

Молекулярная биология, 2022, T. 56, № 3, стр. 428-438
Выбор донорной молекулы в экспериментах по геномному редактированию в клетках животных
О. В. Володина a, С. А. Смирнихина a, *
a Медико-генетический научный центр имени академика Н.П. Бочкова
115522 Москва, Россия
* E-mail: smirnikhinas@gmail.com
Поступила в редакцию 15.07.2021
После доработки 08.10.2021
Принята к публикации 12.11.2021
- EDN: BHDVXO
- DOI: 10.31857/S0026898422030168
Аннотация
Редактирование генома стало мощным инструментом изучения свойств генов или изменения нуклеотидной последовательности. За последние несколько десятилетий разработаны программируемые нуклеазы, которые могут вносить двухцепочечный разрыв в интересующий участок молекулы ДНК. Репарация ДНК после инициации двухцепочечного разрыва может пойти по пути негомологичного соединения концов (NHEJ), которое приводит к различным ошибкам и нокауту гена. Другие варианты репарации – направленная гомологичная репарация (HDR) или репарация с помощью одноцепочечной матрицы (SSTR) – позволяют вносить желаемые изменения в ген. В природе HDR происходит в присутствии донорной матрицы – сестринской хроматиды. При редактировании генома животных клеток эффективность HDR и SSTR значительно ниже, чем NHEJ. Чтобы искусственно повысить их эффективность, а также ввести донорную молекулу для нужного изменения геномной ДНК используют двухцепочечные ДНК, одноцепочечные олигодезоксинуклеотиды и длинные одноцепочечные ДНК. В данном обзоре рассмотрены донорные молекулы, которые используют для репарации двухцепочечных разрывов с помощью HDR или SSTR при редактировании генома, их применение и модификации, повышающие эффективность HDR и SSTR.
ВВЕДЕНИЕ
Целевое редактирование генома стало мощным инструментом изучения свойств генов или изменения нуклеотидной последовательности. За последние несколько десятилетий разработаны программируемые нуклеазы – нуклеазы цинковых пальцев (ZFN) [1], мегануклеазы [2], эффекторные нуклеазы, подобные активаторам транскрипции (TALEN) [3], а также система, основанная на коротких палиндромных повторах, регулярно расположенных группами (CRISPR) и CRISPR-ассоциированных белках (Cas) [4], которые позволяют вносить двухцепочечный разрыв (ДЦР) в интересующую геномную последовательность [5].
Репарация молекулы ДНК, в которой возник ДЦР, может проходить несколькими путями, основные из которых – негомологичное соединение концов (NHEJ) и направленная гомологичная репарация (HDR) [6]. К более редкому механизму репарации ДЦР можно отнести отжиг одиночной цепи (SSA – single strand annealing), частным случаем которого является репарация с помощью одноцепочечной матрицы (SSTR).
При NHEJ часто возникают ошибки, представленные в основном короткими вставками и делециями (инделами). В многоэтапном процессе NHEJ участвуют белки, которые распознают, укорачивают и лигируют концы ДНК. После укорачивания образуются концы ДНК различной конфигурации, а поврежденные концы цепи соединяются лигазой без использования молекулярных доноров. В результате исправленная ДНК часто содержит мутации [7]. В исследовательских целях путь NHEJ в сочетании с геномным редактированием можно использовать для направленного нокаута генов [8].
Оставшиеся два механизма – HDR и SSTR – менее ошибочны, однако требуют введения донорной молекулы, с которой должна произойти рекомбинация для репарации ДЦР [9, 10]. При этом эффективность HDR и SSTR при редактировании генома клеток животных ниже, чем NHEJ [11–13], однако только эти два механизма открывают возможность внесения в геном целенаправленных изменений, таких как вставка фрагмента или целого гена.
Низкая эффективность HDR и SSTR обусловлена несколькими факторами. Во-первых, HDR прямо конкурирует с путем репарации NHEJ. NHEJ активен на протяжении всего клеточного цикла, тогда как HDR ограничена лишь поздней фазой G2 и S клеточного цикла, когда происходит репликация ДНК перед делением клетки. HDR конкурирует также с еще одним механизмом репарации – микрогомологичным соединением концов (MHEJ), активным в S (и ранней M) фазах [14]. Во-вторых, на HDR могут отрицательно влиять соматические или спорадические мутации в таких генах, как RAD51, BRCA1 или BRCA2, белковые продукты которых непосредственно участвуют в репарации ДЦР. В случае организма или клеточной линии с уже существующей мутацией в этих генах, а также при попытке их редактирования с помощью CRISPR-Cas работа будет существенно осложнена [15]. Показано, что задержка клеточного цикла в фазах G2/M повышает эффективность геномного редактирования [16]. Наилучших результатов удалось добиться при использовании нокодазола, который эффективно и обратимо блокирует клеточный цикл в фазе G2/M, когда ДНК уже полностью реплицировалась, а ядерная оболочка остается разрушенной, что улучшает доступ Cas9 к ДНК и повышает эффективность HDR [17].
МЕХАНИЗМЫ РЕПАРАЦИИ
Репарация ДЦР может пойти разными путями в зависимости от таких факторов, как фаза клеточного цикла, наличие и близость донорной молекулы, активность факторов, участвующих в разных путях репарации.
Путь NHEJ не зависит от донорной молекулы и фазы клеточного цикла, поэтому он является доминирующим. В ходе NHEJ гетеродимер Ku70-Ku80 соединяет концы ДНК, а затем комплекс LIG4-XRCC4 осуществляет лигирование. NHEJ делится на каноническое негомологичное соединение концов (к-NHEJ) и альтернативное негомологичное соединение концов (а-NHEJ), также называемое микрогомологичным соединением концов (MHEJ) (рис. 1). ДЦР репарируются посредством спаривания оснований одиночных цепей ДНК с последующим укорачиванием концевых участков, заполнением пробелов и лигированием ДНК [18].
Рис. 1.
Канонический и альтернативный пути NHEJ. а – Канонический путь NHEJ, включающий белки KU и XRCC4, может консервативным способом соединять концы молекулы ДНК в месте разрыва, даже дистальные и не полностью комплементарные; б – основное событие альтернативного пути – делеция на стыке, обычно связанная с использованием внутренних микрогомологий. XRCC1 и ДНК-лигаза-III лигируют концы цепи в месте ДЦР. Адаптировано из работы [19], лицензия Creative Common CC BY. ДЦР – двухцепочечный разрыв ДНК, NHEJ – негомологичное соединение концов, ДНК-ПК – ДНК-протеинкиназа.

HDR с использованием двухцепочечной матрицы проходит по каноническому пути, при котором RAD51 образует филаменты на 3'-концах цепи ДНК-мишени, чтобы обеспечить инвазию одиночной цепи поврежденной ДНК в донорную молекулу. При этом необходимы факторы, которые обеспечивают взаимодействие RAD51 с ДНК, включая BRCA2, PALB2 и DSS1. Эффективность HDR с использованием двухцепочечной матрицы снижается при истощении или инактивации этих факторов [20]. В ходе HDR комплекс MRN узнает ДЦР, гидролизует 5′-концы ДНК по обе стороны от него на расстоянии 15–20 п.н. [6]. Завершают резекцию белки SGS1, DNA2 и EXO1 [21]. Затем комплекс MRN перемещается к фланкирующим участкам двухцепочечной матрицы и привлекает протеинкиназу ATM – ключевой фермент сигнальной системы ДЦР [22]. MRN также связывает концы ДНК, что увеличивает его локальную концентрацию и таким образом способствует активации ATM [23]. Белок RPA покрывает одноцепочечные участки ДНК. После этого белок RAD51 образует нуклеопротеиновые филаменты с одноцепочечными 3′-концами ДНК, которые затем выполняют поиск комплементарных участков с образованием структуры Холлидея [24].
HDR может происходить двумя путями: синтеззависимым отжигом цепи (SDSA) или восстановлением ДЦР. SDSA – предпочтительный путь направленной гомологичной репарации в соматических клетках. Он приводит к облигатному некроссоверному исходу, что позволяет избежать потенциальных хромосомных перестроек и потери гетерозиготности [25]. В процессе SDSA сначала одна из цепей поврежденной ДНК внедряется в молекулу-матрицу, в результате чего образуются D-петля и структура Холлидея [26]. ДНК-полимераза продлевает внедренную цепь комплементарно молекуле-матрице. Одновременно с этим комплементарно вытесненной цепи синтезируется остальная часть поврежденной ДНК. При этом возможен процесс миграции точки ветвления, когда точка пересечения цепей, принадлежащих рекомбинирующим ДНК, начинает перемещаться между ними. Иногда в процессе слияния синтезированных цепей с молекулой ДНК могут возникать гетеродуплексы, другие пробелы и бреши, которые устраняются при лигировании, после чего рекомбинацию можно считать завершенной [24].
При восстановлении ДЦР происходит укорочение 5′-концов и инвазия 3′-концов в гомологичную матрицу – этому способствуют белки DMC1 и RAD51. Комплексы стабилизируются, образуется D-петля и затем структура Холлидея. В разрезании структуры Холлидея участвуют резолвазы – ферменты, обладающие эндонуклеазной активностью [27]. В зависимости от исхода разрезания образуется либо кроссоверная последовательность (в большинстве случаев), либо некроссоверная [24].
Если матрицей для репарации ДЦР служит одноцепочечная молекула, то репарация проходит по альтернативному пути – с помощью одноцепочечной матрицы (SSTR) (рис. 2) [10]. SSTR не зависит от RAD51/BRCA2, поскольку RAD51 может способствовать повторному отжигу двух цепей геномной ДНК. В этом случае сразу после разрыва укорачиваются 5′-концы и остаются одноцепочечные 3′-концы, которые участвуют в отжиге с экзогенным донором [28]. Этот вид репарации зависит от RAD52, RAD59, SRS2 и комплекса MRE11–RAD50–XRS2 (MRX) [29]. Как только MRX связывается с молекулой ДНК и инициирует укорочение концов ДЦР, донорная молекула оцДНК с помощью белка RAD52 может отжигаться с доступными концами геномной ДНК. Отжигу способствуют Rad59 и Srs2, они предотвращают взаимодействие концов ДНК с Rad51 и блокируют отжиг, опосредованный Rad52. Матрица оцДНК копируется ДНК-полимеразой (предположительно ДНК-полимеразой δ), а затем отжигается на противоположной стороне ДЦР, вероятно, через Rad52. Несовпадения на 5'-конце донора оцДНК создают облигатный гетеродуплекс, который исправляется Msh2. Участки, которые остаются после SSTR, часто заполняются полимеразой ζ [30].
Рис. 2.
Репарация ДЦР с использованием одноцепочечных матриц. MRX показан фиолетовым, RAD52 – голубым, Msh2 – зеленым, полимераза ζ – оранжевым и δ – розовым. Адаптировано из работы [30] с разрешения издательства.

ВИДЫ И СИНТЕЗ ДОНОРНЫХ МОЛЕКУЛ
На данный момент при геномном редактировании в качестве донорных молекул используют одноцепочечные (одноцепочечные олигодезоксирибонуклеотиды, оцОДН) и длинные оцДНК (доцДНК) [31, 32]), а также двухцепочечные – плазмиды и продукты амплификации [33, 34].
дцДНК могут быть как специально сконструированными донорными плазмидами, так и продуктами амплификации. Плазмиды используют чаще, поскольку обычно они значительно меньше традиционных линейных донорных молекул, а получать их проще и дешевле [35]. Донорная плазмида – кольцевая дцДНК, на которой располагаются последовательность для вставки и плечи гомологии. Длина плазмиды может достигать 15 т.п.н., длина плеча гомологии обычно находится в диапазоне от 500 до нескольких тысяч пар нуклеотидов [36]. Гомологичная последовательность может быть амплифицирована из интересующего гена и затем вставлена в остов плазмиды [37]. Существует опосредованная CRISPR/Cas9 стратегия интеграции с использованием донорной плазмиды, несущей два одинаковых сайта расщепления, фланкирующих кассету с обеих сторон. После доставки донорной плазмиды вместе с мРНК Cas9 и гидовой РНК (гРНК) в клетке разрезаются обе стороны экзогенной кассеты, а в нужный хромосомный сайт вносится также ДЦР. Такой способ, как полагали авторы метода, должен приводить к точной интеграции без плазмидного остова, однако точность этого метода ниже, чем при классическом применении донорных плазмид [38].
Двухцепочечные линейные матрицы обычно создают с помощью ПЦР. Для повышения эффективности HDR при их использовании применяют модификацию 5′-концов. Модифицированные донорные молекулы меньше образуют внехромосомные конкатемеры, что повышает доступность клетки для донорной молекулы, а также могут снижать доступность 5′-концов геномной ДНК для NHEJ.
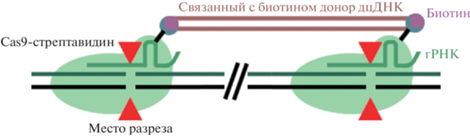
Важно отметить, что нуклеаза, используемая при редактировании с помощью системы CRISPR-Cas, может быть доставлена в клетки как в виде ДНК (например, в составе плазмиды), так и в виде рибонуклеопротеинового комплекса (РНП-комплекса) вместе с гРНК. В последнем случае нуклеазу можно модифицировать. Разработан метод, в котором длинная двухцепочечная матрица была биотинилирована с 5′-концов. Ее конъюгировали с двумя белками Cas9 (рис. 3). С помощью такой конструкции проведена замена двух экзонов гена A на два экзона гена B для создания модельных мышей. Достигнута 87% целевая эффективность и 12.5% эффективность создания гомозигот при редактировании зигот [39]. Вероятно, эффективность внесения изменений в геном возрастает, поскольку матрица находится недалеко от места разрыва, что способствует повышению вероятности HDR. Этот метод потенциально можно использовать для замены длинного фрагмента ДНК, например, целого гена или энхансера.
Рис. 3.
дцДНК, конъюгированная с белками Cas9. Адаптировано из работы [39] с разрешения издательства.
Длина доцДНК, служащих матрицами для репарации, может достигать 3 т.н. Для синтеза этих ДНК амплифицируют нужный участок и прилегающие плечи гомологии с помощью ПЦР с двойным биотинилированным прямым праймером. Используют либо химерную ПЦР подходящей ДНК-матрицы, либо субклонирование в случае более сложных матриц [31]. доцДНК, полученная с помощью биотинилирования асимметричных ПЦР-продуктов, нуждается в дальнейшей биотиновой аффинной очистке с использованием взаимодействия биотин-стрептавидин. Создать доцДНК можно также с помощью обратной транскрипции. В этом методе кДНК генерируется процессивной обратной транскриптазой, например, TGIRT-III [40]. Подходы, основанные на обратной транскрипции, могут эффективно использоваться для получения доноров оцДНК длиной до 3.5 т.н. [41–43]. Однако надо учитывать, что используемые для генерации линейной доцДНК ферменты, осуществляющие обратную транскрипцию, могут допускать ошибки при копировании [44], что затрудняет получение точной последовательности ДНК. Кроме того, эти ферменты часто генерируют укороченные продукты доцДНК [45]. оцОДН – небольшие одноцепочечные молекулы длиной до 200 н., как и любые другие донорные молекулы, состоят из последовательности для вставки и двух плеч гомологии. Длина плеча гомологии может варьировать от 30 до 60 н. (оцОДН можно заказать у биотехнологических компаний [46]). Важно понимать, что стандартный амидофосфитный метод синтеза олигонуклеотидов, используемый в том числе в российских биотехнологических компаниях, сопряжен с большим количеством ошибок при синтезе длинных (более 100) олигонуклеотидов, что ведет к повышению ошибок в экспериментах по редактированию. Ряд ведущих зарубежных компаний гарантируют высокое качество синтеза олигонуклеотидов длиной до 200 н.
ПОДБОР оцОДН ДЛЯ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ SSTR
В зависимости от цепи, на которой отжигается гРНК для CRISPR-Cas9, возможны две различные ориентации оцДНК (“целевая” и “нецелевая”). Целевая оцДНК имеет ту же последовательность, с которой отжигается гРНК, тогда как нецелевая комплементарна ей и содержит последовательность PAM (рис. 4). Предыдущие исследования, в которых в качестве донорных матриц использовали короткие оцДНК, предполагают, что отбор цепи критически влияет на эффективность включения. Это обусловлено тем, что Cas9 высвобождает нецелевую PAM-дистальную цепь геномной ДНК (с которой отжигается гРНК), но плотно захватывает три другие цепи. Свободная цепь становится доступной для отжига с комплементарным ей оцОДН. Донорный оцОДН, комплементарный ненаправленной цепи, повышает частоту SSTR в 2.6 раза по сравнению с оцОДН, комплементарным направленной цепи [47, 48].
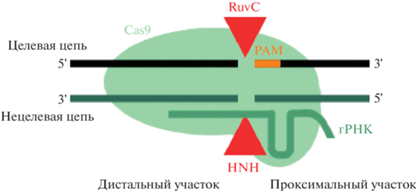
Кроме того, в этих исследованиях установлено, что длина плеч гомологии также критически важна для эффективности SSTR при использовании доноров оцДНК. Сконструированы оцОДН с разными длинами плеч на 5′- и 3′-стороне. Наибольшую эффективность (57 ± 5%) SSTR обеспечивало использование асимметричной оцДНК, оптимизированной для отжига с перекрытием сайта разрезания Cas9 (36 п.н.) на PAM-дистальной стороне и с удлинением (91 п.н.) на PAM-проксимальной стороне разрыва [47, 48].
Считается, что более короткое 3'-плечо позволяет оцОДН отжигаться с таргетным локусом ДНК без необходимости инвазии 3'-конца поврежденной цепи в ДНК, служащей матрицей [49].
С целью повышения эффективности SSTR с использованием оцДНК с белком Cas9 конъюгировали несколько молекул оцОДН (рис. 5). В ходе исследования доказано, что множественная конъюгация оцОДН с Cas9 значительно увеличивает эффективность точного редактирования генома, и такая платформа совместима с оцОДН различной длины [50].
Рис. 5.
Конъюгация оцОДН с Cas9. Адаптировано из работы [50], лицензия Creative Commons Attribution 4.0 International License.

В одном из экспериментов с оцОДН мутацию расположили ближе к концу плеч гомологии и проверили, сможет ли ингибирование деградации концов оцОДН посредством включения фосфотиоатных связей повысить эффективность редактирования. Введение фосфотиоатных связей позволило повысить эффективность редактирования с мутацией на 3′-конце на 4%. Защита 5′‑конца не дала никакого эффекта, как и локализация мутантного нуклеотида рядом с ДЦР и на расстоянии 61 н. от 3′-конца. Защита с помощью фосфотиоатных связей увеличивала эффективность редактирования мутации на расстоянии 15 н. от разрыва и 27 н. у 3'-конца [51].
СРАВНЕНИЕ ДОНОРНЫХ МОЛЕКУЛ
В качестве донорных матриц можно использовать плазмидные дцДНК, с их помощью эффективно проводили модификацию генов в процессе HDR в нескольких эукариотических системах [52]. дцДНК обычно применяют для интеграции больших участков генов, например, флуоресцентных репортерных генов или генов резистентности к антибиотикам, что позволяет затем осуществить селекцию клеток, в которых произошли изменения. Исследования, проведенные на рыбах (Danio rerio), показали, что плазмидная ДНК в качестве донорной молекулы более эффективна (46%), чем линейные дцДНК (1%) [53]. Однако у двухцепочечных доноров есть ряд минусов. Их создание может быть дорогим и трудоемким [35], а использование может приводить к нежелательным событиям: дцДНК могут встраиваться в ДНК клетки в процессе NHEJ, что приводит к дублированию плеч гомологии или частичному включению матрицы дцДНК [54]. Также дцДНК могут встраиваться в нецелевые ДЦР, которые встречаются в ДНК клетки независимо от воздействия нуклеаз [55]. Кроме того, матрицы дцДНК могут оказывать цитотоксическое действие на культивируемые клетки [42, 50, 56–59]. Еще одно неудобство заключается в том, что молекулы дцДНК способны формировать большие внехромосомные массивы посредством как концевых соединений, так и путей гомологичной рекомбинации [58, 60]. Сцепление донорных молекул в большие массивы ведет к уменьшению количества отдельных молекул, служащих матрицами для репарации. Более того, если введенная ДНК образует сцепления, будучи изолированной от ядерной ДНК [60], то это может препятствовать репарации с ее участием, пока изолированные сцепленные донорные молекулы не получат доступ к ядру после разрушения ядерной оболочки [61].
ОцОДН в основном используются для исправления точечных мутаций [50]. В отличие от дцДНК готовые оцОДН можно получить за несколько дней, что позволяет ускорить подготовку к редактированию и снизить затраты на синтез матриц [37]. Основной недостаток оцОДН-доноров – их размер, равный примерно 200 н. [62]. Такое ограничение обусловлено повышением количества ошибок в более длинных олигонуклеотидных последовательностях, синтезируемых стандартным амидофосфитным методом, а также сложностью очистки конечного продукта, так как добавляется примесь не полностью синтезированных продуктов. Это ограничение делает невозможным вставку более длинного фрагмента ДНК. Также выяснилось, что опосредованная оцОДН репарация может привести к ошибкам – интеграции части донорной молекулы в неправильном направлении, введении нескольких фрагментов оцОДН с разной ориентацией [63]. По данным недавних исследований оцДНК могут индуцировать вставки или делеции при репарации ДЦР, созданных с использованием системы CRISPR-Cas9 [64, 65].
Недавно стали использовать третий вид матриц – доцДНК [43], подходящий для вставки достаточно большого участка ДНК – от 200 до 5000 н. [66]. При этом доцДНК меньше по размеру и менее цитотоксичны, чем плазмидные матрицы. Впервые длинные одноцепочечные матрицы использовали в методе под названием Easi-CRISPR (эффективная вставка с помощью оцДНК-CRISPR). Easi‑CRISPR разработан в качестве одноэтапного метода создания целевой вставки в молекулу ДНК с высокой эффективностью [43]. В этой стратегии использовали доцДНК с предварительно собранными РНП-комплексами crRNA + + tracrRNA + Cas9 (RNP) для двух сайтов CRISPR-Cas9 в одном локусе [67]. Проведенное на Т-клетках человека сравнение доцДНК и дцДНК, выявило меньшую цитотоксичность доцДНК. Кроме того, доцДНК реже, чем оцДНК встраивались в нецелевые участки. Эффективность редактирования при использовании доцДНК и дцДНК была сопоставимой – 50–60% [54]. Однако данные об эффективности и надежности этого метода все еще ограничены. В исследовании на мышах установлено, что использование доцДНК привело не только к исправлению целевой мутации, но и ко многим нежелательным событиям – инделам, точечным мутациям, частичной интеграции донора [68]. Химический синтез доцДНК может быть достаточно дорогим по сравнению с синтезом оцОДН. Кроме того, синтез доцДНК с помощью Easi-CRISPR и плазмидного разрезания с последующей денатурацией считается трудоемким [44, 69], поэтому доцДНК относительно редко применяют для исправления мутаций или вставки фрагментов ДНК, несмотря на потенциальные преимущества этого метода [70].
В опытах на D. rerio сравнили все четыре варианта донорных молекул. С этой целью использовали специально подготовленные доцДНК, симметричные и асимметричные оцОДН, плазмидную донорную молекулу и линеаризованную дцДНК (ПЦР-продукт). Ген tyr кодирует тирозиназу, которая превращает тирозин в меланин, а рыбки данио с мутацией в этом гене (tyr25del/25del) имеют фенотип альбиносов. В отредактированных клетках наблюдалось восстановление гена tyr с последующим появлением пигментации. При редактировании плазмидой только у 5.4% головастиков появлялось небольшое количество пигмента. При использовании симметричных оцОДН пигмент проявился у 39.1% головастиков, тогда как в случае асимметричных – только у 1.3%. При редактировании с использованием доцДНК 98.5% головастиков имели заметную пигментацию. HDR с применением ПЦР-продуктов дцДНК была неэффективной [46].
ЗАКЛЮЧЕНИЕ
На данный момент в редактировании генома с дальнейшей репарацией HDR или SSTR используют одноцепочечные и двухцепочечные донорные молекулы. Нет единого мнения, какой из типов донорных молекул наиболее эффективный; каждый из них применяется в разных целях и имеет свои преимущества и недостатки. Двухцепочечные донорные молекулы выбирают в основном для вставки больших участков, например генов-репортеров, в геном, что может быть полезным для отбора отредактированных клонов клеток по экспрессии маркерного гена (флуоресцентного репортера или устойчивости к определенным антибиотикам) с целью получения клональной культуры. В дальнейшем при вырезании генов-репортеров можно получить клоны клеток с измененным геномом (редактированной мутацией), пригодных, например, для ex vivo терапии ряда заболеваний человека. оцОДН используют для небольших изменений генома, таких как исправление точковых мутаций. Такой подход можно использовать, к примеру, для коррекции мутаций, приводящих к наследственным заболеваниям, разработки терапии in vivo. Промежуточный вариант – доцДНК – имеет достаточную длину для интеграции больших фрагментов ДНК, однако отличается высокой стоимостью, сопоставимой с использованием плазмидной ДНК, а также схожей эффективностью, что нивелирует преимущества такого типа матриц. Этот подход пока не нашел широкого применения в экспериментах по геномному редактированию. Несмотря на то, что поставленные задачи зачастую определяют тип используемых донорных молекул, необходимы дополнительные эксперименты, позволяющие определить оптимальные длины и состав донорных молекул в каждом конкретном случае.
Работа выполнена в рамках государственного задания Минобрнауки России для ФГБНУ “МГНЦ”.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Durai S., Mani M., Kandavelou K., Wu J., Porteus M.H., Chandrasegaran S. (2005) Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucl. Acids Res. 33(18), 3978–5990. https://doi.org/10.1093/nar/gki912
Silva G., Poirot L., Galetto R., Smith J., Montoya G., Duchateau Ph., Pâques F. (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr. Gene Therapy. 11(1), 11–27. https://doi.org/10.2174/156652311794520111
Cermak T., Doyle E. L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucl. Acids Res. 39(12), e82. https://doi.org/10.1093/nar/gkr218
Jinek M., Chylinski K., Fonfara I., Hauer M., Dou-dna J.A., Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337(6096), 816–821. https://doi.org/10.1126/science.1225829
Guha T.K., Wai A., Hausner G. (2017) Programmable genome editing tools and their regulation for efficient genome engineering. Comp. Struct. Biotechnol. J. 15, 146–160. https://doi.org/10.1016/j.csbj.2016.12.006
Symington L.S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. https://doi.org/10.1146/annurev-genet-110410-132435
Chang H., Pannunzio N.R., Adachi N., Lieber M.R. (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18(8), 495–506. https://doi.org/10.1038/nrm.2017.48
Li T., Huang S., Zhao X., Wright D.A., Carpenter S., Spalding M.H., Weeks D.P., Yang B. (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucl. Acids Res. 39(14), 6315–6325. https://doi.org/10.1093/nar/gkr188
Devkota S. (2018) The road less traveled: strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Casmediated transgenesis. BMB Rep. 51(19), 437–443. https://doi.org/10.5483/BMBRep.2018.51.9.187
Richardson C.D., Kazane K.R., Feng S.J., Zelin E., Bray N.L., Schäfer A.J., Floor S.N., Corn J.E. (2018) CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 50(8), 1132–1139. https://doi.org/10.1038/s41588-018-0174-0
Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B., Meng X., Miller J.C., Zhang L., Rebar E.J., Gregory Ph.D., Urnov F.D., Jaenisch R. (2009) Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857. https://doi.org/10.1038/nbt.1562
Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C., Zeitler B., Cherone J.M., Meng X., Hinkley S.J., Rebar E.J., Gregory Ph.D., Urnov F.D., Jaenisch R. (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–734. https://doi.org/10.1038/nbt.1927
Sommer D., Peters A., Wirtz T., Mai M., Ackermann J., Thabet Y., Schmidt J., Weighardt H., Wunderlich F.T., Degen J., Schultze J.L., Beyer M. (2014) Efficient genome engineering by targeted homologous recombination in mouse embryos using transcription activator-like effector nucleases. Nat. Commun. 5, 3045. https://doi.org/10.1038/ncomms4045
Heyer W.D., Ehmsen K.T., Liu J. (2010) Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139. https://doi.org/10.1146/annurev-genet-051710-150955
Yanik M., Ponnam S., Wimmer T., Trimborn L., Mül-ler C., Gambert I., Ginsberg J., Janise A., Domicke J., Wende W., Lorenz B., Stieger K. (2018) Development of a reporter system to explore MMEJ in the context of replacing large genomic fragments. Mol. Therapy. Nucl. Acids. 11, 407–415. https://doi.org/10.1016/j.omtn.2018.03.010
Smirnikhina S.A., Anuchina A.A., Lavrov A.V. (2019) Ways of improving precise knock-in by genome-editing technologies. Hum. Genet. 138(1), 1–19. https://doi.org/10.1007/s00439-018-1953-5
Lin S., Staahl B.T., Alla R.K., Doudna J.A. (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, e04766. https://doi.org/10.7554/eLife.04766
Seol J.H., Shim E.Y., Lee S.E. (2018) Microhomology-mediated end joining: good, bad and ugly. Mutat. Res. 809, 81–87.
González-Marín C., Gosálvez J., Roy R. (2012) Types, causes, detection and repair of DNA fragmentation in animal and human sperm cells. Internat. J. Mol. Sci. 13(11), 14026–14052. https://doi.org/10.3390/ijms131114026
Liu M., Rehman S., Tang X., Fan Q., Chen D., Ma W. (2019) Methodologies for improving HDR efficiency. Front. Genet. 9(691). https://doi.org/10.3389/fgene.2018.00691
Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 34(6), 981‒994. https://doi.org/10.1016/j.cell.2008.08.037
Falck J., Coates J., Jackson S.P. (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 434(7033), 605‒611. https://doi.org/10.1038/nature03442
Dupré A., Boyer-Chatenet L., Gautier J. (2006) Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 13(5), 451–457. https://doi.org/10.1038/nsmb1090
Helleday T., Lo J., van Gent D.C., Engelward B.P. (2007) DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair. 6(7), 923–935. https://doi.org/10.1016/j.dnarep.2007.02.006
Liu J., Ede C., Wright W. D., Gore S.K., Jenkins S.S., Freudenthal B.D., Washington M.T., Veaute X., Heyer W.-D. (2017) Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase δ-extending D-loops. eLife. 6, e22195. https://doi.org/10.7554/eLife.22195
McMahill M.S., Sham C.W., Bishop D.K. (2007) Synthesis-dependent strand annealing in meiosis. PLoS Biol. 5(11), e299. https://doi.org/10.1371/journal.pbio.0050299
Agmon N., Yovel M., Harari Y., Liefshitz B., Kupiec M. (2011) The role of Holliday junction resolvases in the repair of spontaneous and induced DNA damage. Nucl. Acids Res. 39(16), 7009–7019. https://doi.org/10.1093/nar/gkr277
Davis L., Zhang Y., Maizels N. (2018) Assaying repair at DNA nicks. Methods Enzymol. 601, 71–89. https://doi.org/10.1016/bs.mie.2017.12.001
Gallagher D.N., Pham N., Tsai A.M., Janto A.N., Choi J., Ira G., Haber J.E. (2020) A Rad51-independent pathway promotes single-strand template repair in gene editing. PLoS Genet. 16(10), e1008689. https://doi.org/10.1371/journal.pgen.1008689
Gallagher D.N., Haber J.E. (2021) Single-strand template repair: key insights to increase the efficiency of gene editing. Curr. Genet. 67, 747–753. https://doi.org/10.1007/s00294-021-01186-z
Hu Z., Zhou M., Wu Y., Li Z., Liu X., Wu L., Liang D. (2019) ssODN-mediated in-frame deletion with CRISPR/Cas9 restores FVIII function in hemophilia A-patient-derived iPSCs and ECs. Mol. Therapy. Nucl. Acids. 17, 198–209. https://doi.org/10.1016/j.omtn.2019.05.019
Bennett H., Aguilar-Martinez E., Adamson A.D. (2021) CRISPR-mediated knock-in in the mouse embryo using long single stranded DNA donors synthesised by biotinylated PCR. Methods (San Diego, Calif.). 191, 3–14. https://doi.org/10.1016/j.ymeth.2020.10.012
Cristea S., Freyvert Y., Santiago Y. (2013) In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioengin. 110(3), 871–880. https://doi.org/10.1002/bit.24733
Fueller J., Herbst K., Meurer M., Gubicza K., Kurtulmus B., Knopf J.D., Kirrmaier D., Buchmuller B.C., Pereira G., Lemberg M.K., Knop M. (2020) CRISPR-Cas12a-assisted PCR tagging of mammalian genes. J. Cell Biol. 219(6), e201910210. https://doi.org/10.1083/jcb.201910210
Chen F., Pruett-Miller S.M., Davis G.D. (2015) Gene editing using ssODNs with engineered endonucleases. Methods Mol. Biol. 1239, 251–265. https://doi.org/10.1007/978-1-4939-1862-1_14
Мягкая Н.О., Марочкин Н.А., Норбобаева М.Б., Серба Т.В., Стародуб М.А., Курбединов Р.А., Костенко С.А., Заяц И.В. (2020) Обзор метода CRISPR/Cas9 для редактирования генома. Молодой ученый. 12(302), 81–85.
Designing Homologous Repair Templates. New England BioLabs Inc. https://international.neb.com/applications/genome-editing/designing-homologous-repair-templates
Mao C.Z., Zheng L., Zhou Y.M., Wu H.-Y., Xia J.-B., Liang C.-Q., Guo X.-F., Peng W.-T., Zhao H., Cai W.-B., Kim S.-K., Park K.-S., Cai D.-Q., Qi X.-F. (2018) CRISPR/Cas9-mediated efficient and precise targeted integration of donor DNA harboring double cleavage sites in Xenopus tropicalis. FASEB J. fj201800093. Advance online publication. https://doi.org/10.1096/fj.201800093
Pineault K.M., Novoa A., Lozovska A., Wellik D.M., Mallo M. (2019) Two CRISPR/Cas9-mediated methods for targeting complex insertions, deletions, or replacements in mouse. MethodsX. 6, 2088–2100. https://doi.org/10.1016/j.mex.2019.09.003
Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P. J., Hiatt J., Saco J., Krystofinski P., Li H., Tobin V., Nguyen D.N., Lee M.R., Putnam A.L., Ferris A.L., Chen J.W., Schickel J.-N., Pellerin L., Carmody D., Alkorta-Aranburu G., Del Gaudio D., Matsumoto H., Morell M., Mao Y., Cho M., Quadros R.M., Gurumurthy C.B., Smith B., Haugwitz M., Hughes S.H., Weissman J.S., Schumann K., Esensten J.H., May A.P., Ashworth A., Kupfer G.M., Greeley S.A.W., Bacchetta R., Meffre E., Roncarolo M.G., Romberg N., Herold K.C., Ribas A., Leonetti M.D., Marson A. (2018) Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 559(7714), 405–409. https://doi.org/10.1038/s41586-018-0326-5
Quadros R.M., Miura H., Harms D.W., Akatsuka H., Sato T., Aida T., Redder R., Richardson G.P., Inagaki Y., Sakai D., Buckley S.M., Seshacharyulu P., Batra S.K., Behlke M.A., Zeiner S.A., Jacobi A.M., Izu Y., Thoreson W.B., Urness L.D., Mansour S.L., Ohtsuka M., Gurumurthy C.B. (2017) Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18(1), 92. https://doi.org/10.1186/s13059-017-1220-4
Miura H., Gurumurthy C.B., Sato T., Sato M., Ohtsuka M. (2015) CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci. Rep. 5, 12799. https://doi.org/10.1038/srep12799
Leonetti M.D., Sekine S., Kamiyama D., Weissman J.S., Huang B. (2016) A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. USA. 113(25), E3501–E3508. https://doi.org/10.1073/pnas.1606731113
Miura H., Quadros R.M., Gurumurthy C.B., Ohtsuka M. (2018) Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protocols. 13(1), 195–215. https://doi.org/10.1038/nprot.2017.153
Harrison G.P., Mayo M.S., Hunter E., Lever A.M. (1998) Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5' and 3' of the catalytic site. Nucl. Acids Res. 26(14), 3433–3442. https://doi.org/10.1093/nar/26.14.3433
Bai H., Liu L., An K., X. Lu, Harrison M., Zhao Y., Yan R., Lu Z., Li S., Lin S., Liang F., Qin W. (2020) CRISPR/Cas9-mediated precise genome modification by a long ssDNA template in zebrafish. BMC Genomics. 21(1), 67. https://doi.org/10.1186/s12864-020-6493-4
Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Kaini P., Sander J.D., Joung J.K., Peterson R.T., Yeh J.-R.J. (2013) Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One. 8(7). e68708 https://doi.org/10.1371/journal.pone.0068708
Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E. (2016) Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34(3), 339–344. https://doi.org/10.1038/nbt.3481
O'Brien A.R., Wilson L., Burgio G., Bauer D.C. (2019) Unlocking HDR-mediated nucleotide editing by identifying high-efficiency target sites using machine learning. Sci. Rep. 9(1), 2788. https://doi.org/10.1038/s41598-019-39142-0
Lim D., Sreekanth V., Cox K.J., Law B.K., Wagner B.K., Karp J.M., Choudhary A. (2020) Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun. 11(1), 4043. https://doi.org/10.1038/s41467-020-17725-0
Harmsen T., Klaasen S., van de Vrugt H., Te Riele H. (2018) DNA mismatch repair and oligonucleotide end-protection promote base-pair substitution distal from a CRISPR/Cas9-induced DNA break. Nucl. Acids Res. 46(6), 2945–2955. https://doi.org/10.1093/nar/gky076
Shao Y., Guan Y., Wang L., Qiu Z., Liu M., Chen Y., Wu L., Li Y., Ma X., Liu M., Li D. (2014) CRISPR/ Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat. Protocols. 9(10), 2493–2512. https://doi.org/10.1038/nprot.2014.171
Irion U., Krauss J., Nüsslein-Volhard C. (2014) Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development. 141(24), 4827–4830. https://doi.org/10.1242/dev.115584
Wang B., Karaçay B. Megamer single-stranded donor templates (ssDNA or ssODNs) for successful homology-directed repair (HDR) in genome editing applications. IDT. https://eu.idtdna.com/pages/education/ decoded/article/megamer-single-stranded-donor-templates-(ssdna-or-ssODNs)-for-successful-homology-directed-repair-(HDR)-in-genome-editing-applications
Yoshimi K., Oka Y., Miyasaka Y., Kotani Y., Yasumura M., Uno Y., Hattori K., Tanigawa A., Sato M., Oya M., Nakamura K., Matsushita N., Kobayashi K., Mashimo T. (2021) Combi-CRISPR: combination of NHEJ and HDR provides efficient and precise plasmid-based knock-ins in mice and rats. Hum. Genetics. 140(2), 277–287. https://doi.org/10.1007/s00439-020-02198-4
Highly specific gene knockins of long sequences using CRISPR/Cas9 and a single-stranded DNA donor template. Takara Bio USA, Inc. https://catalog.takara-bio.co.jp/PDFS/Highly-specific-gene-knockins-of-long-sequences-using-CRISPR_Cas9-and-a-single-stranded-DNA-donor-template.pdf
Zhao Y., Zheng Z., Cohen C.J., Gattinoni L., Palmer D.C., Restifo N.P., Rosenberg S.A., Morgan R.A. (2006) High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Therapy: J. Am. Soc. Gene Therapy. 13(1), 151–159. https://doi.org/10.1016/j.ymthe.2005.07.688
Mello C.C., Kramer J.M., Stinchcomb D., Ambros V. (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10(12), 3959–3970.
Forbes D.J., Kirschner M.W., Newport J.W. (1983) Spontaneous formation of nucleus-like structures around bacteriophage DNA microinjected into Xenopus eggs. Cell. 34(1), 13–23. https://doi.org/10.1016/0092-8674(83)90132-0
Stinchcomb D.T., Shaw J.E., Carr S.H., Hirsh D. (1985) Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol. Cell. Biol. 5(12), 3484–3496. https://doi.org/10.1128/mcb.5.12.3484-3496.1985
Ghanta K.S., Mello C.C. (2020) Melting dsDNA donor molecules greatly improves precision genome editing in Caenorhabditis elegans. Genetics. 216(3), 643–650. https://doi.org/10.1534/genetics.120.303564
Levi T., Sloutskin A., Kalifa R., Juven-Gershon T., Gerlitz O. (2020) Efficient in vivo introduction of point mutations using ssODN and a Co-CRISPR approach. Biol. Proc. Online. 22, 14. https://doi.org/10.1186/s12575-020-00123-7
Boel A., De Saffel H., Steyaert W., Callewaert B., De Paepe A., Coucke P.J., Willaert A. (2018) CRISPR/ Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis. Models Mech. 11(10), dmm035352. https://doi.org/10.1242/dmm.035352
Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O’Connor-Giles K.M. (2013) Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194(4), 1029–1035. https://doi.org/10.1534/genetics.113.152710
Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153(4), 910–918. https://doi.org/10.1016/j.cell.2013.04.025
Guide-it long ssDNA production system v2. Takara Bio USA, Inc. https://www.takarabio.com/products/gene-function/gene-editing/crispr-cas9/long-ssdna-for-knockins
Bassett A.R., Tibbit C., Ponting C.P., Liu J.L. (2013) Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 4(1), 220–228. https://doi.org/10.1016/j.celrep.2013.06.020
Codner G.F., Mianné J., Caulder A., Loeffler J., Fell R., King R., Allan A.J., Mackenzie M., Pike F.J., McCabe C.V., Christou S., Joynson S., Hutchison M., Stewart M.E., Kumar S, Simon M.M., Agius L., Anstee Q.M., Volynski K.E., Kullmann D.M., Wells S., Teboul L. (2018) Application of long single-stranded DNA donors in genome editing: generation and validation of mouse mutants. BMC Biol. 16(1), 70. https://doi.org/10.1186/s12915-018-0530-7
Yoshimi K., Kunihiro Y., Kaneko T. (2016) ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 7, 10431. https://doi.org/10.1038/ncomms10431
Ranawakage D.C., Okada K., Sugio K., Kawaguchi Y., Kuninobu-Bonkohara Y., Takada T., Kamachi Y. (2021) Efficient CRISPR-Cas9-mediated knock-in of composite tags in zebrafish using long ssDNA as a donor. Front. Cell Dev. Biol. 8, 598634. https://doi.org/10.3389/fcell.2020.598634
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология