

Молекулярная биология, 2022, T. 56, № 3, стр. 510-515
Новые аналоги уридина как возможные противовирусные агенты, специфичные к SARS-CoV-2
А. А. Маслова a, *, Е. С. Матюгина a, Е. Ю. Шустова b, В. П. Волок b, Л. И. Козловская b, c, С. Н. Кочетков a, А. Л. Хандажинская a
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Федеральный научный центр исследований и разработки иммунобиологических препаратов им. М.П. Чумакова Российской академии наук (Институт полиомиелита)
108819 Москва, Россия
c Первый Московский государственный медицинский университет им. И.М. Cеченова Министерства здравоохранения Российской Федерации (Сеченовский университет)
119435 Москва, Россия
* E-mail: maslova_anna94@mail.ru
Поступила в редакцию 24.11.2021
После доработки 06.12.2021
Принята к публикации 07.12.2021
- EDN: XPWGGW
- DOI: 10.31857/S0026898422030107
Аннотация
Разработка лекарственных средств против SARS-CoV-2 – важнейшая задача, стоящая перед мировой наукой и здравоохранением. На сегодняшний день основным подходом к созданию средств, эффективных при COVID-19, остается репозиционирование, т.е. перепрофилирование препаратов, допущенных к клиническому применению, например, Ремдесивира (препарат против лихорадки Эбола), Фавипиравира (против гриппа) и др. Однако очевидно, что эти препараты недостаточно специфичны и эффективны. Другой перспективный подход предполагает создание новых молекул, однако нужно отметить, что реализация этого подхода требует гораздо больше времени и затрат. Тем не менее, продолжается поиск новых противовирусных агентов, специфичных к SARS-CoV-2. Цель нашей работы состояла в получении новых 5-замещенных производных уридина – потенциальных ингибиторов РНК-зависимой РНК-полимеразы коронавируса. Такие вещества получены с высоким выходом по реакции Сузуки–Мияуры и охарактеризованы с помощью современных физико-химических методов. Однако тестирование антивирусной активности этих веществ в отношении SARS-CoV-2 не выявило существенного ингибиторного эффекта.
ВВЕДЕНИЕ
COVID-19 – высококонтагиозное респираторное заболевание, вызываемое бетакоронавирусом SARS-CoV-2. После того, как в конце декабря 2019 года в Китае были зарегистрированы первые случаи заболевания, SARS-CoV-2 быстро распространился по миру, что привело к развитию пандемии, в результате которой количество умерших приближается к 5 млн. человек [1]. Несмотря на многочисленные исследования, которые позволили лучше понять механизмы патогенеза SARS-CoV-2, этот вирус остается общемировой проблемой. Наблюдаются новые вспышки этого заболевания, обусловленные появлением и распространением мутантных вариантов вируса.
Структура и жизненный цикл SARS-CoV-2 хорошо изучены [2, 3]. Главный фермент репликации вируса – РНК-зависимая РНК-полимераза (RdRp, nsp12), функционирует в составе репликазно-транскриптазного комплекса (RTC), контролирующего синтез вирусной РНК [3]. Ключевая роль RdRp в репликации SARS-CoV-2 делает этот фермент привлекательной терапевтической мишенью, а отсутствие ее аналогов в клетках млекопитающих предполагает высокую селективность ингибирования. В настоящее время интенсивно разрабатываются ингибиторы RdRp, которые потенциально могут обладать выраженной противовирусной активностью. Существующие в настоящее время ингибиторы RdRp делятся на два основных класса – нуклеозидные (НИ) и ненуклеозидные ингибиторы (ННИ), связывающиеся в активном центре и аллостерическом сайте соответственно [4].
Создание НИ считается перспективной стратегией, направленной на остановку репликации вирусной РНК, обусловленной высокой степенью консервативности активных сайтов RdRp и относительно низкой скоростью мутаций в этих областях, что обеспечит широкую противовирусную активность и высокий порог резистентности. Основной механизм действия нуклеозидных аналогов включает их внутриклеточное превращение в соответствующие трифосфаты, которые, конкурируя с трифосфатами природных нуклеозидов, становятся терминаторными субстратами вирусных полимераз. Для эффективного ингибирования репликации вирусной РНК нуклеотидный аналог, встроенный в растущую цепь, не должен выщепляться за счет корректирующей активности, характерной для значительной доли полимераз, либо скорость его включения должна многократно превышать скорость выщепления. Противовирусные НИ делятся на три типа: облигатные терминаторы цепи, необлигатные терминаторы цепи и мутагенные. Облигатные терминаторы цепи не содержат 3'-гидроксильную группу в рибозидной части молекулы, что делает невозможным дальнейшее удлинение нуклеотидной цепи. Необлигатные терминаторы цепи содержат природное основание и 3'-гидроксил на остатке сахара, но они имеют дополнительный заместитель в положениях C-1' или C-2' рибозного кольца, блокируя последующее образование фосфодиэфирной связи с подходящим нуклеозидтрифосфатом [5]. Механизм летального мутагенеза заключается в неспособности распознавать аналоги нуклеозидов в качестве обычных нуклеотидных оснований, что вызывает несоответствие в спаривании оснований и увеличение количества мутаций, и приводит в конечном итоге к появлению нежизнеспособных геномов.
Оценена способность многих препаратов, первоначально нацеленных на RdRp других РНК-вирусов, таких как Ремдесивир (вирус Эбола), Фавипиравир (вирус гриппа), NHD EIDD-2801 (широкий спектр действия) и Софосбувир (вирус гепатита C), ингибировать SARS-CoV-2 [6]. Софосбувир – высокоактивный уридиновый нуклеотид, специфично ингибирующий репликацию вируса гепатита С. Сходство циклов репликации вируса гепатита С и коронавируса позволило предположить, что Софосбувир можно будет применять в терапии COVID-19 [7]. Первым специфическим противовирусным препаратом, одобренным Ассоциацией университетов Европы для лечения пациентов с тяжелым течением COVID-19, стал Ремдесивир. Один из основных недостатков Ремдесивира – необходимость внутривенного введения, ограничивает широкое применение этого препарата и вынуждает использовать его в условиях больничного стационара. Для изменения ситуации создана ингаляционная форма, которая в настоящее время проходит 1 фазу клинических испытаний. Ряд клинических исследований показал эффективность Фавипиравира в терапии COVID-19, однако эксперименты на животных выявили потенциальную тератогенную активность этого препарата [8]. Тем не менее, Фавипиравир одобрен к применению в ряде стран. Молнупиравир (EIDD-2801) представляет собой пероральное биодоступное пролекарство – аналог нуклеозида β-D-N4-гидроксицитидина, действующего по механизму летального мутагенеза с широким спектром действия. Показано, что N4-гидроксицитидин более чем в 100 раз активен, чем Рибавирин или Фавипиравир в отношении коронавируса SARS-CoV-2, причем его противовирусная активность коррелирует с уровнем мутагенеза в вирусной РНК. Молнупиравир оказался безопасным в 1 фазе клинических испытаний, в настоящее время продолжаются его клинические испытания. Однако показана также способность N4-гидроксицитидина вызывать геномные мутации в клетках человека in vitro [9].
Таким образом, в настоящий момент отсутствует высокоэффективное и специфичное лекарственное средство против SARS-CoV-2, поэтому создание новых агентов, нацеленных на ингибирование активности RdRp коронавируса, представляется крайне важным и требующим незамедлительного решения. В нашей работе описан синтез и определение противовирусной активности шести 5-замещенных производных уридина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали 5-бромуридин (“Sigma”, США), бороновые кислоты и растворители высокой степени чистоты (“Acros”, Бельгия). Колоночную хроматографию проводили на обращенно-фазовом носителе LiChroprep RP-18, элюировали водой. Спектры ЯМР регистрировали на спектрометре AMX III-400 (“Bruker”, США) с рабочей частотой 400 МГц для 1H (Me4Si в качестве внутреннего стандарта для органических растворителей и 3-(триметилсилил)-1-пропансульфонат натрия ((DSS) для D2O) и 100.6 МГц для 13С-ЯМР (с подавлением углерод-протонного взаимодействия). Контроль за ходом реакций осуществляли методом ТСХ на пластинах Кизельгель 60 F254 (“Merck”, Германия), элюировали системой диоксан-25% водный NH3 (4 : 1).
Общая методика получения соединений 1a–g. К 5-бромуридину, растворенному в смеси воды (4 мл) и метанола (2 мл), приливали раствор бороната (1 экв) в тетрагидрофунане (TГФ) (4 мл), продували аргоном. Добавляли катализатор тетракси(трифенилфосфин)палладий (0.1 экв) и гидроксид натрия (10 экв). Реакционную массу кипятили в течение 7 ч, затем упаривали, растворяли в хлороформе (15 мл) и экстрагировали водой (15 мл). Водную фракцию промывали хлороформом (2 × 5 мл), концентрировали и выделяли целевое вещество колоночной хроматографией на RP-18, элюировали водой. Выход продуктов 1a–g составил 37–59%.
1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-5-(пиридин-2-ил)пиримидин-2,4(1H,3H)-дион (1a) светло-желтые кристаллы, 41% 1H ЯМР (CD3OD): 3.76–3.95 (2H, м, H1), 4.08–4.06 (5'H, м, H4'), 4.25–4.28 (2H, м, H3′, H2'), 5.98–5.99 (1H, д, H1′), 7.43–7.47 (1H, м, H5 Py), 8.06–8.10 (1H, м, H4 Py), 8.44–8.46 (1H, д, H6 Py), 8.49 (1H, с, H6), 8.77–8.78 (1H, д, H2 Py). 13C ЯМР (D2O): 60.13, 68.90, 74.08, 83.73, 90.03, 112.46, 123.94, 130.12, 137.44, 139.19, 147.62, 147.98, 155.05, 169.10.
5-(3-аминофенил)-1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)пиримидин-2,4(1H,3H)-дион (1b) серые кристаллы, 37% 1H NMR (CD3OD): 3.77–3.87 (2H, м, H5′), 4.02–4.04 (1H, м, H4′), 4.19–4.27 (2H, м, H3′, H2′), 5.96–5.98 (1H, д, H1′), 6.63–6.67 (1H, м, Ph), 6.89–6.96 (2H, м, Ph), 7.06–7.11 (1H, д, Ph), 7.92 (1H, с, H6). 13C ЯМР (D2O): 60.52, 69.24, 73.91, 83.65, 89.93, 115.55, 116.35, 116.54, 129.41, 135.44, 138.07, 146.09, 157.63, 172.64.
5-(4-аминофенил)-1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)пиримидин-2,4(1H,3H)-дион (1c) бежевые кристаллы, 39% 1H ЯМР (CD3OD): 3.71–3.90 (2H, м, H5′), 4.00–4.02 (1H, м, H4′), 4.18–4.27 (2H, м, H3′, H2′), 5.95–5.97 (1H, д, H1′), 6.72–6.75 (2H, м, Ph), 7.28–7.33 (2H, м, Ph), 7.82 (1H, с, H6). 13C ЯМР (CD3OD): 60.38, 69.26, 74.00, 83.81, 90.15, 114.39 × 2, 122.53, 128.55 × 2, 130.78, 135.88, 148.32, 150.88, 165.91.
4-(1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)бензамид (1d) желтые кристаллы, 53% 1H ЯМР (D2O): 3.72–3.91 (2H, м, H5′), 4.06–4.10 (1H, м, H4′), 4.20–4.24 (1H, т, H3′), 4.30–4.33 (1H, т, H2′), 5.93–5.94 (1H, д, H1′), 7.52–7.56 (2H, м, Ph), 7.77–7.80 (2H, м, Ph), 7.92 (1H, с, H6). 13C ЯМР (D2O): 56.95, 59.84, 66.08, 73.54, 83.07, 89.60, 114.88, 127.01, 128.33, 130.84 × 2, 138.32, 138.45, 157.62, 163.24, 172.51.
N-(4-(1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)фенил)ацетамид (1e) белые кристаллы, 57% 1H ЯМР (D2O): 2.13 (3H, с, CH3 ), 3.71–3.88 (2H, м, H5′), 4.04–4.08 (1H, м, H4′), 4.18–4.22 (1H, т, H3′), 4.29–4.32 (1H, т, H2′), 5.92–5.94 (1H, д, H1′), 7.39 (4H, с, Ph), 7.78 (1H, с, H6). 13C ЯМР (D2O): 22.34, 56.93, 60.00, 68.70, 73.40, 83.08, 89.46, 115.41, 121.28, 128.25, 128.87, 129.25, 135.52, 137.57, 158.13, 165.77, 172.49.
4-(1-((2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-N-метилбензамид (1g) белые кристаллы, 54% 1H ЯМР (D2O): 2.91 (3H, с, CH3), 3.73–3.92 (2H, м, H5′), 4.06–4.11 (1H, м, H4′), 4.21–4.25 (1H, т, H3′), 4.31–4.34 (1H, т, H2′), 5.94–5.95 (1H, д, H1′), 7.51–7.55 (2H, м, Ph), 7.71–7.73 (2H, м, Ph), 7.91 (1H, с, H6). 3C ЯМР (D2O) : 26.39, 48.90, 60.31, 69.05, 74.05, 83.57, 90.08, 115.38, 127.01, 127.52, 128.84, 138.25, 138.79, 157.81, 163.24, 171.13, 172.67.
Исследование противовирусной активности. Стоковые растворы тестируемых соединений в концентрации 5 мкМ готовили в 100% ДМСО.
Противовирусную активность оценивали на штамме ПИК35 вируса SARS-CoV-2 (GISAID ID EPI_ISL_428851). Линия клеток почки зеленой мартышки Vero получена из “Biologicals”, ВОЗ, Швейцария (RCB 10-87). Клетки культивировали в среде ДМЕМ (ФГБНУ “ФНЦИРИП им. М.П. Чумакова РАН”, Россия) с фетальной сывороткой крупного рогатого скота (“Gibco”, США, 5%), стрептомицином (0.1 мг/мл) и пенициллином (100 ед./мл) (“ПанЭко”, Россия).
Методика оценки противовирусной активности описана ранее [10]. Готовили восемь двукратных разведений стоковых растворов соединений в среде ДМЕМ (“ФНЦИРИП им. М.П. Чумакова РАН”), которые смешивали с равными объемами вирусной суспензии, содержащей 50–200 ТЦД50 на лунку, инкубировали в течение 1 ч при 37°C. Далее смеси вирус–соединение добавляли к монослоям клеток Vero в двух повторностях. Цитопатический эффект (ЦПЭ) оценивали микроскопически после 5-дневной инкубации при 37°C. Значения ЕС50 рассчитывали по методу Кербера. Эксперимент повторяли не менее 2 раз с каждым соединением. В качестве положительного контроля использовали N4-гидроскицитидин, в качестве отрицательного – ДМСО, дозу вируса контролировали титрованием.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
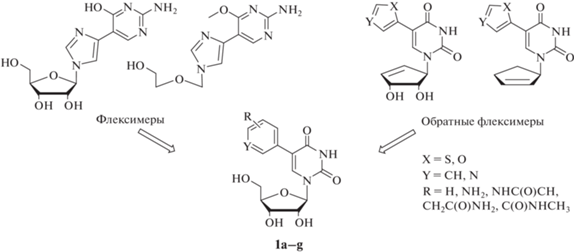
Ранее группой ученых под руководством К. Seley-Radtke был разработан новый тип модифицированных нуклеозидов, несущих гибкие или “расщепленные” аналоги пуриновых оснований, названные флексимерами [10]. Гетероциклические основания флексимеров состоят из имидазольного и пиримидинового колец, связанных одной углерод-углеродной связью, а не сконденсированных, как в обычных пуринах (рис. 1). В результате азотистое основание сохраняет необходимые водородные связи и ароматические свойства, которые требуются для распознавания, получая при этом гибкость для адаптации к сайту связывания фермента и потенциальным мутациям. Показано, что такие соединения способны ингибировать репликацию коронавирусов HCoV-NL63 и MERS-CoV в культуре клеток [11, 12]. Идея флексимерных соединений получила развитие в виде синтеза “обратных” флексимеров (рис. 1), в которых каркас пуринового основания соединен с сахарным фрагментом по N-3 пиримидинового кольца, а не N-9 имидазольного кольца. Это меняет конформацию флексимерного основания, которое также можно рассматривать как C5-замещенный пиримидин [13]. В продолжение этих работ мы решили синтезировать серию 5-замещенных производных уридина (1a–g), которые можно рассматривать как обратные флексимеры, в надежде получить вещества, обладающие противовирусной активностью в отношении SARS-CoV-2. Для получения целевых соединений мы рассмотрели различные современные методы создания С-С-связи.
Ключевое место в синтезе сложных органических молекул с помощью реакций кросс-сочетания занимает палладиевый катализ. Многолетний интерес к этой области объясняется двумя основными причинами. Во-первых, такие каталитические системы позволяют легко создавать углерод–углеродные и углерод–азотные связи в устойчивых условиях с отличным выходом. Во-вторых, мягкие условия реакции позволяют осуществить синтез, требующий селективного связывания молекул в определенных положениях без воздействия на другие функциональные группы [14].
Хотя катализируемые палладием реакции кросс-сочетания хорошо известны для широкого диапазона соединений, в случае нуклеозидов возникает ряд трудностей. Помимо проблем, обусловленных перекрестным связыванием многофункциональных гетероциклических субстратов, полярная природа производных нуклеозидов часто приводит к тому, что они плохо растворяются в типичных органических растворителях [15]. Один из распространенных подходов к устранению этого недостатка – защита гидроксильных групп углеводного фрагмента с целью получения более гидрофобного субстрата. Этот метод включает две дополнительные стадии синтеза, что приводит к снижению выхода. Более привлекательным представляется использование незащищенных нуклеозидных, нуклеотидных или олигонуклеотидных производных в реакциях кросс-сочетания. Это может быть достигнуто с помощью полярных органических растворителей. Нуклеозиды и производные нуклеотидов эффективно растворяются в воде или в ее сочетании с органическими растворителями, что позволяет проводить реакции в гомогенных условиях без необходимости применения защитных групп [15].
Описаны варианты постановки реакции Сузуки–Мияуры с незащищенными нуклеозидгалогенидами в различных системах из воды и сорастворителей. Так Western Е.С. и соавт. [16] арилировали 5-йод-2-дезоксиуридин в присутствии катализаторов (три-(4,6-диметил-3-сульфонатофенил)фосфина и ацетата палладия) в системе растворителей вода–ацетонитрил 2 : 1. Описано также проведение первой стадии синтеза производного уридина с помощью реакции кросс-сочетания Сузуки между 5-йод-2-дезоксиуридином и 4-формилфенилбороновой кислотой в смеси метанол–вода 5 : 1 в присутствии катализатора тетракис(трифенилфосфин)палладия [17]. Несмотря на нерастворимость трифенилфосфина в воде, показано [18], что реакция между арилбороновыми кислотами и 5-йод-2-дезоксиуридином в воде в качестве единственного растворителя успешно катализируется Pd(OAc)2/PPh3 при 120°C с использованием микроволнового излучения.
Схема 1 . Синтез 5-замещенных производных уридина 1a–g.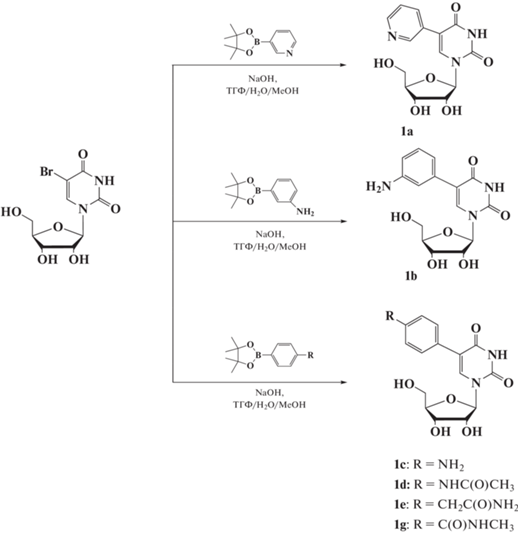
На основании анализа опубликованных данных для проведения реакции Сузуки–Мияуры между 5-бромуридином и различными боронатами (схема 1 ) была подобрана система растворителей Н2О/МеОН/ТГФ = 2 : 1 : 2. После выделения и очистки с помощью ионообменной хроматографии оценена биологическая активность и цитотоксичность синтезированных веществ. Противовирусную активность определяли по способности исследуемых соединений ингибировать гибель клеток Vero, зараженных штаммом ПИК35 SARS-CoV-2. В качестве положительного контроля использовали N4-гидроксицитидин (ЕС50 > 5 мкМ). Однако соединения 1a–g не проявили ожидаемого ингибирующего эффекта (ЕС50 > 100 мкМ). Очевидно, что для эффективного подавления SARS-CoV-2 требуется модификация структуры соединений. В данный момент изучается активность соединений 1a–g в отношении других вирусных патогенов.
Исследование выполнено при финансовой поддержке РФФИ в рамках проекта № 20-04-60414 и Министерства науки и высшего образования Российской Федерации (государственное задание по теме “Аналоги компонентов нуклеиновых кислот как потенциальные ингибиторы коронавирусов”). Исследование физико-химических свойств продуктов проведено при поддержке РНФ № 19-74-10048.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Cascella M., Rajnik M., Aleem A., Dulebohn S., Di Napoli R. (2021). Features, evaluation, and treatment of coronavirus (COVID-19). StatPearls.
Santos I.A., Grosche V.R., Bergamini F.R.G., Sabino-Silva R., Jardim A.C.G. (2020) Antivirals against coronaviruses: candidate drugs for SARS-CoV-2 treatment? Front. Microbiol. 11, 1818.
Lei T., Qiang T., Liang C., Ren X., Jia M., Zhang J., Li J., Wan M., YuWen X., Li H., Cao W., Liu H. (2021) RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 213, 113201.
Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. (2020) Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chemi. acs.jmedchem.0c01140. https://doi.org/10.1021/acs.jmedchem.0c01140
De Clercq E., Neyts J. (2009) Antiviral agents acting as DNA or RNA chain terminators. Antiviral Strategies. 189, 53‒84.
Buonaguro L., Tagliamonte M., Tornesello M.L., Buonaguro F.M. (2020) SARS-CoV-2 RNA polymerase as target for antiviral therapy. J. Transl. Med. 18(1), 185. https://doi.org/10.1186/s12967-020-02355-3
Ju J., Li X., Kumar S., Jockusch S., Chien M., Tao C., Morozova I., Kalachikov S., Kirchdoerfer R.N., Russo J.J. (2020) Nucleotide analogues as inhibitors of SARS-CoV polymerase. Pharmacol. Res. Perspect. 8(6), e00674.
Al-Horani R.A., Kar S. (2020) Potential anti-SARS-CoV-2 therapeutics that target the post-entry stages of the viral life cycle: a comprehensive review. Viruses. 12(10), 1092‒1134.
Zhou S., Hill C.S., Sarkar S., Tse L.V., Woodburn B.M.D., Schinazi R.F., Sheahan T.P., Baric R.S., Heise M.T., Swanstrom R. (2021). β-DN 4-hydroxycytidine (NHC) inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 224(3), 415‒419.
Seley-Radtke K. (2018) Flexibility – not just for yoga anymore! Antivir. Chem. Chemother. 26, 2040206618756788.
Матюгина Е.С., Новиков М.С., Козловская Л.И., Волок В.П., Шустова Е.Ю., Ишмухаметов А.А., Кочетков С.Н., Хандажинская А.Л. (2021). Оценка противовирусного потенциала модифицированных гетероциклических оснований и 5′-норкарбоциклических аналогов нуклеозидов в отношении SARS-CoV-2. Acta Naturae, 13(4), 51‒54.
Peters H.L., Jochmans D., de Wilde A.H., Posthuma C.C., Snijder E.J., Neyts J., Seley-Radtke K.L. (2015). Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg. Med. Chem. Lett. 25(15), 2923‒2926.
Zimmermann S.C., Sadler J.M., O’Daniel P.I., Kim N.T., Seley-Radtke K.L. (2013) “Reverse” carbocyclic fleximers: synthesis of a new class of adenosine deaminase inhibitors. Nucleosides, Nucleotides Nucl. Acids. 32(3), 137‒154.
Polshettiwar V., Decottignies A., Len C., Fihri A. (2010) Suzuki–Miyaura cross-coupling reactions in aqueous media: green and sustainable syntheses of biaryls. ChemSusChem: Chemistry & Sustainability Energy & Materials. 3(5), 502–522.
Shaughnessy K.H. (2015). Palladium-catalyzed modification of unprotected nucleosides, nucleotides, and oligonucleotides. Molecules. 20(5), 9419–9454.
Western E.C., Daft J.R., Johnson E.M. 2nd, Gannett P.M., Shaughnessy K.H. (2003) Efficient one-step Suzuki arylation of unprotected halonucleosides, using water-soluble palladium catalysts. J. Org. Chem. 68(17), 6767–6774.
Okamoto A., Inasaki T., Saito I. (2005) Synthesis and ESR studies of nitronyl nitroxide-tethered oligodeo-xynucleotides. Tetrahedron Lett. 46(5), 791–795.
Fresneau N., Hiebel M.A., Agrofoglio L.A., Berteina-Raboin S. (2012). Efficient synthesis of unprotected C-5-aryl/heteroaryl-2'-deoxyuridine via a Suzuki–Miyaura reaction in aqueous media. Molecules. 17(12), 14409–14417.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология