию мРНК генов селенопротеинов-резидентов ЭПР
Методом ПЦР в реальном времени проанализированы изменения уровней экспрессии генов семи селенопротеинов, локализующихся преимущественно в ЭПР, в клетках MCF7, обработанных селенсодержащим (СН) и серусодержащим (ДТТ) индукторами ЭПР-стресса.
Результаты, представленные на рис. 2а, б, позволяют считать, что при обработке клеток СН в минимальной концентрации (0.01 мкМ) количество мРНК SELS, SELТ и SELN не меняется существенно по сравнению с интактными клетками. Значительно увеличилась экспрессия гена SELК (в 2 раза, р < 0.05), а уровни мРНК SELМ, SEР15 и DIO2, наоборот, уменьшились. Повышение концентрации СН до 0.1 мкМ привело к значительному росту уровня экспрессии генов всех исследуемых белков, что особенно заметно для DIO2 (в 3.2 раза, р < 0.05) и SELT (в 2.5 раза, р < 0.05). Высокая концентрация СН (1 мкМ) практически не повлияла на экспрессию генов SELT, SELK, SELS, SEP15, но значительно усилила экспрессию мРНК SELM (в 2.6 раза, р < 0.05), SELN (в 2.1 раза, р < < 0.05) и DIO2 (в 1.9 раза, р < 0.05).
Рис. 2.
Экспрессия мРНК селенопротеинов, резидентных в ЭПР – SELT, SELM, SEP15, SELК, SELS, SELN и DIO2 – в линии клеток MCF7 после обработки различными концентрациями СН (а) и ДТТ (б) в течение 24 ч. Данные получены с помощью ПЦР в реальном времени и представлены как среднее значение ± стандартное отклонение, вычисленные по результатам не менее трех независимых экспериментов. В качестве внутреннего контроля для нормирования использовали ген GAPDH. За единицу (черная пунктирная линия) принят уровень каждой из исследуемых мРНК в необработанных клетках.
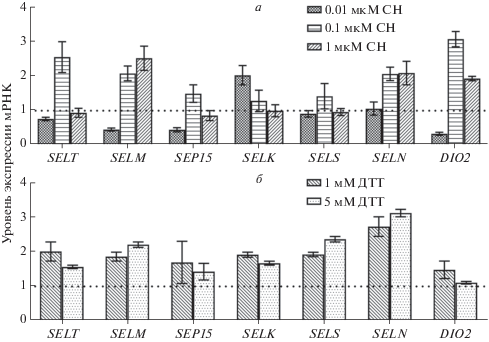
Воздействие ДТТ в концентрации 1 и 5 мМ на клетки в течение 24 ч повысило экспрессию генов почти всех исследуемых селенопротеинов по сравнению с интактными клетками, за исключением DIO2 и SEP15, уровень экспрессии которых не изменялся статистически значимо (рис. 2б).
Изменение экспрессии генов ключевых участников ЭПР-стресса при обработке клеток MCF7 СН и ДТТ в разных концентрациях
С помощью ПЦР в реальном времени выявлены значимые различия в уровнях мРНК исследуемых белков в клетках MCF7, обработанных в течение 24 ч СН или ДТТ в разных концентрациях (рис. 3а, б)
Рис. 3.
Экспрессия мРНК ключевых участников ЭПР-стресса и апоптоза в линии клеток MCF7, обработанных СН (а) и ДТТ (б) в течение 24 ч. Данные получены с помощью ПЦР в реальном времени и представлены как среднее значение ± стандартное отклонение, вычисленное по результатам не менее трех независимых экспериментов. Данные нормировали с использованием GAPDH в качестве внутреннего контроля. За единицу (черная пунктирная линия) принят уровень каждой из мРНК в необработанных клетках.
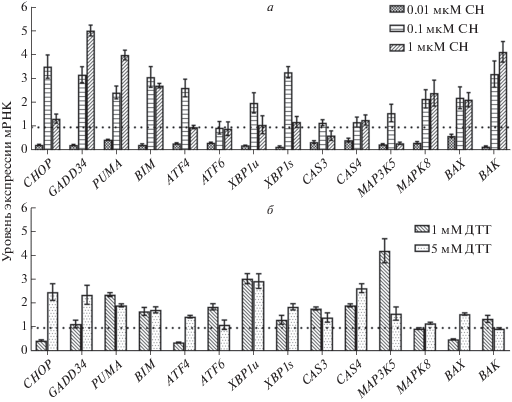
Интересно, что СН в низкой концентрации (0.01 мкМ) вызывает значительное снижение мРНК всех исследуемых генов, ассоциированных с ЭПР-стрессом (рис. 3а). Добавка СН в более высокой концентрации – 0.1 мкМ, приводит к инверсии описанного эффекта, уровень мРНК возрастает по сравнению с контрольными клетками. Выявлено значительное увеличение экспрессии следующих генов: CHOP (3.6 раза, р < 0.05), GADD34, BIM, XBSs, BAK и MAPK8 примерно в 3 раза (р < 0.05) (рис. 3а).
Увеличение концентрации СН до 1 мкМ приводит к максимальному росту уровня мРНК GADD34 – в 5 раз (р < 0.05), BAK и PUMA – в 4 раза (р < 0.05), а уровень мРНК MAP3K5, наоборот, значительно снизился. Стоит отметить, что при воздействии 1 мкМ СН на клетки в течение 24 ч экспрессия генов CHOP, ATF4, ATF6, XBP, CAS3, CAS4 практически не отличалась от значений в контроле (рис. 3а).
ДТТ стимулировал усиление экспрессии почти всех изученных генов в клетках MCF7: мРНК XBPu – в 3 раза при добавке 1 и 5 мМ, MAP3K5 – в 4 раза, при добавке 1 мМ ДТТ, CHOP, CAS4 и GADD34 – более чем в 2 раза в присутствии 5 мМ ДТТ. Исключение составили гены CHOP, BAX, ATF4 – уровень их экспрессии снизился почти в 2 раза при инкубации клеток с 1 мМ ДТТ в течение 24 ч по сравнению с интактными клетками (рис. 3б).
Изменение экспрессии на уровне белка ключевых маркеров ЭПР-стресса (ATF4, ATF6, XBP1s, XBP1u) при обработке клеток MCF7 СН и ДТ в разных концентрациях в течение 24 ч
Так как белки ATF4, ATF6, XBP1s, XBP1u являются ключевыми маркерами основных сигнальных путей UPR, мы изучили изменение их уровня при добавлении СН и ДТТ к клеткам MCF7.
Обнаружено повышение экспрессии мРНК и количественного содержания ATF4, ATF6 и XBP1s в клетках MCF7, обработанных 0.1 и 1 мкМ СН (рис. 4в). Уровни экспрессии генов ATF4 и ATF6 и сплайсированных форм XBP1 возрастали статистически значимо (примерно в 2 раза по сравнению с контролем), что подтверждено данными Вестерн-блотинга (рис. 4а). При воздействии СН во всех использованных концентрациях уровень экспрессии белка XBP1s значимо не менялся (рис. 4а).
Рис. 4.
Экспрессия генов ключевых маркеров ЭПР-стресса: ATF4, ATF6, XBP1s, XBP1u в клетках MCF7, обработанных СН и ДТТ в течение 24 ч. а – Вестерн-блот-анализ содержания белков ATF4, ATF6, XBP1s и XBP1u в клетках MCF7, обработанных СН (0.01, 0.1 или 1 мкМ). б – Вестерн-блот-анализ содержания белков ATF4, ATF6, XBP1s и XBP1u в клетках MCF7, обработанных 1 или 5 мМ ДТТ. в – Уровень экспрессии мРНК ATF4, ATF6, XBP1s и XBP1u относительно мРНК GAPDH (отн. ед.) в клетках MCF7, обработанных СН в разной концентрации. г – Уровень экспрессии мРНК ATF4, ATF6, XBP1s и XBP1u относительно мРНК GAPDH (отн. ед.) в клетках MCF7, обработанных ДТТ в разной концентрации. Представлены средние значения ± стандартное отклонение (SD), вычисленные по результатам трех независимых экспериментов.
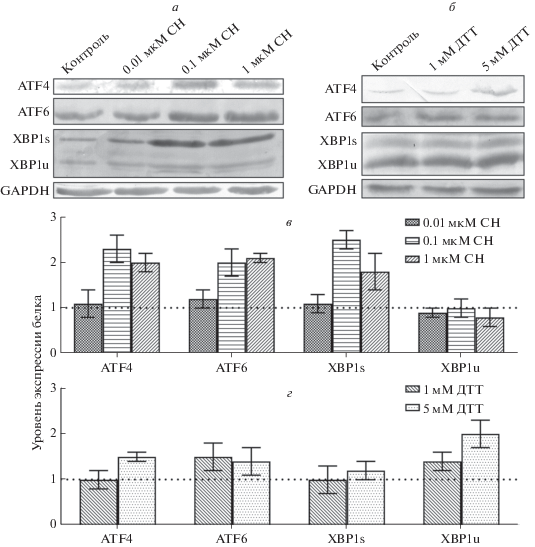
Влияние ДТТ (1 и 5 мM) на уровень экспрессии белков отличалось от воздействия СН на клетки MCF7. Уровень мРНК ATF4, ATF6, XBP1s при инкубации клеток с 1 и 5 мМ ДТТ практически не изменялся и не отличался от уровня в контроле (рис. 4г). Исключением стал белок XBP1u, уровень экспрессии которого увеличился почти в 2 раза при воздействии 5 мМ ДТТ. Таким образом, мы обнаружили противоположное действие СН (во всех концентрациях) и ДТТ на уровень экспрессии белка XBP1u – незначительное снижение в случае СН и повышение в случае ДТТ, что подтверждено результатами Вестерн-блотинга (рис. 4б).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Нарушение состояния и нормального функционирования клеток приводит к развитию стресса ЭПР, который инициирует UPR-ответ, направленный на восстановление гомеостаза ЭПР, т.е. в клетке начинают работать адаптивные сигнальные пути, способствующие выживанию клеток, в противном случае происходит переключение с адаптивных процессов на проапоптотические.
Эти процессы происходят как в нормальных, так и в онкотрансформированных клетках. Следовательно, блокирование адаптивного пути стресса ЭПР или стимуляция пути апоптоза можно рассматривать как потенциальную противоопухолевую стратегию. Раковые клетки активируют UPR для облегчения стрессового состояния ЭПР как стратегию выживания и прогрессии канцерогенеза. UPR играет важную роль в обеспечении устойчивости опухолевых клеток к химио- или лучевой терапии [37, 38]. По-видимому, существует возможность терапии рака, направленной на индукцию ЭПР-стресса, а также путем ингибирования компонентов, участвующих в адаптивном пути UPR и стимуляции апоптотического пути. В нашей работе мы использовали хорошо известные индукторы ЭПР-стресса – СН, одно из наиболее распространенных соединений селена, которое используют в качестве потенциального противоопухолевого препарата, и ДТТ. Оба индуктора ЭПР-стресса обладают высокой активностью, благодаря чему вызывают глобальные модификации, изменяют конформацию белков и, как следствие, приводят к накоплению неправильно свернутых белков в просвете ЭПР. Мы сравнили влияние СН и ДТТ на развитие ЭПР-стресса. Эти вещества вызывают противоположные модификации белков: так, СН образует в белках дисульфидные связи, а ДТТ, наоборот, восстанавливает их. Важно подобрать такие концентрации индукторов ЭПР, которые способствовали бы пролонгированному ЭРП-стрессу и гибели раковых клеток, не оказывая при этом цитотоксического эффекта на здоровые клетки, окружающие опухоль. В качестве раковых клеток мы выбрали клетки линии MCF7, эпителиоподобной клеточной линии, полученной из инвазивной аденокарциномы протоков молочной железы человека. Клетки линии MCF7 часто используют для изучения молекулярной биологии рака и цитотоксичности противоопухолевых фармпрепаратов in vitro. Цитотоксические свойства СН и ДТТ в возрастающих концентрациях мы определяли при помощи МТТ-анализа, который показал, что обработка опухолевых клеток 1 и 5 мМ ДТТ, а также 0.01 мкМ СН в течение 24 ч практически не влияет на их пролиферацию и жизнеспособность. Дальнейшее увеличение концентрации СН приводило к снижению выживаемости клеток MCF7. Критической концентрацией СН, близкой к LD50, оказалась 1 мкМ, так как мы наблюдали гибель около 40% клеток. Очевидно, что именно эта доза СН приводит к однозначному переключению механизмов ЭПР-стресса с адаптивных на проапоптические и вызывает гибель опухолевых клеток, предположительно по пути апоптоза. Далее мы анализировали молекулярные механизмы наблюдаемых явлений.
Большой интерес представляет роль селенопротеома, т.е. совокупности всех селенсодержащих белков организма, в развитии, росте и прогрессии рака [25, 27, 28]. Мы сделали акцент на семи селенопротеинах, резидентно присутствующих в ЭПР, проанализировав изменение их экспрессии при воздействии на клетки MCF7 двух индукторов ЭПР-стресса. Эти селенопротеины, несмотря на сходное строение, обладают структурными особенностями, различиями в топологии и выполняют различные функции: участвуют в распознавании и регуляции окислительно-восстановительного статуса и правильного фолдинга белков, в контроле потока Ca2+ и регуляции Са2+-зависимой передачи сигналов и др. [29, 39]. Сниженная экспрессия селенопротеинов-резидентов ЭПР связана с повышенным клеточным стрессом и воспалением. Нами показано, что экспрессия мРНК практически всех исследуемых селенопротеинов увеличивается как при воздействии 1 и 5 мМ ДТТ, так и 0.1 мкМ СН. Вероятно, эта концентрация СН ассоциирована с развитием ЭПР-стресса по адаптивному сигнальному пути UPR, как и обе концентрации ДТТ.
Воздействие СН в низкой концентрации (0.01 мкМ) приводило к снижению уровня экспрессии генов трех селенопротеинов – SELМ, SEР15, DIO2. Единственным селенопротеином, уровень мРНК которого значительно увеличился, был SELК (в 2 раза, р <0.05). Это позволяет предположить, что большую роль в развитии адаптационного пути на начальных стадиях инициации ЭПР-стресса играет именно SELК. Показано, что подавление экспрессии SELK путем РНК-интерференции усугубляло гибель клеток HepG2 и апоптоз. Предполагается, что белок SELК регулируется ЭПР-стрессом и играет важную роль в защите клеток HepG2 от апоптоза, вызванного индукторами стресса ЭПР [40]. Показано также, что подавление экспрессии белка SELK сопровождается окислительным стрессом и апоптозом в мышцах курицы [41]. Напротив, избыточная экспрессия SELK защищает кардиомиоциты от апоптоза благодаря своим антиоксидантным свойствам [42]. Кроме того, SELK относится к тем селенопротеинам, которые сами не обладают ферментативной активностью, но используют остаток селеноцистеина альтернативным образом, в основном при взаимодействии с другими белками ЭПР [43]. Основные клеточные функции SELK – участие в пути деградации белка, связанного с ЭПР (ERAD), регуляция потока Са2+ из ЭПР, а также восстановление мембраны ЭПР. SELK входит в состав мембранных комплексов ЭПР, связывается с SELS, валозин-содержащим белком (VCP; p97) и Derlin, компонентами комплекса ERAD, который облегчает транслокацию неправильно свернутых белков из ЭПР в процессе UPR. SELK обладает более высоким сродством к трансмембранному белку Derlin-1, в то время как SELS имеет более высокое сродство к Derlin-2, что указывает на потенциальную способность этих селенопротеинов распознавать и регулировать транслокацию различных субстратов через канал Derlin в мембране ЭПР [44, 45]. SELK также важен для гомеостаза кальция, так как участвует в высвобождении внутриклеточного кальция из ЭПР. Можно предположить, что те селенопротеины, экспрессия которых снизилась при воздействии на клетки MCF7 0.01 мкМ СН, незначимы на данном этапе, и клетка экономит свои метаболические и энергетические ресурсы, чтобы максимально быстро и безболезненно восстановить гомеостаз ЭПР.
Селенопротеины М и N, как следует из характера изменения их экспрессии, вовлечены в эти процессы, развиваемые как по адаптивному сценарию, так и по апоптическому пути. Мы наблюдали повышение уровня мРНК в клетках MCF7, обработанных как ДТТ (1 и 5 мМ), так и 1 мкМ СН, при котором происходит гибель около 40% клеток. Активно обсуждается участие SELМ и SELN в проапоптическом пути развития ЭПР-стресса. Увеличение экспрессии этих белков способствует выживаемости клеток, обеспечивая антиоксидантную защиту и регулируя гомеостаз кальция [46–49]. Особый интерес представляет повышение экспрессии DIO2 (в 3 и 2 раза) при обработке клеток 0.1 и 1 мкМ СН соответственно. Объяснить этот результат довольно сложно, так как подробно описана в основном роль данного селенопротеина в активации тиреоидных гормонов [50, 51]. Показано, что экспрессия DIO2 у грызунов наиболее высока вскоре после рождения, что указывает на его потенциальное участие в развитии мышц. DIO2 действует под контролем фактора транскрипции Forkhead box O3 (FoxO3), который регулирует такие клеточные функции, как рост, развитие и метаболизм мышц. Интересно, что FoxO3 играет важную роль в метаболизме скелетных мышц, регулируя гликолитический и липолитический поток, а также метаболизм митохондрий [42]. Дозозависимое усиление экспрессии мРНК DIO2 сложно объяснить исходя из функциональной роли этого белка. Этот феномен требует дальнейшего изучения. Экспрессия ключевых участников ЭПР-стресса и маркеров апоптоза на уровне как мРНК, так и белка существенно менялась в зависимости от природы индуктора ЭПР-стресса и его концентрации.
Учитывая данные ПЦР в реальном времени и Вестерн-блотинга, мы можем предположить, что 1 мМ ДТТ активирует сигнальные пути IRE1 и ATF6 UPR с участием ASK1 (MAP3K5) киназы, которая способствует увеличению экспрессии белков апоптоза – BIM, PUMA и BAK. IRE1, один из трех основных сенсорных белков UPR, представляет собой бифункциональную молекулу, содержащую в своей цитоплазматической части домен с киназной активностью и С-концевой домен с РНКазной активностью. Активация IRE1 включает ее олигомеризацию и трансаутофосфорилирование киназных доменов. IRE1, после его активации по каноническому пути, удаляет с помощью РНКазного домена интрон из полноразмерной мРНК XBP1, образуя фактор транскрипции XBP1s, который активирует гены шаперонов. Кроме того, IRE1 способен взаимодействовать с адаптерным белком TRAF2 (фактор 2, ассоциированный с рецептором фактора некроза опухоли). Это взаимодействие инициирует каскад фосфорилирования киназой ASK1 и завершается активацией N-концевой киназы c-JUN (JNK). Воздействие на клетки 5 мМ ДТТ приводило к активации PERK сигнального пути UPR. Это подтверждается увеличением экспрессии мРНК ATF4 и нижерасположенных белков-маркеров апоптоза. Так ATF4 обеспечивает индукцию CHOP, активированный CHOP индуцирует ряд генов, кодирующих белки, участвующие в процессах апоптоза, такие как GADD34, PUMA, BIM, BAX, CAS4 [52–54]. Также, вероятно, запускается путь IRE1, что подтверждается возросшим уровнем мРНК сплайсированной формы XBP. Следует отметить довольно странные результаты, полученные нами при анализе экспрессии мРНК ключевых участников UPR в клетках, обработанных низкой дозой СН (0.01 мкМ). Эти результаты, а именно, снижение уровней мРНК всех исследуемых белков, участников ЭПР-стресса, сложно объяснить. Можно предположить, что СН в концентрации 0.01 мкМ запускает IRE1-сигнальный путь UPR. При этом IRE1 способен расщеплять множество мРНК без специфичной выборки, в том числе и мРНК изучаемых нами белков. Этот процесс известен как IRE1α-зависимый регулируемый распад (RIDD) [54, 55]. Другим возможным объяснением снижения уровня мРНК, как это не удивительно, может быть увеличение количества селенопротеина К, которое мы наблюдали при воздействии 0.01 мкМ СН. Это воздействие приводило к значительному увеличению в клетках MCF7 уровня мРНК ATF4 и всех исследуемых селенопротеинов, локализующихся в ЭПР. Возможно, фактор транскрипции ATF4 в данном случае стимулирует транскрипцию генов селенопротеинов ЭПР, неактивных в нормальном физиологическом состоянии, но транскрибируемых в условиях стресса, что мы и наблюдали. Воздействие на клетки MCF7 как 0.1, так и 1 мкМ СН приводило к увеличению экспрессии ключевых маркеров ЭПР-стресса на уровне как белка, так и мРНК. Мы предполагаем, что СН в этих концентрациях способствует активации сразу двух проапоптотических сигнальных путей UPR: PERK и IRE1. Это подтверждается увеличением уровня мРНК таких маркеров апоптоза, как PUMA, BIM, BAX и BAК. Если сравнить использованные нами индукторы ЭПР-стресса (СН и ДТТ), можно сделать следующие выводы. Используемые нами соединения имеют разное строение, одно из них содержит селен, другое серу, но оба действуют сходным образом, провоцируя стресс ЭПР: вызывают продукцию АФК, нарушают конформацию белков. Конформационные изменения в случае СН возникают за счет образования дисульфидных связей, в случае ДТТ – их восстановления. Нами показано, что данные соединения затрагивают различные адаптивные и проапоптотические пути UPR. Для развития ЭПР-стресса имеет значение также концентрация индуктора. Нами показано, что СН является более сильным стрессором, нарушающим гомеостаз ЭПР, а ДТТ – более слабым. Таким образом, полученные нами данные важны для понимания молекулярных механизмов регуляции ЭПP-cтресса, вызванного индукторами разной природы, и роли в этих процессах селенопротеинов, локализующихся строго в ЭПР. Мы полагаем, что подобные исследования позволят проводить поиск потенциальных мишеней для онкотерапевтических препаратов и стратегий воздействия при онкологических заболеваниях.
Авторы выражают благодарность сектору Оптической микроскопии и спектрофотометрии ЦКП ПНЦБИ РАН (http://www.ckp-rf.ru/ckp/ 670266/).
Работа выполнена в рамках Госзадания № 0191-2019-0019.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Azamjah N., Soltan-Zadeh Y., Zayeri F. (2019) Global trend of breast cancer mortality rate: a 25-year study. Asian. Pac. J. Cancer Prev. 20, 2015–2020.
Tang Y., Wang Y., Kiani M.F., Wang B. (2016) Classification, treatment strategy, and associated drug resistance in breast cancer. Clin. Breast Cancer. 16, 335–343.
Fisusi F.A., Akala E.O. (2019) Drug combinations in breast cancer therapy. Pharm. Nanotechnol. 7, 3–23.
Comşa Ş., Cîmpean A.M., Raica M. (2015) The story of MCF 7 breast cancer cell Line: 40 years of experience in research. Anticancer Res. 35, 3147–3154.
Hotamisligil G.S., Davis R.J. (2016) Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 8, a006072.
Almanza A., Carlesso A., Chintha C., Creedican S., Doultsinos D., Leuzzi B., Luís A., McCarthy N., Montibeller L., More S., Papaioannou A., Püschel F., Sassano M. L., Skoko J., Agostinis P., Belleroche J., Eriksson L.A., Fulda S., Gorman A.M., Healy S., Kozlov A., Muñoz-Pinedo C., Rehm M., Chevet E., Samali A. (2019) Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS J. 286(2), 241–278.
Lemmer I.L., Willemsen N., Hilal N., Bartelt A. (2021) A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 20, 101169.
Cui Z., Li C., Li X., Zhang Q., Zhang Y., Shao J., Zhou K. (2015) Sodium selenite (Na2SeO3) induces apoptosis through the mitochondrial pathway in CNE-2 nasopharyngeal carcinoma cells. Int. J. Oncol. 46(6), 2506–2514.
Jiang Q., Li F., Shi K., Wu P., An J., Yang Y., Xu C. (2013) ATF4 activation by the p38MAPK–eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett. 587(15), 2420–2429.
Liu T., Sun Y., Yang S., Liang X. (2020) Inhibitory effect of selenium on esophagus cancer cells and the related mechanism. J. Nutr. Sci. Vitaminol. 66(5), 456–461.
Bonfim N.E.S., Baranoski A., Mantovani M.S. (2020) Cytotoxicity of sodium selenite in HaCaT cells induces cell death and alters the mRNA expression of PUMA, ATR, and mTOR genes. J. Trace Elem. Med. Biol. 62, 126605.
Yang L., Cai Y.-S., Xu K., Zhu J.-L., Li Y.-B., Wu X.-Q., Sun J., Lu S.-M., Xu P. (2018) Sodium selenite induces apoptosis and inhibits autophagy in human synovial sarcoma cell line SW982 in vitro. Mol. Med. Rep. 17(5), 6560–6568.
Гольтяев М.В., Варламова Е.Г., Новосёлов С.В., Фесенко Е.Е. (2020) Активация сигнальных путей апоптоза в условиях пролонгированного ЭР-стресса, вызванного действием селен-содержащих соединений на клетки тератокарциномы семенников мыши. ДАН. 490(1), 9–11. https://doi.org/10.1134/S160767292001007X
Кузнецова Ю.П., Гольтяев М.В., Горбачева О.С., Новосёлов С.В., Варламова Е.Г., Фесенко Е.Е. (2018) Влияние селенита натрия на экспрессию мРНК генов селеноцистеин-содержащих белков млекопитающих в раковых клетках семенников и предстательной железы. ДАН, 480(1), 107–111. https://doi.org/10.1134/S1607672918030018
Takahashi K., Suzuki N., Ogra Y. (2017) Bioavailability comparison of nine bioselenocompounds in vitro and in vivo. Int. J. Mol. 18, 506.
Misra S., Boylan M., Selvam A., Spallholz J.E., Björnstedt M. (2015) Redox-active selenium compounds – from toxicity and cell death to cancer treatment. Nutrients. 7, 3536–3556.
Guan L., Han B., Li Z., Hua F., Huang F., Wei W., Yang Y., Xu C. (2009) Sodium selenite induces apoptosis by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction in human acute promyelocytic leukemia NB4 cells. Apoptosis. 14, 218–225.
Okuno T., Honda E., Arakawa T., Ogino H., Ueno H. (2014) Glutathione–dependent cell cycle G1 arrest and apoptosis induction in human lung cancer A549 cells caused by methylseleninic acid: comparison with sodium selenite. Biol. Pharm. Bull. 37, 1831–1837.
Palsamy P., Bidasee K.R., Shinohara T. (2014) Selenite cataracts: activation of endoplasmic reticulum stress and loss of Nrf2/Keap1-dependent stress protection. Biochim. Biophys. Acta. 1842(9), 1794–1805.
Cui Z., Li C., Li X., Zhang Q., Zhang Y., Shao J., Zhou K. (2015) Sodium selenite (Na2SeO3) induces apoptosis through the mitochondrial pathway in CNE-2 nasopharyngeal carcinoma cells. Int. J. Oncol. 46(6), 2506–2514.
Badr D.M., Hafez H.F., Agha A.M., Shouman S.A. (2016) The combination of α-tocopheryl succinate and sodium selenite on breast cancer: a merit or a demerit? Oxid. Med. Cell Longev. 2016, 4741694.
Tartier L., McCarey Y.L., Biaglow J.E., Kochevar I.E., Held K.D. (2000) Apoptosis induced by dithiothreitol in HL-60 cells shows early activation of caspase 3 and is independent of mitochondria. Cell Death Differ. 7(10), 1002–1010.
Xiang X.Y., Yang X.C., Su J., Kang J.S., Wu Y., Xue Y.N., Dong Y.T., Sun L.K. (2016) Inhibition of autophagic flux by ROS promotes apoptosis during DTT-induced ER/oxidative stress in HeLa cells. Oncol. Rep. 35, 3471–3479.
Hughes D.J., Kunická T., Schomburg L., Liška V., Swan N., Souček P. (2018) Expression of selenoprotein genes and association with selenium status in colorectal adenoma and colorectal cancer. Nutrients. 10(11), 1812.
Jia Y., Dai J., Zeng Z. (2020) Potential relationship between the selenoproteome and cancer. Mol. Clin. Oncol. 13(6), 83.
Capone F., Polo A., Sorice A., Budillon A., Costantini S. (2020) Integrated analysis to study the relationship between tumor-associated selenoproteins: focus on prostate cancer. Int. J. Mol. Sci. 21(18), 6694.
Li J., Zhu Y., Zhou Y., Jiang H.G., Chen Z.H., Lu B.H., Shen X.N. (2020) The SELS rs34713741 polymorphism is associated with susceptibility to colorectal cancer and gastric cancer: a meta-analysis. Genet. Test. Mol. Biomarkers. 24(12), 835–844.
Callejón-Leblic B., Arias-Borrego A., Rodríguez-Moro G., Roldán F.N., Pereira-Vega A., Gómez-Ariza J.L., García-Barrera T. (2021) Advances in lung cancer biomarkers: the role of (metal-) metabolites and selenoproteins. Adv. Clin. Chem. 100, 91–137.
Varlamova E.G. (2020) Protein–protein interactions of ER-resident selenoproteins with their physiological partners. Biochimie. 171–172, 197–204. https://doi.org/10.1016/j.biochi.2020.03.012
Rocca C., Pasqua T., Boukhzar L., Anouar Y., Angelone T. (2019) Progress in the emerging role of selenoproteins in cardiovascular disease: focus on endoplasmic reticulum-resident selenoproteins. Cell Mol. Life Sci. 76(20), 3969–3985.
Zhang C., Ge J., Liv M., Zhang Q., Talukder M., Li J.L. (2020) Selenium prevent cadmium-induced hepatotoxicity through modulation of endoplasmic reticulum-resident selenoproteins and attenuation of endoplasmic reticulum stress. Environ. Pollut. 260, 113873.
Pitts M.W., Hoffmann P.R. (2018) Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium. 70, 76–86.
Wang P., Lu Z., He M., Shi B., Lei X., Shan A. (2020) The effects of endoplasmic-reticulum-resident selenoproteins in a nonalcoholic fatty liver disease pig model induced by a high-fat diet. Nutrients. 12(3), 692.
Varlamova E.G. (2018) Participation of selenoproteins localized in the ER in the processes occurring in this organelle and in the regulation of carcinogenesis-associated processes. J. Trace Elem. Med. Biol. 48, 172–180.
Варламова Е.Г., Гольтяев М.В., Фесенко Е.Е. (2019) Белки-партнеры селенопротеина SELM и роль соединений селена в регуляции его экспрессии в раковых клетках человека. ДАН. 488(2), 212–216.
Goltyaev M.V., Mal’tseva V.N., Varlamova E.G. (2020) Expression of ER-resident selenoproteins and activation of cancer cells apoptosis mechanisms under ER-stress conditions caused by methylseleninic acid. Gene. 755, 144884.
Kim C., Kim B. (2018) Anti-cancer natural products and their bioactive compounds inducing ER stress–mediated apoptosis: a review. Nutrients. 10(8), 1021.
Harnoss J., Thomas A., Reichelt M., Guttman O., Wu T.D., Marsters S.A., Shemorry A., Lawrence D., Kan D., Segal E., Merchant M., Totpal K., Crocker L.M., Mesh K., Dohse M., Solon M., Modrusan Z., Rudolph J., Koeppen H., Walter P., Ashkenazi A. (2020) IRE1α disruption in triple-negative breast cancer cooperates with antiangiogenic therapy by reversing ER stress adaptation and remodeling the tumor microenvironment. Cancer Res. 80(11), 2368–2379.
Zhang Y., Roh Y.J., Han S.J., Park I., Lee H.M., Ok Y.S., Lee B.C., Lee S.R. (2020) Role of selenoproteins in redox regulation of signaling and the antioxidant system: a review. Antioxidants (Basel). 9(5), 383.
Du S., Zhou J., Jia Y., Huang K. (2010) SelK is a novel ER stress-regulated protein and protects HepG2 cells from ER stress agent-induced apoptosis. Arch. Biochem. Biophys. 502(2), 137–143.
Fan R.F., Cao C.Y., Chen M.H., Shi Q.X., Xu S.W. (2018) Gga–let–7f–3p promotes apoptosis in selenium deficiency-induced skeletal muscle by targeting selenoprotein K. Metallomics. 10(7), 941–952.
Addinsall A.B., Wright C.R., Andrikopoulos S., Poel C., Stupka N. (2018) Emerging roles of endoplasmic reticulum-resident selenoproteins in the regulation of cellular stress responses and the implications for metabolic disease. Biochem J. 475(6), 1037–1057.
Marciel M.P., Hoffmann P.R. (2019) Molecular mechanisms by which selenoprotein K regulates immunity and cancer. Biol. Trace Elem. Res. 192(1), 60–68.
Lee J.H., Park K.J., Jang J.K., Jeon Y.H., Ko K.Y., Kwon J.H., Lee S.R., Kim I.Y. (2015) Selenoprotein S-dependent selenoprotein K binding to p97(VCP) protein is essential for endoplasmic reticulum-associated degradation. J. Biol. Chem. 290(50), 29941–29952.
Shchedrina V.A., Everley R.A., Zhang Y., Gygi S.P., Hatfield D.L., Gladyshev V.N. (2011) Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J. Biol. Chem. 286(50), 42937–42948.
Huang J.Q., Ren F.Z., Jiang Y.Y., Lei X. (2016) Characterization of selenoprotein M and its response to selenium deficiency in chicken brain. Biol. Trace Elem. Res. 170(2), 449–458.
Gong T., Hashimoto A.C., Sasuclark A.R., Khadka V.S., Gurary A., Pitts M.W. (2019) Selenoprotein M promotes hypothalamic leptin signaling and thioredoxin antioxidant activity. Antioxid. Redox Signal. https://doi.org/10.1089/ars.2018.7594
Jiang H., Shi Q.Q., Ge L.Y., Zhuang Q.F., Xue D., Xu H.Y., He X.Z. (2019) Selenoprotein M stimulates the proliferative and metastatic capacities of renal cell carcinoma through activating the PI3K/AKT/mTOR pathway. Cancer Med. 8(10), 4836–4844.
Varone E., Pozzer D., Di Modica S., Chernorudskiy A., Nogara L., Baraldo M., Cinquanta M., Fumagalli S., Villar-Quiles R.N., De Simoni M.G., Blaauw B., Ferreiro A., Zito E. (2019) SELENON (SEPN1) protects skeletal muscle from saturated fatty acid-induced ER stress and insulin resistance. Redox Biol. 24, 101176.
AlRasheed M.M., AlAnzi A., AlShalhoub R., Abanmy N., Bakheet D. (2019) A study of the role of DIO1 and DIO2 polymorphism in thyroid cancer and drug response to therapy in the Saudi population. Saudi Pharm. J. 27(6), 841–845.
Zevenbergen C., Groeneweg S., Swagemakers S.M.A., de Jong A., Medici-Van den Herik E., Rispens M., Klootwijk W., Medici M., de Rijke Y.B., Meima M.E., Larsen P.R., Chavatte L., Venter D., Peeters R.P., Van der Spek P.J., Visser W.E. (2019) Functional analysis of genetic variation in the SECIS element of thyroid hormone activating type 2 deiodinase. J. Clin. Endocrinol. Metab. 104(5), 1369–1377.
Sovolyova N., Healy S., Samali A., Logue S.E. (2014) Stressed to death – mechanisms of ER stress-induced cell death. Biol. Chem. 395(1), 1–13.
Hetz C., Zhang K., Kaufman R.J. (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell. Biol. 21(8), 421–438.
Sano R., Reed J.C. (2013) ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 1833(12), 3460–3470.
Maurel M., Chevet E., Tavernier J., Gerlo S. (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39(5), 245–254.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология