

Молекулярная биология, 2022, T. 56, № 5, стр. 710-731
Мышиные модели хронических вирусных инфекций и ассоциированных с ними опухолей
Д. В. Авдошина a, А. С. Кондрашова a, М. Г. Беликова a, b, c, Е. О. Баюрова a, b, *
a Федеральный научный центр исследований и разработки иммунобиологических препаратов им. М.П. Чумакова Российской академии наук (Институт полиомиелита)
108819 Москва, Россия
b Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи Минздрава России
123098 Москва, Россия
c Российский университет дружбы народов
117198 Москва, Россия
* E-mail: bayurova_eo@chumakovs.su
Поступила в редакцию 18.03.2022
После доработки 12.04.2022
Принята к публикации 13.04.2022
- EDN: JGKEDY
- DOI: 10.31857/S0026898422050020
Аннотация
В настоящее время вирусы признаны одними из этиологических факторов развития опухолей человека. К онкогенным вирусам относятся вирус Эпштейна–Барр, папилломавирусы человека высокого канцерогенного риска, вирусы гепатита В и С, вирус Т-клеточного лейкоза человека типа I, вирус иммунодефицита человека типа 1 (опосредованно) и еще несколько предположительно онкогенных вирусов. Показано, что в патогенезе примерно 15% диагностируемых во всем мире опухолей человека участвуют вирусы. Онкогенные вирусы вызывают длительные персистирующие инфекции, при этом опухоль является случайным побочным продуктом стратегии вирусной репликации. Вирусы, как правило, не способны к индукции быстрого канцерогенеза, что подтверждает концепцию о развитии опухолей в результате совокупности множества накладывающихся друг на друга событий, в которых онкогенные вирусы человека играют разные, часто противоположные, роли. Одной из лучших экспериментальных in vivo систем для моделирования патологии человека, включая вирусные инфекции и образование опухолей, считается мышь. Однако мыши невосприимчивы к инфицированию известными онкогенными вирусами. Для преодоления этого ограничения и изучения различных аспектов вирус-ассоциированного канцерогенеза разработано множество мышиных моделей, начиная с ксенотрансплантатов тканей и клеток человека, включая вирус-инфицированные и опухолевые, и заканчивая генетически модифицированными мышами, восприимчивыми к вирусным инфекциям и вирус-ассоциированному канцерогенезу. В представленном обзоре рассмотрены основные известные на данные момент модели, проанализированы их преимущества и недостатки, описаны области их применения и намечены перспективы дальнейшего развития таких моделей.
ВВЕДЕНИЕ
Домовая мышь (Mus musculus) – одна из лучших экспериментальных in vivo систем для моделирования патологии человека, включая вирусные инфекции и образование опухолей. К преимуществам мышей в качестве моделей относятся небольшой размер, короткая продолжительность жизни, простота разведения, хорошо охарактеризованная иммунная система, большое молекулярное и физиологическое сходство с человеком и полностью расшифрованный геном, что подразумевает возможность обширных генетических модификаций. Мышиные модели оказались полезными для понимания биологических процессов, происходящих во время роста опухоли, и для проведения доклинических испытаний противоопухолевой терапии [1]. Однако мыши не могут быть инфицированы ни одним из вирусов, вызывающих хронические инфекции человека, связанные с развитием опухолей, включая вирусы гепатита В (ВГВ) и С (ВГС), вирус иммунодефицита человека типа 1 (ВИЧ-1), вирус Эпштейна–Барр (ВЭБ), вирус Т-клеточного лейкоза человека типа 1 (ВТЛЧ-I), папилломавирусы человека высокого канцерогенного риска (ВПЧ ВКР) и другие. Для изучения различных аспектов вирус-ассоциированного канцерогенеза разработано большое количество мышиных моделей вирус-ассоциированных опухолей, начиная с ксенотрансплантатов, полученных из эксплантов тканей человека, включая опухоли, и заканчивая генетически модифицированными мышами, восприимчивыми к вирусным инфекциям и вирус-ассоциированным опухолям. В настоящем обзоре рассмотрены принципы конструирования моделей, приведены примеры их использования для изучения хронических вирусных инфекций, вирус-ассоциированного онкогенеза и разработки методов их терапии.
МЫШИ С КСЕНОТРАНСПЛАНТАЦИЕЙ КЛЕТОК ЧЕЛОВЕКА – МОДЕЛИ КСЕНОГРАФТОВ
Ксенотрансплантаты, полученные от пациентов, в иммуносупрессированных мышах
В мышиных моделях трансплантированные клетки и ткани человека выполняют те же функции, что и в организме человека. Такие модели чрезвычайно полезны для прямого изучения патогенов человека и механизмов заражения.
В моделях опухолей, ассоциированных с вирусной инфекцией, опухолевые клетки человека или биоптаты опухолей (ксенотрансплантаты, полученные от пациентов, patient derived xenografts – PDX) трансплантируют мышам с иммунодефицитом. В течение последних двух десятилетий PDX-модели были основным инструментом для доклинического скрининга при разработке новых методов лечения опухолей. Эти модели помогли выявить клинически эффективные химиотерапевтические агенты, на настоящий момент они остаются основными моделями для фармацевтической промышленности [2], главным образом потому, что позволяют быстро и легко оценить эффекты, вызываемые различными агентами в опухолевых тканях и клетках in vivo [1]. Получение аналогичных моделей для отдельных пациентов стало шагом вперед в персонализации схем лечения [3].
Линии мышей, используемые для создания PDX-моделей. Клетки и ткани человека можно трансплантировать только иммунодефицитным/иммунокомпрометированным мышам. С этой целью разработана целая панель линий мышей с ослабленным иммунитетом. Наиболее широко используемые линии перечислены ниже в соответствии с уровнем иммунодефицита. Бестимусные голые мыши (nude) несут спонтанную делецию в гене Foxn1, которая нарушает развитие и/или вызывает отсутствие тимуса, что приводит к угнетению иммунной системы с уменьшением количества Т-клеток. Это тяжелый, но не абсолютный иммунодефицит, так как у мышей остается небольшое количество Т-клеток на периферии и интактный врожденный иммунитет с повышенной активностью NK-клеток. Это может ограничивать приживаемость ксенографтов (т.е. процент успешно приживленных опухолей), рост и способность к метастазированию большинства первичных солидных опухолей и делает невозможным приживление злокачественных гемопоэтических клеток [4]. У мышей с тяжелым комбинированным иммунодефицитом (SCID) в результате редкого врожденного заболевания практически отсутствует B- и T-клеточный иммунитет. У этих мышей наблюдается нормальная дифференцировка и функции нелимфоидных клеток крови, включая моноциты, гранулоциты, мегакариоциты, эритроциты и NK-клетки [5]. Хорошими реципиентами клеток человека оказались мыши NOD/LtSz-SCID, полученные Grenier и соавт. [6]. Высокие показатели приживаемости клеток человека у мышей NOD/LtSz-SCID объясняются множественной иммунной дисфункцией, которая включает снижение функции макрофагов, комплементзависимой гемолитической активности и активности NK-клеток [6], однако у этих мышей сохраняется остаточная активность NK-клеток [7]. Чтобы обойти эту проблему, были разработаны линии мышей NOD/SCID/β2mnull и NOD/SCID/γсnull с выраженным снижением продукции интерферона-γ (ИФН-γ) дендритными клетками. Обе линии лишены активности NK-клеток, что обеспечивает высокую приживаемость клеток человека [7, 8] Получены также варианты NOD.Cg-PrkdcscidIl2rgtm1Sug (NOG), NOD.Cg-PrkdcscidIl2rgtm1Wjll (NSG) и NOD.Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ (NRG) – линии иммунодефицитных мышей IL2rgnull на основе линии NOD. Мыши NOG и NSG имеют мутированный ген Prkdc. У мышей NOG укорочен цитоплазматический домен общей гамма-цепи рецептора интерлейкина 2 (Il2rg), у NSG гамма-цепь делетирована; мыши NRG имеют таргетные мутации в гене Rag1, вызывающие нарушение функций кодируемого белка [9]. В результате этих модификаций у мышей NOG критически нарушен врожденный и адаптивный иммунитет. У мышей NSG отсутствуют Т-, В- и NK-клетки, и они поддерживают высокие уровни приживления гемопоэтических стволовых клеток из пуповинной крови человека [10]. Более подробное обсуждение особенностей этих моделей выходит за пределы данного обзора.
PDX-модели инфекции вирусом Эпштейна–Барр (ВЭБ). К настоящему времени разработаны PDX-модели опухолей, связанных с рядом вирусных инфекций, начиная с ВЭБ. В 1999 г. Murakami и соавт. [11] впервые показали, что эпителиальные клеточные линии, инфицированные ВЭБ, принадлежащему к семейству герпесвирусов, при подсадке мышам SCID продуцируют опухоли. В этих экспериментах ВЭБ-положительные эпителиальные клеточные линии GT38 и GT39, полученные из незлокачественных участков карциномы желудка человека, имплантировали под кожу мышам SCID. У всех мышей примерно через 2 мес. после подсадки развились опухоли, которые представляли собой недифференцированную карциному с детектируемой кольцевой, но не линейной ДНК ВЭБ, экспрессирующую малую РНК-1, кодируемую ВЭБ [11]. В аналогичном эксперименте условно иммортализованные эндотелиальные клетки человека (HuARLT), пермиссивные к заражению другим герпесвирусом – вирусом герпеса, ассоциированным с саркомой Капоши (KSHV), инфицированные либо in vitro до приживления, либо in vivo после приживления, образовывали опухоли у мышей с иммунодефицитом (Rag2–/–γc–/–) [12]. В более поздних исследованиях обнаружено развитие ВЭБ-ассоциированных лимфоцитарных опухолей из образцов опухолей толстой кишки, желудка, молочной железы и легких, трансплантированных подкожно мышам NOG [13]. Опухоли, состоящие из CD45+/CD20+ ВЭБ+ В-клеток, быстро росли и образовывали большие метастатические поражения в лимфатических узлах, печени, легких и селезенке мышей, тогда как первично привитые карциномы росли медленно и не метастазировали [14]. Суммируя эти данные, можно сказать, что ВЭБ+ лимфоцитарные ксенографты представляют собой удобную модель для изучения ВЭБ-ассоциированных опухолей.
PDX-модели ВПЧ-инфекции и рака шейки матки. Разработана панель моделей рака шейки матки (cervical cancer, CC), основанная на ксенотрансплантатах образцов CC (CC-PDX), полученных от пациентов. В общей сложности от 98 пациентов-доноров получена 61 CC-PDX с приживаемостью до 75% [15]. В CC-PDX сохранились многие характеристики первичной опухоли, такие как геномные и гистологические особенности, чувствительность к противоопухолевым препаратам и даже морфологические признаки (при окрашивании гематоксилином и эозином), такие как центры митотически активных клеток, сходные структурные отложения коллагена, паттерны цитоплазматического иммунного окрашивания на ВПЧ и распространенное ядерное окрашивание на p16INK4a, которые наблюдали в первичных биопсиях и в серийно трансплантированных ксенотрансплантатах [15, 16]. Показано, что частота успешных имплантаций в CC-PDX-моделях была выше при использовании мышей с тяжелым иммунодефицитом, таких как SCID, NOD или NSG [15].
PDX-модели ВГВ- и ВГС-инфекций. PDX-модели позволяют изучать клинико-патологическую картину инфицирования ВГВ. Первой появилась модель Trimera, представляющая собой мышей SCID, которым трансплантировали ex vivo инфицированные ВГВ фрагменты печени человека [17]. Приживление фрагментов печени человека, оцениваемое по окрашиванию гематоксилином и эозином и по экспрессии мРНК сывороточного альбумина человека, отмечено у 85% трансплантированных животных через 1 мес. после имплантации. ДНК ВГВ впервые обнаруживали через неделю после трансплантации печени, а пик виремии приходился на 2–3 нед. после трансплантации. Модель ВГВ-Trimera использовали для оценки терапевтических эффектов поликлональных анти-HBs-антител человека и ингибиторов обратной транскриптазы, которые снижали как число инфицированных животных, так и вирусную нагрузку в их сыворотках [17]. В более позднем исследовании мышам NOD/SCID трансплантировали уникальную панель образцов гепатоцеллюлярной карциномы (ГЦК) и соответствующих тканей стромы, уже положительных по ВГВ [18]. Обнаружено, что все клетки ГЦК продуцировали HBcAg и обладали высокой пролиферативной активностью, тогда как положительную реакцию на HBsAg наблюдали только у доброкачественных гепатоцитов. Используя PDX-модели ВГВ/ГЦК в мышах, удалось фенокопировать вирусологические и клеточные особенности тканей пациентов. Это исследование показало применимость PDX-моделей для изучения как ВГВ, так и вызываемого ВГВ канцерогенеза [18].
С использованием мышей с тяжелым иммунодефицитом на основе PDX разработана также модель ГЦК, связанной с ВГС [19]. NSG-мышам имплантировали первичную опухоль из печени пациента с хроническим гепатитом С и после нескольких циклов in vivo пассирования получили культуру ксенотрансплантатной опухоли мыши. Первичная опухоль пациента и ксенотрансплантаты мыши были гистологически сходными. Генетическое профилирование подтвердило идентичность ВГС-ГЦК-PDX клиническому образцу; в исходных опухолевых тканях человека и в ксенотрансплантированных опухолях детектировали альбумин человека, α1-антитрипсин, глипикан 3, α-SMA и маркеры коллагена типа 1A2. Модель ВГС-ГЦК-PDX успешно использовали при тестировании химиотерапевтических агентов для эффективной схемы лечения ГЦК. Эта модель остается перспективной для оценки новых или дополнительных таргетных химиотерапевтических агентов против ГЦК. Однако примечательно, что в отличие от ВГВ-ГЦК-PDX, пассирование приводило к исчезновению РНК ВГС [19], что делает модель не пригодной ни для изучения ВГС, ни для изучения механизмов ВГС-ассоциированного канцерогенеза, ни для иммунотерапии ВГС-ассоциированных ГЦК.
Преимущества и недостатки PDX-моделей. В целом, PDX-модели имеют сходные с первичной опухолью гистопатологические особенности, включая характеристики генома клеток, статус микроокружения, меж- и внутриопухолевую гетерогенность [20]. Их чувствительность к химиотерапевтическим агентам сходна с чувствительностью, наблюдаемой при клиническом применении противоопухолевых препаратов. В настоящий момент ведется интенсивная разработка мышиных “аватаров” на основе PDX для оптимизации лечения хронических вирусных и онкологических заболеваний человека и персонализированной терапии [21]. Приживаемость может быть сравнительно низкой (см., например, [15]), однако ее можно увеличить за счет использования иммортализованных или опухолевых клеточных линий, полученных из тканей пациентов [16]. Это позволяет применять PDX-модели на различных этапах развития диагностики и терапии опухолей, в совмещенных доклинических/клинических испытаниях для оценки эффективности терапии и преодоления лекарственной устойчивости, идентификации биомаркеров и прецизионной медицины [20].
В то же время PDX-модели имеют ряд недостатков и ограничений по сравнению с аутохтонными моделями опухолей мышей. Во-первых, подавление иммунной системы мыши не позволяет использовать эти модели для проверки эффективности вакцин и чувствительности к иммунотерапии. Кроме того, из-за иммунокомпрометированности хозяина PDX-модели могут неадекватно отражать процесс роста опухоли, особенно метастазирования, в котором существенную роль играет воспаление, определяемое клетками иммунной системы [16]. Специфическим ограничением ксенотрансплантатов иммортализованных или трансформированных клеточных линий человека является тот факт, что они представляют собой экспансию определенного клонального компонента поликлональных опухолей вследствие селективного давления процессов культивирования клеток или эксплантации тканей [22]. В результате этих процессов ксенотрансплантаты могут изменить или полностью утратить признаки аутохтонных опухолей, такие как окружающие их нормальные ткани, стромальные клетки, сосудистая и лимфатическая циркуляция и инфильтрация иммунных клеток (детально описано в обзорах [1, 23]). В силу этих особенностей и ограничений предикативное значение моделей ксенотрансплантатов неоднозначно и оценивается как достаточно высоко [24], так и критически низко [25].
Кроме того, эти модели имеют ряд практических недостатков. Так, обнаружено, что значительная часть PDX, полученных из гетерогенных образцов опухолей пациентов, включая опухоли молочной железы, толстой кишки, поджелудочной железы, мочевого пузыря и почки, прогрессирует в ВЭБ+ и высокометастатические лимфоцитарные опухоли, что ставит под угрозу результаты экспериментов [14] и указывает на необходимость систематического анализа опухолевых тканей и/или клеток человека перед имплантацией для исключения контаминации “посторонними” вирусами (в данном случае ВЭБ). Эксперименты с ВГВ-ГЦК-PDX показали также, что интенсивность пролиферации клеток в ксенотрансплантатах значительно выше, чем в исходных тканях человека (оценивали по окрашиванию ki67) [18], что может означать отбор во время приживления субпопуляции опухолевых клеток с повышенной склонностью к размножению в мышах [26], что в конечном итоге приводит к отличиям PDX от исходных опухолевых тканей. И последний, но не менее важный фактор: эксперименты с ВГС-ГЦК-PDX выявили временный характер экспрессии вируса в ксенотрансплантатах, что ограничивает использование PDX-моделей для изучения вирусных инфекций и вирус-ассоциированного онкогенеза, по крайней мере, в применении к ВГС.
Ксенотрансплантаты тканей человека, искусственно зараженные вирусом или продуцирующие отдельные вирусные белки
Создание моделей ксенотрансплантатов опухолей пациентов проходило параллельно с созданием моделей, в которых мышам имплантировали иммортализованные или трансформированные клетки человека, искусственно инфицированные вирусом, или сначала имплантировали клетки человека, а затем инфицировали их тем или иным вирусом. Эксперименты по созданию подобных моделей проведены для ВПЧ ВКР-, ВГС-, ВГВ-ассоциированных опухолей.
Для моделирования патологий, связанных с ВПЧ ВКР, мышам nude прививали искусственную кожу человека, полученную с использованием первичных кератиноцитов, продуцирующих онкобелок Е7 ВПЧ 16. Трансплантаты стабильно продуцировали Е7 в течение 6 мес. после подсадки. У трансплантированных мышей образовались поражения, гистологически напоминающие аногенитальные поражения человека, вызванные онкогенными ВПЧ. Иммунодетекция или количественная ПЦР мРНК ВПЧ и микроРНК основных биомаркеров показали, что привитая кожа, модифицированная Е7 ВПЧ 16, имеет общие молекулярные характеристики с предопухолевыми и опухолевыми поражениями, связанными с инфекциями ВПЧ ВКР у человека [27]. Эта модель стала ценной платформой для фундаментальных исследований ВПЧ-ассоциированного онкогенеза.
Первые успехи достигнуты и в отношении ВГС. Разработана панель моделей ксенотрансплантатных опухолей для ВГС-инфекции в SCID мышах. В этой панели использовали адаптированные к мышам гепатоциты человека, несущие геномные или субгеномные репликоны ВГС и репортерные гены люциферазы или зеленого флуоресцентного белка (GFP) [28, 29]. Имплантированные мышам SCID клеточные линии образовывали подкожные опухоли и метастазы в печени, реплицирующие ВГС. Сходным образом у мышей с иммунодефицитом вызывали образование опухолей, вводя клетки линии Huh7, полученной из гепатокарциномы человека, а затем инфицировали ВГС [30]. Оба варианта позволяют изучать репликацию ВГС и тестировать противовирусные препараты против ВГС, но не тестировать вакцины или изучать иммунопатогенез хронической ВГС-инфекции, включая развитие ГЦК (поскольку у мышей SCID отсутствует В- и Т-клеточный иммунный ответ). Аналогичные модели для ВГВ, созданные на основе трансгенных мышей SCID с активатором плазминогена урокиназного типа (мыши uPA/SCID), рассмотрены в разделе “Трансгенные мыши, которым имплантировали ксенотрансплантаты, полученные от пациентов”.
Подход с использованием клеточных линий помог устранить перечисленные выше практические ограничения PDX-моделей, такие как контаминация гетерогенными вирусами и/или опухолями, селекция отдельных клонов, отличающихся по свойствам от остальных клеток, а также спонтанный клиренс вирусной инфекции. Однако, как и в случае с PDX-моделями, этот подход не позволял и не позволяет проводить испытания вакцин и методов иммунотерапии.
Приживление элементов иммунной системы человека
Описанные модели можно дополнительно усовершенствовать путем прививки иммунных клеток человека, что позволяет хотя бы частично разрешить проблему иммунодефицита.
На основе иммунокопрометированных мышей создано несколько гуманизированных мышиных моделей с привитыми иммунными клетками человека:
hu-PBL-SCID (human peripheral blood lymphocyte) с привитыми зрелыми периферическими мононуклеарными клетками человека, полученными из крови, селезенки или лимфатических узлов (преимущественно привитые CD3+ Т-клетки человека);
hu-SRC-SCID (human SCID repopulating cell) с подсаженными гемопоэтическими стволовыми клетками, что приводит к развитию гемопоэтической и наивной иммунной системы человека;
SCID-hu с трансплантированной печенью и тимусом плода человека. У этих мышей развивается новый очаг кроветворения и дополнительный органоид, выполняющий функции тимуса человека;
мыши hu-Tg с трансгенной экспрессией генов человека. Таким образом, модели hu-mouse нового поколения воспроизводят созревание широкого спектра гемопоэтических клеток человека, включая Т-клетки, В-клетки, макрофаги и дендритные клетки, необходимые для выработки специфического иммунного ответа. Модели этого типа позволяют воспроизвести как инфекцию, так и выработку иммунного ответа на эту инфекцию [31]. Эти модели можно использовать для исследования опухолей [2, 32, 33], аутоиммунитета [34] и вирусных инфекций, специфичных для человека, что осуществлено в применении к ВИЧ-1 [35], вирусу денге, ВЭБ, вирусу саркомы Капоши [36–38] и ряду других вирусных инфекций [34].
ВЭБ инфицирует исключительно В-лимфоциты человека и имеет двухфазный латентный цикл и литический цикл. Эти особенности в комплексе с необходимостью как врожденного, так и адаптивного иммунного ответа для борьбы с инфекцией, сделали разработку подходящей мышиной модели сложной задачей. Для ВЭБ широко используются гуманизированные мыши с Т-клеточным иммунодефицитом. В этих моделях ВЭБ индуцирует летальную лимфому с паттерном экспрессии генов латентного типа III, сходным с посттрансплантационными лимфопролиферативными заболеваниями. Эти модели применяют для тестирования вакцин, адаптивных клеточных подходов и разработки новых терапевтических стратегий. Модели hu-PBL-SCID широко использовались в доклинических испытаниях терапии ВЭБ (обзор таких моделей см. в [39]). Получение нового поколения гуманизированных мышей, таких как NOG, NSG, с реконструированной иммунной системой человека, привело к созданию уникальных иммунокомпетентных моделей ВЭБ-инфекции, которые позволяют исследовать ВЭБ-индуцированные злокачественные новообразования и механизмы предотвращения их образования (см. обзор [39]).
Аналогичные модели на основе мышей hu-PBL-SCID использовали для изучения пассивной иммунизации против ВИЧ-1. Репопуляция мышей с иммунодефицитом иммунными клетками человека делает их восприимчивыми к заражению лимфотропным вирусом ВИЧ-1 [40, 41].
Тем не менее, эти модели не полностью соответствуют иммунокомпетентным животным, а довольно высокая стоимость этих моделей ограничивает их широкое использование.
ТРАНСГЕННЫЕ МЫШИ, ЭКСПРЕССИРУЮЩИЕ ГЕНЫ ЧЕЛОВЕКА
Преодолеть ограничения, налагаемые иммунодефицитом, позволяет подход, направленный на использование трансгенных гуманизированных мышей, экспрессирующих гены человека, что позволяет получить представление о контексте вирус-индуцированного канцерогенеза in vivo на фоне функциональной иммунной системы. Мышей часто используют из-за их геномного и физиологического сходства с человеком и простоты генетической модификации. В частности, трансгенных мышей применяют для моделирования прогрессирования вирусной инфекции и связанных с ней патологий. Поскольку у трансгенных мышей сохранена иммунная система, на них можно успешно изучать как химиотерапевтические, так и иммунотерапевтические препараты [42].
Трансгенные мыши могут быть созданы с использованием ряда методов, таких как рекомбиназные системы Cre и Tat, обычные и условные нокауты, RCAS (компетентный к репликации ALV LTR с акцептором сплайсинга) и других [43].
Для конструирования трансгенных мышей чрезвычайно важен выбор базовой линии, поскольку генетический фон может играть как положительную, так и отрицательную роль. Например, линия C3H/HeN(C3H) имеет более высокую частоту спонтанной и химически индуцированной гепатоцеллюлярной карциномы, чем линия C57BL/6, хотя это затрудняет интерпретацию данных о влиянии гена, связанного с канцерогенезом, в то время как линия мышей C57BL/6 имеет относительно более низкую частоту формирования спонтанных опухолей [44]. В моделях на основе мышей линии C57BL/6, BALB/c и SSIN/SENCAR у 100% трансгенных животных, экспрессирующих ранние гены ВПЧ 16 под контролем промотора гена кератиноцитов К14, развиваются гиперпластические и/или диспластические поражения. При этом только у мышей, полученных в результате обратного скрещивания с линией FVB/n, развиваются злокачественные плоскоклеточные карциномы двух патологических степеней – высокодифференцированной и умеренно/низкодифференцированной, каждая из которых имеет характерные паттерны злокачественного поведения [45].
Мыши, трансгенные по факторам, позволяющим заражение вирусами человека
Вирусы, патогенные для человека, не всегда способны заражать мышей. Решить эту проблему можно, создавая линии мышей, трансгенных по человеческому рецептору вируса, что делает клетки мыши восприимчивыми к вирусной инфекции. На этой основе созданы in vitro и in vivo модели для исследования процесса вирусной инфекции и вирус-ассоциированного патогенеза.
Трансгенные мыши, экспрессирующие ген рецептора ангиотензин I-превращающего фермента 2 человека (ACE2) под контролем промотора гена цитокератина-18 (K18) (K18-hACE2), служат моделью для изучения инфекции SARS-CoV-2. Интраназальная инокуляция SARS-CoV-2 мышам K18-hACE2 приводит к высокому уровню репликации вируса в легких с распространением инфекции на другие органы [46]. Ген рецептора полиовируса человека (PVR) использовали для создания трансгенных мышей, которые экспрессируют транскрипты PVR и сайты связывания полиовируса в широком диапазоне тканей: внутримозговая инъекция полиовируса типа 1 (штамм Mahoney) мышам, трансгенным по PVR, приводит к репликации вируса в головном и спинном мозге и развитию паралитического полиомиелита [47].
Однако восприимчивость мышей к вирусам человека определяется не только присутствием вирусных рецепторов на поверхности клетки. Имеющиеся у мыши и человека генетические различия определяют различия в репликации вирусов, восприимчивости организма к определенным вирусам, а также в патологических особенностях вирусной инфекции у мыши и человека. Сравнительное секвенирование геномов человека и мыши определило уникальность 300 генов, но на нуклеотидном уровне только 40% генома мыши выравнивается с геномом человека [48 ] . Репликация вирусов – облигатных внутриклеточных паразитов – зависит от функционирования аппарата клетки-хозяина и ее метаболизма, во многом различающихся у человека и мыши. Множество факторов-клетки хозяина, участвующих в репликации вирусов, делает этот путь чрезвычайно сложным для большинства вирусов. Так, например, заражение гепатоцитов человека ВГС зависит от поверхностных белков-рецепторов вируса – CD81, окклюдина (OCLN), белка плотных контактов клаудина-1 (CLDN1) и белка суперсемейства скавенджер-рецепторов клеточной поверхности класса В типа 1 (scavenger receptor class B type 1, SR-BI). Для заражения клеток мыши абсолютно необходимы CD81 и OCLN [49]. Однако при этом зараженные клетки мыши не поддерживают репликацию ВГС и хроническую ВГС-инфекцию, что обусловлено различиями в целом ряде факторов, определяющих репликацию вируса в клетках человека. В число этих факторов входят рецептор липопротеинов низкой плотности, гликозаминогликаны, рецептор эфрина А2, рецепторные тирозинкиназы, рецептор эпидермального фактора роста, кадгерин Е, индуцирующий клеточную смерть DFFA-подобный эффектор B (cell death-inducing DFFA-like effector b), C1-подобный белок 1 синдрома Ниманна–Пика, рецептор трансферрина 1 (см. обзор [50]). В качестве еще одного примера можно привести экзогенную экспрессию NTCP человека, которая может сделать исходно нечувствительные клеточные линии HepG2 (человек), Huh7 (человек), Hepa1–6 (мышь), AML-12 (мышь) и первичные клетки гепатоцитов мыши (PMH), восприимчивыми к вирусу гепатита D (ВГD), который использует белки оболочки ВГВ. NTCP человека может вызвать чувствительность к ВГВ только в линиях клеток HepG2 и Huh7 человека, но не в линиях клеток Hepa1–6, AML-12 или PMH мыши. Эти данные свидетельствуют о том, что, хотя NTCP человека и является функциональным рецептором, который опосредует инфекцию ВГВ в клетках человека, он не позволяет поддерживать инфекцию ВГВ в гепатоцитах мыши, для этого требуются другие внутриклеточные факторы [51]. Репликация ВИЧ-1 в клетках человека зависит от целого ряда факторов, включая циклин Т1, ингибитор сплайсинга р32, APOBEC (цитозиндезаминаза, редактирующая мРНК аполипопротеина В), Fut-2, TRIM5α, Lv-1, Ref-1 и циклофилин А. Отсутствие этих факторов делает невозможной репликацию ВИЧ-1 в клетках мыши [52]
Кроме того, существует множество различий в структуре и функциях врожденной иммунной системы у человека и мыши, влияющих на процесс вирусной инфекции. В первую очередь это касается – баланса лейкоцитарных популяций, структуры и функций дефензинов, Toll-подобных рецепторов, индуцируемых NO-синтаз, рецепторов подавления цитотоксичности Ly49 и KIR, Fc-рецептора, подмножества иммуноглобулинов, компонентов B-клеточного (BLNK, Btk, и λ5) и Т‑клеточного (ZAP70 и общая γ-цепь) сигнальных каскадов, Thy-1, γδT-клеток, цитокинов, хемокинов и их рецепторов, поляризации Th1/Th2, продукции и функций костимуляторных молекул и антигенпредставляющих функций эндотелиальных клеток [53]. Совокупность этих факторов определяет противовирусный иммунный ответ и, соответственно, возможность и особенности репликации вируса в мышиных клетках в сравнении с человеческими. Кроме того, различия могут наблюдаться и между различными линиями лабораторных мышей. Так, например, линии лабораторных мышей, восприимчивых или устойчивых к вирусу гриппа А, различаются по гену Mx [54]. Ген Mx, регулируемый интерфероном, играет важную роль во врожденной иммунной реакции организма на вирус гриппа А.
Мыши, трансгенные по экспрессии вирусов или отдельных вирусных генов
Если мышь невозможно инфицировать вирусом человека даже при экспрессии необходимого рецептора, то ее можно сделать трансгенной по экспрессии отдельных вирусных генов или даже всего вирусного генома. Большинство из описанных к настоящему моменту трансгенных мышей этого типа созданы для изучения механизмов развития патологий печени, индуцированных инфекцией гепатотропными вирусами.
Полный геном ВГВ или его специфические субгеномные фрагменты ввели в пронуклеусы оплодотворенного одноклеточного эмбриона мыши с помощью микроинъекции. Эти мыши не могут элиминировать вирус, но их можно использовать для изучения иммунного ответа, вызванного ВГВ, и ВГВ-ассоциированного канцерогенеза. Показано развитие ГЦК у трансгенных мышей со стабильной продукцией HBx, полноразмерного HBsAg и мутантов pre-S [55, 56]. На этих моделях показана важность вирусных онкогенных белков, которые как сами, так в сочетании с онкогенами человека способствуют развитию ГЦК, индуцируя окислительный стресс, нарушая регуляцию экспрессии генов хозяина и активируя пути передачи онкогенного сигнала [56]. Трансгенных по ВГВ мышей использовали также для изучения эффектов лекарственных средств, включая цитокины, способных предотвратить прогрессирование ВГВ, а также для разработки стратегий преодоления иммунной толерантности к ВГВ [55].
Для изучения влияния ВГС на патологию печени, стеатоз и индукцию ГЦК также созданы трансгенные мыши, продуцирующие полноразмерный полипротеин ВГС или его отдельные белки [54]. Трансгенных мышей, продуцирующих белок нуклеокапсида (кора) ВГС, использовали для анализа механизма индуцированного ВГС канцерогенеза. Показано, что длительная продукция корового белка ВГС приводит к развитию ГЦК у трансгенных мышей. Развитие ГЦК предположительно обусловлено нарушением белком нуклеокапсида ВГС баланса оксидантов/антиоксидантов в печени без индукции воспаления [57]. Не ясно, однако, применимы ли данные, полученные на этих моделях, для объяснения патологии, наблюдаемой у человека. Необходимо отметить, что трансгенные мыши характеризуются сверхпродукцией белков ВГС, что разительно отличается от низкого уровня белков ВГС при естественной инфекции. Кроме того, в отличие от моделей, трансгенных по ВГВ, экспрессия генома ВГС в мышиных клетках не приводит к продукции вирусных частиц или репликации вирусного генома. Таким образом, на этих моделях нельзя изучать вирусную инфекцию и моделировать весь естественный процесс заражения клеток/организма и распространения вируса, наблюдаемый в тканях человека [54].
Еще одно ограничение этих моделей инфекции ВГВ и ВГС – экспрессия вируса во всех тканях организма, в отличие от естественного процесса инфекции, происходящего преимущественно (ВГС) или исключительно (ВГВ) в печени. По совокупности этих свойств трансгенные по ВГВ и ВГС мыши не подходят для оценки противовирусных препаратов или методов терапии/иммунотерапии, а также для отработки методов контроля инфекционного процесса, в частности, гистологического наблюдения процесса клиренса в тканях печени [58]. Эти ограничения можно отчасти преодолеть, задавая экспрессию вируса (вирусных генов) под контролем экзогенных промоторов, специфичных для органа (ткани), например, конститутивного промотора гена альбумина или индуцибельного промотора металлотионеина, определяющих экспрессию в печени [56].
Предпринято также несколько попыток создать трансгенных мышей, экспрессирующих весь геном ВИЧ-1 [52, 59, 60]. Например, сконструированы трансгенные мыши, содержащие интактные копии провириона ВИЧ-1. Эти мыши не имели признаков инфекции в течение 9 мес. наблюдения, но одна из них дала потомство (F1), у которого развилась болезнь, сходная с синдромом приобретенного иммунодефицита, и оно погибло на 25-й день жизни. Из селезенки, лимфатических узлов и кожи пораженных животных (пять из пяти) был выделен вирус ВИЧ-1, неотличимый от родительского вируса [59]. Cозданы также мыши, трансгенные по отдельным генам ВИЧ-1, таким как Tat [61, 62]. Как и в других моделях мыши, трансгенные по отдельным вирусным генам, не имели ни рецепторов, необходимых для вирусной инфекции, ни кофакторов, необходимых для эффективной репликации вируса, и были толерантными к продукции вирусных белков. Таким образом, ни одна из этих моделей не годилась для изучения репликации ВИЧ-1, разработки стратегий противовирусной терапии или новых подходов к вакцинации [54].
Для изучения онкогенных свойств белков KSHV, разработана панель трансгенных мышей, продуцирующих белки этого вируса. Эти генетически модифицированные мыши продуцировали один или несколько белков KSHV, обладавших онкогенными свойствами в культуре клеток, они предоставили новые возможности изучения патогенеза KSHV не только для понимания механизмов онкогенеза, связанного с KSHV, но и для оценки эффективности терапии инфекций, связанных с KSHV, путем направленного воздействия на эти вирусные белки [63].
Разработаны трансгенные мыши K14E7, в кератиноцитах которых экспрессируется ген Е7 ВПЧ 16 под промотором K14. В этой модели экспрессия E7 приводила к гиперплазии эпителия, инфильтрации иммунных клеток и иммуномодуляции, сходной с ВПЧ-ассоциированной неоплазией у человека. Сигнатура экспрессии мРНК генов в коже была сходной с сигнатурой цервикальной интраэпителиальной неоплазии 3-й стадии у человека. Интересно, что при трансплантации кожи мышей K14E7 мышам дикого типа и мышам, трансгенным по Т-клеточному рецептору с увеличенным количеством E7-специфических цитотоксических Т-клеток E7TCR269, кожа мышей К14Е7 приживалась, в то время как аналогичная кожа, экспрессирующая овальбумин, отторгалась. Хорошую приживаемость кожных трансплантатов К14Е7 объяснили экспрессией Е7 ВПЧ 16, приводящей к иммуносупрессии, в частности, к подавлению экспрессии интерферона-γ и понижению активности NK-клеток и тучных клеток [64], участвующих в процессе отторжения.
MHC-гуманизированные мыши
Классические антигены главного комплекса гистосовместимости класса I (MHC I) представляют собой тримерные молекулы, обнаруживаемые на поверхности ядерных клеток у всех челюстных позвоночных. Молекулы MHC I распознаются двумя семействами рецепторов: клонотипическими Т-клеточными рецепторами, которые экспрессируются на поверхности CD8+ цитотоксических Т-лимфоцитов (ЦТЛ), и мономорфными рецепторами, экспрессируемыми как NK-клетками, так и ЦТЛ. Продукция молекул MHC I в клетках представляет собой последовательный процесс, осуществляемый с помощью белков: протеаз, шаперонов, транспортеров и ряда других.
Несмотря на то, что MHC I человека и мыши в значительной степени гомологичны по своей структуре, организации и функциям, в механизме процессинга и представления антигена в контексте MHC I человека и мыши имеются различия. Технологии трансгенеза и нокаута или нокина позволяют добавлять соответствующие гены человека или заменять гены мыши ортологичными генами человека для получения иммунологически гуманизированных мышей [65]. Такие экспериментальные животные особенно важны в качестве доклинических инструментов для идентификации эпитопов пептидов, представляемых в контексте HLA I при инфекционных заболеваниях и опухолях, а также для сравнительной оценки эффективности иммунотерапии. В моделях, в которых сохранялись интактные молекулы MHC I мыши, цитолитические ответы не ограничивались ответами, рестриктированными HLA I человека. Чтобы лучше использовать трансгенные молекулы HLA I и генерировать более функциональные HLA-рестриктированные ответы CD8+ T-клеток, Pascolo S. и соавт. создали мышей с двойным нокаутом H-2 Db и β2-m, экспрессирующих химерную α3-H-цепь HLA-A*02:01, ковалентно связанную с β2-m человека с помощью пептидного плеча (мыши HHD II) [66]. Цитолитические ответы этих мышей опосредовались исключительно моноцепью HLA-A*02:01. Для дальнейшей оптимизации моделей с трансгенными молекулами HLA I, Boucherma R. и соавт. создали семь новых линий трансгенных мышей с моноцепью HLA I в контексте тройного нокаута H-2 Kb, Db и β2-m мыши (H-2 I null) [67]. В этих мышах соответствующие домены H-цепи α1α2, наиболее часто встречающиеся во всех популяциях человека молекул HLA: HLA-A*01:03, -A*24:02, -B*08:01, -B*27:05, -B*35:01, -B*44:02 или -C*07:01, были слиты с доменом α3 мыши и ковалентно связаны с β2-микроглобулином человека [67].
Позднее были разработаны и другие полностью MHC-гуманизированные мыши, позволяющие изучать HLA-рестрицированное распознавание пептидов без вмешательства представления эпитопов в контексте молекул MHC мыши [68]. В гуманизированной модели A2.DR1 молекулы HLA, наиболее часто встречающиеся у представителей европеоидной расы – HLA-A*0201, представляющие эпитоп-связывающие домены α1 и α2 HLA-A0201 с доменом α3 H-2D*b, ковалентно связанным с β2m человека (HDD), а также HLA-DR1 экспрессируются при нокауте всех генов MHC мыши. Эту модель использовали для тестирования вакцин против ВПЧ 16, моделируя ВПЧ-инфекцию присадкой сингенных клеточных линий, экспрессирующих белки Е6 и Е7 [68]. Так, путем трансфекции фибробластов сердца и легких мышей, трансгенных по HLA-A*0201, онкогеном H-Ras V12 и генами Е6 и Е7 ВПЧ 16 получена трансгенная клеточная линия, онкогенная для мышей HLA-A*0201. При этом из белка Е7 ВПЧ 16 был удален доминантный эпитоп, распознаваемый иммунной системой мышей H-2D(b), чтобы гарантировать, что противоопухолевый иммунный ответ будет направлен исключительно на HLA-A*0201-рестрицированные эпитопы [69]. Эту модель использовали для тестирования эффективности двух кандидатных вакцин против ВПЧ 16, основанных на плазмидной ДНК и рекомбинантном вирусе венесуэльского энцефалита лошадей [69].
Трансгенные мыши, которым имплантировали ксенотрансплантаты, полученные от пациентов
Этот подход применяли преимущественно для создания моделей заражения гепатотропными вирусами ВГВ и ВГС (подробности см. в [70]). При этом наиболее широко использовалась и используется модель, основанная на мышах SCID, трансгенных по активатору плазминогена урокиназного типа (Alb-uPA)/SCID), или мыши, uPA/трансгенные по белку, активирующему рекомбинацию гена 2 (RAG-2), с острой или подострой печеночной недостаточностью у новорожденных мышат. Печень этих мышей можно “воссоздать” путем постепенного заселения гепатоцитами человека [71, 72]. Тяжелая печеночная недостаточность необходима для того, чтобы клетки человека имели свободное пространство и условия для пролиферации и достижения высокой степени химеризма. В таких системах гепатоциты человека остаются функциональными в течение как минимум 2 мес. после трансплантации и после прохождения множественных клеточных делений (подтверждено продукцией альбумина человека). Инфицирование мышей с трансплантированными гепатоцитами человека ВГВ из сыворотки крови человека или выделенным из in vitro культуры приводило к продуктивной инфекции с виремией до ≈1010 копий/мл, образованию функциональной кольцевой ковалентно замкнутой ДНК (ккзДНК) ВГВ и распространению вируса [72]. Вирусная нагрузка возрастала с увеличением доли клеток печени мыши, замещенных клетками человека, сохраняя возможность контролировать как степень печеночной недостаточности, так и степень ее восстановления. Виремия наблюдалась до 22 нед. с момента инфицирования (до наступления смерти или планового окончания эксперимента). Пассирование вируса показало, что сыворотки крови мышей содержат инфекционный ВГВ. Экспериментально установлено, что HBeAg незаменим как для активной продукции вируса, так и для его передачи [73]. Таким образом, химерные мыши могут быть напрямую инфицированы вирионами ВГВ с образованием функциональной ккзДНК ВГВ и с последующим распространением вируса. В аналогичном эксперименте линии клеток человека инъецировали в селезенку мышей FAH–/–RAG2–/–IL2Rγ– (FRG) или трансгенным по тимидинкиназе (TK) мышам NOG (TK-NOG) с последующим инфицированием ВГВ (подробности см. в [70]). ВГВ можно вводить также после инъекции CD34+ гемопоэтических стволовых клеток и гепатоцитов человека в печень новорожденных мышей NOD/SCID/IL2Rγ–/–, трансгенных по HLA-A2 (мышей A2/NSG) [70]. Ни одна из этих моделей не может применяться ни для изучения иммуноопосредованного воспаления, врожденного или адаптивного противовирусного иммунного ответа, ни для тестирования иммунотерапии ВГВ-инфекции или ВГВ-ассоциированных опухолей, ввиду иммунодефицитного статуса мышей-реципиентов.
Чтобы устранить это ограничение, разработаны модели двойных гуманизированных химерных мышей, которые предусматривают пересадку иммунных клеток человека. С этой целью мезенхимальные стволовые клетки костного мозга человека (hBMSCs) трансплантировали мышам Fah–/–Rag2–/–IL-2Rγc–/– SCID (FRGS) с фульминантной печеночной недостаточностью, в результате чего получили модель hBMSC-FRGS, гуманизированную по клеткам печени и иммунной системы [74]. Мыши hBMSC-FRGS характеризовались активной пролиферацией и трансдифференцировкой функциональных гепатоцитов человека и широкого спектра иммунных клеток, включая В-клетки, Т-клетки, NK-клетки, дендритные клетки и макрофаги. После инфицирования ВГВ у этих мышей развивалась устойчивая виремия и специфические иммунные и воспалительные реакции. Через 54 нед. у 55% мышей наблюдалось прогрессирование заболевания до хронического гепатита и цирроза печени [74]. В целом, мыши hBMSC-FRGS оказались уникальной системой для выявления взаимодействий вируса с организмом хозяина, включая патофизиологические повреждения печени при хроническом гепатите с прогрессированием в цирроз, и специфические иммунные и воспалительные реакции организма человека на инфекцию ВГВ и повреждение печени. При этом сохранялся профиль экспрессии генов, ассоциированных с течением болезни, клеточный иммунный ответ, продукция цитокинов и характерные для гепатита В биохимические и патологические изменения [74]. В аналогичном исследовании созданы мышиные модели AFC8-hu HSC/Hep и A2/NSG-hu-HSC/Heр, в которых трансгенным мышам A2 трансплантировали гемопоэтические стволовые клетки человека CD34+ и клетки-предшественники гепатоцитов человека (или клетки печени плода). В обеих моделях ВГВ был способен инфицировать мышей, персистируя при этом в организме мыши не менее 4 мес. [70, 75] и вызывая фиброз печени. Описанные модели оказались очень полезными для изучения механизмов поражения печени при ВГВ-инфекции [75]. Модели типа Trimera и модели на основе uPA/SCID и Fah–/–Rag–/–IL-2–/– мышей успешно использовали для изучения инфекции ВГС, а также при разработке противовирусных препаратов, эффективных при ВГС-инфекции [76–79].
Ограничения трансгенных мышиных моделей
Использование химерных трансгенных моделей имеет ряд ограничений, включая генетическую изменчивость, отсутствие инструментов для изучения иммунного ответа, длительность исследования, сложные лабораторные методы и этические аспекты. Важно также отметить, что трансгенные мыши обладают низким потенциалом к формированию опухолей и метастазированию и низким уровнем мутаций в опухолях [80]. Помимо этого, экспрессия введенного гена в трансгенных мышах чаще всего контролируется конститутивным синтетическим промотором, который не позволяет варьировать экспрессию генов в зависимости от генетического фона модели, что приводит к низкой клеточной гетерогенности и влияет на характер прогрессирования опухоли и метастазирования [81].
Получение мышиных моделей для анализа вирусных инфекций человека хорошо налажено для вирусов, использующих ограниченную панель факторов клетки-хозяина. Однако уровень репликации даже таких вирусов в клетках мыши ниже, чем в клетках человека, что во многом связано с активацией системы врожденного иммунитета и продукцией интерферонов первого типа [82]. В большинстве моделей молекулярные сенсоры нуклеиновых кислот и ИФН I подавляют размножение вирусов и блокируют появление симптомов инфекции, что затрудняет изучение вирус-индуцированных патологий.
Как уже упомянуто, человек и мышь имеют по существу разные иммунные системы. Поэтому мышиные модели не позволяют точно воспроизвести иммунный ответ человека на вирусы или отдельные вирусные антигены, что затрудняет экстраполяцию результатов, полученных на мышиных моделях, на человека [82]. В силу этого для моделирования хронических инфекций, вызванных гепатотропными вирусами, такими как ВГВ и ВГС, весьма актуальны модели, основанные на гуманизированных химерных мышах. Однако эти модели, как и PDX-модели, не годятся для изучения воспаления, врожденного или адаптивного противовирусного иммунного ответа, тестирования иммунотерапии или вакцин. Сохранение в мышиных моделях интактной иммунной системы важно не только для решения задач иммунотерапии, но и при исследовании эффектов химиопрепаратов, так как ранее было показано, что часть химиотерапевтических препаратов индуцирует развитие противоопухолевого иммунного ответа [83].
Двойные гуманизированные химерные модели казалось бы обходят эту проблему, однако врожденные и адаптивные противовирусные иммунные ответы в этих моделях ограничены, а характер опосредованного Т- и В-клетками иммунного ответа на вирусную инфекцию отличается от иммунного ответа человека, в основном из-за клеточного состава микроокружения печени [70, 75]. Кроме того, печень мышей в двойных химерных моделях содержит меньше гепатоцитов, и поддерживает более низкий уровень репликации вируса, чем печень человека. И последнее, но не менее важное: эти модели основаны на узкоспециализированных линиях мышей, технически они очень сложны и дороги. Их создание и поддержание требует много времени, средств, развитой инфраструктуры и высококвалифицированного персонала, что ограничивает их широкое применение. Нужны более простые системы, подходящие для моделирования отдаленных последствий хронической вирусной инфекции, вызванной длительной продукцией вирусных белков, с оценкой их влияния на регуляцию генов, метаболизм экспрессирующих клеток и “невинных” клеток окружения, а также на состояние (функционирование) врожденного и адаптивного иммунного ответа.
МОДЕЛИРОВАНИЕ ВИРУС-АССОЦИИРОВАННОГО ОНКОГЕНЕЗА
Имплантация иммунокомпетентным мышам сингенных линий опухолевых клеток, несущих целые вирусные геномы или отдельные вирусные гены
Простейшие модели вирус-ассоциированного канцерогенеза основаны на использовании мышиных опухолевых (опухолеродных) клеток, экспрессирующих вирусные гены. Клетки имплантируют сингенным мышам, т.е. мышам, имеющим тот же генетический фон, что и имплантируемые клетки. Модели вирус-ассоциированных опухолей, созданные на основе таких клеток, называют аллографтными, или сингенными моделями. Сингенные мышиные модели (СММ) сохраняют интактной иммунную систему мыши, поэтому они хорошо подходят для разработки противоопухолевых и противовирусных препаратов, воздействующих на иммунную систему, таких как ингибиторы контрольных точек, профилактические и иммунотерапевтические противовирусные и противоопухолевые вакцины. Сингенные клеточные линии могут быть получены из спонтанно возникающих опухолей мышей или с использованием мутагенных соединений и транспозонов и модифицированы для экспрессии одного или нескольких вирусных онкогенов в сочетании с известными клеточными онкогенами путем стабильной трансфекции или трансдукции ретро- или лентивирусами, а также путем введения гена с помощью системы CRISPR-Cas. Например, получена панель клеточных линий рака легкого, сингенных мышам линии C57BL/6 [80]. Описаны модификации этого протокола для других видов клеток, например, клеток опухоли мочевого пузыря [84] и прямой кишки [85]. В другом подходе в уже злокачественные клетки путем стабильной трансфекции или трансдукции с использованием ретро- или лентивирусов вводят гены одного или нескольких вирусных белков. В ряде случаев экспрессия вирусных генов может приводить к увеличению онкогенного потенциала уже существующих опухолевых линий [86, 87].
Сингенные модели имеют множество преимуществ, в том числе и то, что опухолевые клетки можно легко поддерживать и размножать in vitro перед имплантацией мышам. Это приводит к образованию воспроизводимых по размеру и темпам роста опухолей со сходной выживаемостью животных. Основные ограничения использования сингенных клеточных линий связаны с их генетической однородностью [83] и ограниченными размерами, проще говоря, “бедностью” панели опухолевых линий, способных стабильно формировать опухоли в иммунокомпетентных мышах. Необходимо также учитывать, что прогрессирование опухоли сложный процесс – эффекты от воздействия отдельного вирусного белка или даже целого вируса у человека и мыши могут различаться. Это ограничивает применимость результатов, полученных в СММ, для трактовки феномена вирус-ассоциированного канцерогенеза человека.
Сингенные модели описаны как в научной литературе, так и на сайтах компаний-производителей (например, https://www.criver.com/products-services/discovery-services/pharmacology-studies/ oncology-immuno-oncology-studies/oncology-models/ syngeneic-mouse-models или https://www.taconic.com/resources/syngeneic-cell-line-reference-database/), поэтому мы не будем рассматривать их подробно.
Инфекции, вызываемые вирусами грызунов, родственными онкогенным вирусам человека
В первую очередь следует упомянуть вирус опухоли молочной железы мышей (MMTV), относящийся к ретровирусам [88]. Однако MMTV не является моделью родственного вируса, это скорее реальный этиологический агент опухолей молочной железы. Об этом свидетельствует анализ накопленных к настоящему моменту данных, включая идентификацию провируса, LTR, белков Gag и нуклеокапсида, обратную транскриптазу и белок оболочки в 13% образцов опухолей молочной железы человека [88].
Наиболее хорошо изучен вирус папилломы мыши (MmuPV1), используемый для моделирования, вирус-ассоциированного канцерогенеза. С помощью MmuPV1 на мышах моделировали ВПЧ-ассоциированный плоскоклеточный рак головы и шеи. Инфицирование вирусом MmuPV1 эпителия языка мышей с иммунодефицитом приводило к развитию плоскоклеточной дисплазии высокой степени с ранними признаками инвазивной карциномы в течение 4 мес. [89]. В сочетании с пероральным канцерогеном 4-нитрохинолин-1-оксидом (4NQO) MmuPV1 вызывал развитие инвазивного плоскоклеточного рака языка у мышей как с иммунодефицитом, так и иммунокомпетентных. Эти опухоли экспрессировали маркеры папилломавирусной инфекции и ВПЧ-ассоциированного канцерогенеза [89]. У иммунокомпетентных мышей MmuPV1, как и ВПЧ, передается половым путем [90], что позволяет изучать механизм заражения, а также использовать его в качестве модели для оценки эффективности профилактических препаратов. Кроме того, установлено, что механизм онкогенеза MmuPV1, по крайней мере отчасти, связан с его интеграцией в геном инфицированных клеток [91].
Наиболее близким к ВЭБ оказался ɣ-герпесвирус 68 мыши (MHV-68). К сожалению, MHV-68 значительно отличается от ВЭБ, различается также иммунный ответ хозяина на эти два вируса [39]. MHV-68 способен иммортализовать фетальные клетки печени мыши in vitro, что приводит к их дифференцировке в плазмабласты, которые, в свою очередь, могут образовывать опухоли в мышах nude и Rag2–/–, но не при подсадке иммунокомпетентным мышам линии C57Bl/6 или мышам с дефицитом В-клеток [92].
Для моделирования ВИЧ-1-инфекции разработаны химерные псевдовирионы ВИЧ-1–вирус лейкоза мышей (MuLV). Эти химеры могут быть получены путем инфицирования ВИЧ-1 Т-клеточной линии, несущей MuLV, или путем замены кодирующей области gp120 ВИЧ-1 на gp80 MuLV [52]. С целью оценки протективных свойств вакцин против ВИЧ-1 проведены эксперименты по иммунизации мышей кандидатными ВИЧ-1 вакцинами с последующим их инфицированием химерным псевдовирусом [52].
В целом, этот подход используется относительно редко в силу узкого спектра вирусов грызунов, аналогичных онкогенным вирусам человека.
In vitro и in vivo моделирование молекулярного фона, способствующего злокачественной трансформации
Более сложным представляется моделирование процесса вирус-ассоциированного онкогенеза в части кооперации онкогенов/онкобелков вирусов и человека. Известно, что злокачественная трансформация клетки и развитие опухоли (опухолей) зависят от сложного взаимодействия между вирусными онкогенами и онкогенами/онкобелками хозяина. Ранние доказательства такого сотрудничества получены с использованием in vitro трансформации клеток с участием онкобелков вируса полиомы. Для трансформации первичных фибробластов эмбриона крысы требовалась экспрессия как большого, так и среднего T-антигена вируса [93]. Подобная “кооперация” генов широко используется при моделировании опухолей, связанных с вирусными инфекциями.
Чтобы обеспечить созревание и выход потомства, вирусы должны в течение достаточно долгого времени подавлять или задерживать процесс апоптоза. Онкогенные вирусы особенно активно манипулируют как внешними, так и внутренними путями индукции апоптоза, подавляют активность проапоптотических белков и сигнальных путей, способствуя тем самым онкогенезу [94]. Чаще всего затрагивается р53-индуцированный апоптоз, опосредованный линейным каскадом, включающим трансактивацию bax, транслокацию белка Bax из цитозоля в мембранный компартмент, высвобождение цитохрома c из митохондрий и последовательную активацию каспаз-9, -3, -6 и -7 [95 ] . Выявление ускоренного образования опухолей у мышей с нуль-мутациями по р53 и с дефицитом р53 [96] способствовало созданию широкой панели моделей опухолей на основе мутированного p53 [97]. Вирусы достигают того же эффекта за счет функциональной инактивации p53. Т-антигены адено- и полиомавирусов связывают и инактивируют р53, что позволяет клетке избегать остановки клеточного цикла и способствует ее переходу в S-фазу. Кроме того, вирусные Т-антигены имитируют структуру ДНК (DNA mimicry), конкурируют с р53 за связывание ДНК, изменяя заряд и конфигурацию ДНК-дуплекса [98]. Это свойство Т-антигенов позволяет им нарушать регуляцию транскрипции генов-мишеней р53 и вызывать злокачественную трансформацию, в том числе клеток, непермиссивных для продуктивной вирусной инфекции [99]. Этот механизм сделал экспрессию Т-антигена вируса SV40 надежным способом создания трансгенных мышиных моделей опухолей [100].
Другой пример – вирусы ВПЧ ВКР, ингибирующие р53 за счет специфической активности онкобелков Е6 и Е7. Е6 рекрутирует E6AP и промотирует убиквитинлигазную активность E6AP E3, вызывая деградацию р53, приводящую к снижению его уровня в инфицированных клетках. Онкобелок Е7 связывается с транскрипционным репрессорным комплексом DREAM, что повышает уровни экспрессии большинства генов клеточного цикла и ингибирует p53 [101]. Кроме того, белок E7 ВПЧ ВКР связывает другой опухолевый супрессор – белок ретинобластомы (Rb). Rb связывается с факторами транскрипции семейства E2F и подавляет экспрессию генов ферментов репликации [102]. E7 нарушает взаимодействие между Rb и E2F, что приводит к высвобождению факторов E2F в их транскрипционно-активных формах, стимулируя репликацию и деление клеток [103]. Однако этих событий недостаточно, чтобы вызвать трансформацию клеток. Чтобы трансформировать первичные клетки человека, ВПЧ ВКР должны взаимодействовать с такими активированными онкогенами человека, как RAS – центральный медиатор клеточной пролиферации и дифференцировки, индуцированной факторами роста [104]. RAS действует через белок RAF и ERK-киназы сигнального пути MAP-киназы. RAS, обладающий митогенной активностью, необходим на протяжении G1-фазы клеточного цикла и для прохождения через S-фазу [105]. Продукция активированного RAS в отсутствие таких негативных регуляторов, как p53 или Rb, приводит к трансформации клеток [106]. Это свойство превратило коэкспрессию E6/E7 ВПЧ ВКР в сочетании с активированным RAS в еще один инструмент злокачественной трансформации клеток, что привело к созданию множества мышиных моделей опухолей человека, продуцирующих белки Е6 и Е7 ВПЧ ВКР [107–109]. Сходный сценарий трансформации может быть реализован и с использованием других кооперативных онкобелков, например, MYC [110].
Клетки (клеточные линии), трансформированные совместным действием онкогенов вирусов и человека, имплантируют сингенным иммунокомпетентным мышам. Аналогичные сингенные мышиные модели описаны в разделе “Имплантация иммунокомпетентным мышам сингенных линий опухолевых клеток, несущих целые вирусные геномы или отдельные вирусные гены”. В последнее время появились работы, описывающие индукцию такого сочетанного онкогенеза in vivo. Ярким примером этого может быть внутривагинальная электропорация мышей плазмидами, кодирующими E6/E7 ВПЧ-16, c-MYC, AKT и транспозазу Sleeping Beauty. Кооперативное действие онкогенов (онкобелков) вируса и человека привело к формированию опухолей, которые спонтанно прогрессировали от плоскоклеточного интраэпителиального поражения высокой степени до рака и продуцировали белки Е6 и Е7 ВПЧ. Клеточные линии, полученные из этих опухолей, способны образовывать карциномы у иммунокомпетентных мышей [110]. Совместное воздействие клеточных факторов MYC, RAS и вирусных онкобелков на клеточную трансформацию опосредуется регуляцией активности циклинкиназ [106].
Модели спровоцированного канцерогенеза
В этом подходе для индукции опухоли используют различные канцерогены на фоне вирусной инфекции или экспрессии вирусных антигенов, т.е. совместное действие вирусных антигенов и химических/физических факторов, способствующих образованию опухолей. Haverkos H. опубликован исчерпывающий обзор [111] взаимодействия вирусов с канцерогенами человека. Сделанные в этом обзоре выводы применимы и к мышиным моделям. Мы сосредоточимся на наиболее важных факторах, связанных с канцерогенезом, таких как прием алкоголя, диета и воздействие канцерогенов.
Синергизм между канцерогенезом печени, вызванным ВГВ и ВГС, и потреблением алкоголя описан давно [57]. Механизм канцерогенеза, связанного с ВГС, заключается в индукции окислительного стресса [112]. Как можно было ожидать, у мышей, трансгенных по белку нуклеокапсида ВГС, употребление алкоголя вызывало заметное повышение уровня гидропероксидов фосфатидилхолина, а длительное употребление алкоголя (алкогольная диета) индуцировало развитие фиброза печени, подтверждая как синергический эффект вирусной инфекции и алкоголя в индукции заболевания печени [113], так и возможность моделирования этого процесса путем сочетания экспрессии вирусных антигенов с длительным воздействием этанола.
На примере мышей, несущих Т-антиген SV40 под промотором эластазы-1, показано, что ассоциированный с вирусом онкогенез можно стимулировать с помощью определенных диет. Оказалось, что пища с низким содержанием клетчатки увеличивает частоту образования опухолей [114]. Интересно, что низкое содержание клетчатки в пищевом рационе вызывает изменения в составе кишечного микробиома, что, в свою очередь, приводит к распространению кишечных патобионтов, таких как адгезивно-инвазивная кишечная палочка (AIEC, связанная с болезнью Крона), с последующим изменением микробного метаболома с усилением воспаления кишечника [115], а также с системной метаболической и иммунной дисрегуляцией [116].
Рассмотренные примеры воздействия, приводящие к усилению роста опухоли, имеют один и тот же механизм, заключающийся в индукции и поддержании воспаления. Индукция воспаления лежит в основе хорошо зарекомендовавших себя моделей канцерогенеза кожи, индуцированного химическими канцерогенами. Показано, что предсуществующее воспаление кожи увеличивает восприимчивость к росту опухоли. Это явление связано с увеличением популяции опухолеспецифических Т-клеток, продуцирующих интерлейкин-17 (IL-17) [117]. В то же время дефицит рецептора IL-17 (IL-17R) усиливал инфильтрацию CD8+ Т-клеток, ингибируя при этом инфильтрацию миелоидных клеток CD11b+ и развитие клеток-супрессоров миелоидного происхождения [118], что препятствовало росту опухоли.
Состояние воспаления тканей может быть достигнуто механическим путем. Isaguliants M. и соавт. опубликовали данные, согласно которым воспаление, вызванное электропорацией, приводит к усиленному росту опухоли в участках, проксимальных к месту электропорации [119].
Описанные методы моделирования онкогенеза, ассоциированного с хроническими вирусными инфекциями, нашли свое применение для объяснения механизма индукции и развития опухолей, но не для создания самих моделей, прежде всего, в силу отсутствия воспроизводимости эффектов и их неоднородности, из-за невозможности ограничить их одним органом или типом ткани, а также длительности экспериментов.
ЗАКЛЮЧЕНИЕ
На сегодняшний день разработан ряд стандартных подходов к созданию моделей вирусной инфекции и вирус-ассоциированного канцерогенеза, нашедших широкое применение в лабораторной практике (рис. 1). Каждый из этих подходов имеет свои особенности, преимущества, недостатки и ограничения (табл. 1). Выбор той или иной модели должен осуществляться с учетом этих характеристик. Наиболее распространено использование ксенотрансплантатов тканей и клеток человека (как опухолевых и/или инфицированных, так и здоровых) с последующей инфекцией и трансформацией. Этот метод позволяет в довольно короткие сроки получить панель инструментов для оценки эффективности противоопухолевых препаратов, однако его применение сильно ограничивается супрессией иммунной системы мыши.
Рис. 1.
Основные подходы к созданию моделей вирусной инфекции и вирус-ассоциированного канцерогенеза.
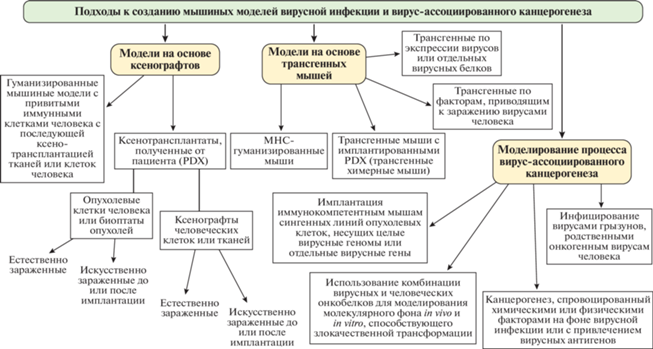
Таблица 1.
Основные преимущества, недостатки и направления использования различных подходов к созданию моделей вирусной инфекции и вирус-ассоциированного канцерогенеза
| Подход | Преимущества | Недостатки | Использование |
|---|---|---|---|
| Гуманизированные мыши с привитыми иммунными клетками человека с последующей ксенотрансплантацией тканей или клеток человека | − Позволяют воспроизвести как инфекцию, так и выработку иммунного ответа на эту инфекцию | − Модели не полностью соответствуют иммунокомпетентным животным. − Высокая стоимость |
− Исследование развития злокачественных новообразований. − Исследование аутоиммунитета. − Исследование вирусных инфекций, специфичных для человека. − Тестирование вакцин |
| Опухолевые клетки человека или биоптаты опухолей | − Подходят для прямых исследований патогенов человека и механизмов заражения. − Позволяют быстро и легко оценить эффекты различных агентов на опухолевые ткани и клетки in vivo. − Имеют сходные с первичной опухолью гистопатологические особенности. − Сходная с человеком чувствительность к химиотерапевтическим агентам. − Управляемая приживаемость |
− Не подходят для тестирования вакцин. − Могут неадекватно отражать процесс роста опухоли, особенно в отношении метастазирования из-за иммунокомпрометированности хозяина. − Могут изменить или полностью утратить признаки аутохтонных опухолей. − Возможна контаминация “посторонними” вирусами. − Временный характер вирусной экспрессии в ксенотрансплантатах. − Склонность к интенсивной пролиферации клеток в ксенографтах |
− Доклинический скрининг новых методов лечения (в том числе персонифицированных) опухолей. − Моделирование развития опухолей и метастазов, связанных с вирусными инфекциями |
| Ксенографты клеток или тканей человека | − Прямые исследования патогенов человека и механизмов заражения. − Моделирование патологий, связанных с вирусными заболеваниями | ||
| MHC-гуманизированные мыши | − Возможность исследования HLA-рестриктированного распознавания пептидов без вмешательства презентации эпитопов в контексте молекул MHC мыши | − Высокая стоимость. − Возможность использования HLA типа 1 в каждой линии мышей |
− Моделирование иммунного ответа на вирусную инфекцию или на вакцинацию/иммунотерапию. − Исследования, направленные на изучение онкологических и аутоиммунных заболеваний |
| Трансгенные мыши с имплантированными PDX (трансгенные химерные мыши) | − Возможность моделирования процесса вирусной инфекции. − При использовании двойных гуманизированных химерных мышей возможность изучать вирусную инфекцию на фоне гуманизированной иммунной системы |
− Генетическая изменчивость. − Отсутствие инструментов для изучения иммунного ответа. − Пролонгированные периоды исследования. − Сложные лабораторные методы и этические аспекты. − Низкий потенциал к формированию опухолей и метастазированию и низкий уровень мутаций в опухолях. − Модели основаны на узкоспециализированных линиях мышей, технически очень сложны и дороги |
− Моделирование хронических заболеваний печени, изучение гепатотропных вирусов, изучение противовирусных препаратов |
| Трансгенные по экспрессии вирусов или отдельных вирусных белков | − Позволяют изучить патогенез хронической вирусной инфекции на фоне компетентной иммунной системы | − Не позволяют анализировать репликацию вируса, противовирусные стратегии или подходы к вакцинации | − Анализ механизмов вирус-индуцированной патологии. − Изучение онкогенных свойств белков in vivo |
| Трансгенные по факторам, приводящим к заражению вирусами человека | − Изучение вирусной инфекции на фоне компетентной иммунной системы | − Ограниченная панель факторов клетки-хозяина. − Не позволяют в точности воспроизвести иммунный ответ человека на вирусы или отдельные вирусные антигены |
− Моделирование всех этапов инфекции вирусами человека |
| Имплантация иммунокомпетентным мышам сингенных линий опухолевых клеток, несущих целые вирусные геномы или отдельные вирусные гены | − Иммунная система мышей интактна. − Легкость поддерживания и размножения опухолевых клеток in vitro перед их имплантацией мышам. − Хорошая воспроизводимость опухолей со сходными темпами роста и выживаемостью мышей |
− Генетическая однородность. − Ограниченная панель опухолевых линий, стабильно формирующих опухоли в иммунокомпетентных мышах |
− Моделирование опухолевого роста. − Доклинический скрининг методов лечения, затрагивающих иммунную систему, в том числе. ингибиторов контрольных точек иммунитета |
| Использование комбинации вирусных и человеческих онкобелков для моделирования молекулярного фона in vivo и in vitro, способствующего злокачественной трансформации | − Моделирование и исследование опухолей, связанных с вирусными инфекциями | ||
| Канцерогенез, спровоцированный химическими или физическими факторами на фоне вирусной инфекции или с привлечением вирусных антигенов | − Изучение вирусного канцерогенеза на фоне компетентной иммунной системы | − Отсутствие воспроизводимости эффектов.
− Неоднородность эффектов. − Невозможности ограничить воздействие одним органом или типом ткани. − Длительность эксперимента |
− Изучение канцерогенеза, индуцируемого химическими или физическими факторами на фоне
вирусной инфекции или кооперацией вирусных антигенов и химических/физических факторов. − Исследование влияния диет и воспаления на развитие опухолей |
| Инфицирование вирусами грызунов, родственными онкогенным вирусам человека | − Изучение вирусной инфекции на фоне компетентной иммунной системы | − Узкий спектр вирусов грызунов, аналогичных онкогенным вирусам человека | − Моделирование вирус-индуцированной патологий, в том числе опухолей, вызываемых вирусами
человека, у лабораторных мышей. − Изучение механизмов заражения. − Оценка эффективности профилактический препаратов |
В случае лимфотропных вирусов, таких как ВИЧ-1 и ВЭБ, наиболее удачными моделями считаются мыши с иммунодефицитом с имплантированными элементами иммунной системы человека, так как эти модели позволяют изучать естественные механизмы реинфекции и патогенеза вирусов этой группы. Однако возможность применения этих моделей для оценки эффективности вакцинных препаратов ограничена, поскольку иммунная система этих мышей не может в полной мере моделировать иммунную систему иммунокомпетентных животных.
Патогенез гепатотропных вирусов изучают на животных, трансгенных по целому вирусному геному или отдельным вирусным генам. Эти модели позволяют изучать механизмы вирусного патогенеза, но они не могут использоваться для тестирования противовирусных препаратов и вакцин, так как не предусматривают возможность элиминации вируса.
Ограниченно в качестве моделей применяют мышей, трансгенных по факторам, необходимым для вирусной инфекции (рецепторы, внутриклеточные кофакторы), так как для успешного инфицирования вирусами человека зачастую необходимо вводить в мышь целую панель клеточных факторов (например, для ВИЧ-1), при том что ряд факторов до сих пор не охарактеризован (например, для ВГС). Редко используется и моделирование процесса вирус-ассоциированного канцерогенеза с помощью вирусов грызунов, родственных онкогенным вирусам человека, прежде всего в силу узости спектра этих вирусов.
Таким образом, несмотря на разнообразие мышиных моделей вирусной инфекции и вирус-ассоциированного патогенеза, до сих пор не создана “идеальная” модель, полностью воссоздающая процесс инфекции, патогенеза и иммунного ответа человека. Исследователям необходимо выбирать модель, наиболее подходящую для решения конкретных задач.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 19-04-01034).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Frese K.K., Tuveson D.A. (2007) Maximizing mouse cancer models. Nat. Rev. Cancer. 7, 645–658.
Morton C.L., Houghton P.J. (2007) Establishment of human tumor xenografts in immunodeficient mice. Nat. Protocols. 2, 247–250.
Rubio-Viqueira B., Jimeno A., Cusatis G., Zhang X., Iacobuzio-Donahue C., Karikari C., Shi C., Danenberg K., Danenberg P.V., Kuramochi H., Tanaka K., Singh S., Salimi-Moosavi H., Bouraoud N., Amador M.L., Altiok S., Kulesza P., Yeo C., Messersmith W., Eshleman J., Hruban R.H., Maitra A., Hidalgo M. (2006) An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res.: An Official J. Am. Ass. Cancer Res. 12, 4652–4661.
Szadvari I., Krizanova O., Babula P. (2016) Athymic nude mice as an experimental model for cancer treatment. Physiol. Res. 65, S441–S453.
Bosma M.J., Carroll A.M. (1991) The SCID mouse mutant: definition, characterization, and potential uses. Annu. Rev. Immunol. 9, 323–350.
Shultz L.D., Schweitzer P.A., Christianson S.W., Gott B., Schweitzer I.B., Tennent B., McKenna S., Mobraaten L., Rajan T.V., Greiner D.L. (1995) Multiple defects in innate and adaptive immunologic function in NOD/LtSz-SCID mice. J. Immunol. 154, 180–191.
Ito M., Hiramatsu H., Kobayashi K., Suzue K., Kawahata M., Hioki K., Ueyama Y., Koyanagi Y., Sugamura K., Tsuji K., Heike T., Nakahata T. (2002) NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 100, 3175–3182.
Christianson S.W., Greiner D.L., Hesselton R.A., Leif J.H., Wagar E.J., Schweitzer I.B., Rajan T.V., Gott B., Roopenian D.C., Shultz L.D. (1997) Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J. Immunol. 158, 3578–3586.
Audige A., Rochat M.A., Li D., Ivic S., Fahrny A., Muller C.K.S., Gers-Huber G., Myburgh R., Bredl S., Schlaepfer E., Scherrer A.U., Kuster S.P., Speck R.F. (2017) Long-term leukocyte reconstitution in NSG mice transplanted with human cord blood hematopoietic stem and progenitor cells. BMC Immunol. 18, 28.
Ishikawa F. (2013) Modeling normal and malignant human hematopoiesis in vivo through newborn NSG xenotransplantation. Internat. I. Hematol. 98, 634–640.
Murakami M., Hoshikawa Y., Satoh Y., Ito H., Tajima M., Okinaga K., Miyazawa Y., Kurata T., Sairenji T. (2000) Tumorigenesis of Epstein–Barr virus-positive epithelial cell lines derived from gastric tissues in the SCID mouse. Virology. 277, 20–26.
Dubich T., Lieske A., Santag S., Beauclair G., Ruckert J., Herrmann J., Gorges J., Busche G., Kazmaier U., Hauser H., Stadler M., Schulz T.F., Wirth D. (2019) An endothelial cell line infected by Kaposi’s sarcoma-associated herpes virus (KSHV) allows the investigation of Kaposi’s sarcoma and the validation of novel viral inhibitors in vitro and in vivo. J. Mol. Med. 97, 311–324.
Fujii E., Kato A., Chen Y.J., Matsubara K., Ohnishi Y., Suzuki M. (2014) Characterization of EBV-related lymphoproliferative lesions arising in donor lymphocytes of transplanted human tumor tissues in the NOG mouse. Exp. Animals. 63, 289–296.
Bondarenko G., Ugolkov A., Rohan S., Kulesza P., Dubrovskyi O., Gursel D., Mathews J., O’Halloran T.V., Wei J.J., Mazar A.P. (2015) Patient-derived tumor xenografts are susceptible to formation of human lymphocytic tumors. Neoplasia. 17, 735–741.
Tanaka T., Nishie R., Ueda S., Miyamoto S., Hashida S., Konishi H., Terada S., Kogata Y., Sasaki H., Tsunetoh S., Taniguchi K., Komura K., Ohmichi M. (2021) Patient-derived xenograft models in cervical cancer: a systematic review. Internat. J. Mol. Sci. 22(17), 9369.
Larmour L.I., Cousins F.L., Teague J.A., Deane J.A., Jobling T.W., Gargett C.E. (2018) A patient derived xenograft model of cervical cancer and cervical dysplasia. PLoS One. 13, e0206539.
Ilan E., Burakova T., Dagan S., Nussbaum O., Lubin I., Eren R., Ben-Moshe O., Arazi J., Berr S., Neville L., Yuen L., Mansour T.S., Gillard J., Eid A., Jurim O., Shouval D., Reisner Y., Galun E. (1999) The hepatitis B virus-trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents. Hepatology. 29, 553–562.
Liu J., Chen S., Zou Z., Tan D., Liu X., Wang X. (2019) Pathological pattern of intrahepatic HBV in HCC is phenocopied by PDX-derived mice: a novel model for antiviral treatment. Translat. Oncol. 12, 1138–1146.
Nazzal M., Sur S., Steele R., Khatun M., Patra T., Phillips N., Long J., Ray R., Ray R.B. (2020) Establishment of a patient-derived xenograft tumor from hepatitis C-associated liver cancer and evaluation of imatinib treatment efficacy. Hepatology. 72, 379–388.
Cho S.Y. (2020) Patient-derived xenografts as compatible models for precision oncology. Lab. Animal Res. 36, 14.
Malaney P., Nicosia S.V., Dave V. (2014) One mouse, one patient paradigm: new avatars of personalized cancer therapy. Cancer Lett. 344, 1–12.
De Both N.J., Vermey M., Groen N., Dinjens W.N., Bosman F.T. (1997) Clonal growth of colorectal-carcinoma cell lines transplanted to nude mice. Internat. J. Cancer. 72, 1137–1141.
Becher O.J., Holland E.C. (2006) Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 66, 3355–3358, discussion 3358–3359.
Fiebig H.H., Berger D.P., Winterhalter B.R., Plowman J. (1990) In vitro and in vivo evaluation of US-NCI compounds in human tumor xenografts. Cancer Treat. Rev. 17, 109–117.
Johnson J.I., Decker S., Zaharevitz D., Rubinstein L.V., Venditti J.M., Schepartz S., Kalyandrug S., Christian M., Arbuck S., Hollingshead M., Sausville E.A. (2001) Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer. 84, 1424–1431.
Byrne A.T., Alferez D.G., Amant F., Annibali D., Arribas J., Biankin A.V., Bruna A., Budinska E., Caldas C., Chang D.K., Clarke R.B., Clevers H., Coukos G., Dangles-Marie V., Eckhardt S.G., Gonzalez-Suarez E., Hermans E., Hidalgo M., Jarzabek M.A., de Jong S., Jonkers J., Kemper K., Lanfrancone L., Maelandsmo G.M., Marangoni E., Marine J.C., Medico E., Norum J.H., Palmer H.G., Peeper D.S., Pelicci P.G., Piris-Gimenez A., Roman-Roman S., Rueda O.M., Seoane J., Serra V., Soucek L., Vanhecke D., Villanueva A., Vinolo E., Bertotti A., Trusolino L. (2017) Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer. 17, 254–268.
Buitrago-Perez A., Hachimi M., Duenas M., Lloveras B., Santos A., Holguin A., Duarte B., Santiago J.L., Akgul B., Rodriguez-Peralto J.L., Storey A., Ribas C., Larcher F., del Rio M., Paramio J.M., Garcia-Escudero R. (2012) A humanized mouse model of HPV-associated pathology driven by E7 expression. PLoS One. 7, e41743.
Hazari S., Hefler H.J., Chandra P.K., Poat B., Gunduz F., Ooms T., Wu T., Balart L.A., Dash S. (2011) Hepatocellular carcinoma xenograft supports HCV replication: a mouse model for evaluating antivirals. World J. Gastroenterol. 17, 300–312.
Meuleman P., Leroux-Roels G. (2008) The human liver-uPA-SCID mouse: a model for the evaluation of antiviral compounds against HBV and HCV. Antiviral Res. 80, 231–238.
Guevin C., Lamarre A., Labonte P. (2009) Novel HCV replication mouse model using human hepatocellular carcinoma xenografts. Antiviral Res. 84, 14–22.
Akkina R. (2013) New generation humanized mice for virus research: comparative aspects and future prospects. Virology. 435, 14–28.
Richmond A., Su Y. (2008) Mouse xenograft models vs GEM models for human cancer therapeutics. Disease Models Mech. 1, 78–82.
Jin K., Teng L., Shen Y., He K., Xu Z., Li G. (2010) Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin. Translat. Oncol.: Official Publ. Fed. Spanish Oncol. Soc. Nat. Canc. Inst. Mexico. 12, 473–480.
Walsh N.C., Kenney L.L., Jangalwe S., Aryee K.E., Greiner D.L., Brehm M.A., Shultz L.D. (2017) Humanized mouse models of clinical disease. Annu. Rev. Pathol. 12, 187–215.
Hatziioannou T., Evans D.T. (2012) Animal models for HIV/AIDS research. Nat. Rev. Microbiol. 10, 852–867.
Brehm M.A., Shultz L.D., Greiner D.L. (2010) Humanized mouse models to study human diseases. Curr. Opin. Endocrinol. Diabetes Obesity. 17, 120–125.
Wang L.X., Kang G., Kumar P., Lu W., Li Y., Zhou Y., Li Q., Wood C. (2014) Humanized-BLT mouse model of Kaposi’s sarcoma-associated herpesvirus infection. Proc. Natl. Acad. Sci. USA. 111, 3146–3151.
Ma S.D., Xu X., Plowshay J., Ranheim E.A., Burlingham W.J., Jensen J.L., Asimakopoulos F., Tang W., Gulley M.L., Cesarman E., Gumperz J.E., Kenney S.C. (2015) LMP1-deficient Epstein–Barr virus mutant requires T cells for lymphomagenesis. J. Clin. Invest. 125, 304–315.
Ahmed E.H., Baiocchi R.A. (2016) Murine models of Epstein–Barr virus-associated lymphomagenesis. ILAR Jl. 57, 55–62.
Gauduin M.C., Parren P.W., Weir R., Barbas C.F., Burton D.R., Koup R.A. (1997) Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat. Med. 3, 1389–1393.
Parren P.W., Ditzel H.J., Gulizia R.J., Binley J.M., Barbas C.F., 3rd, Burton D.R., Mosier D.E. (1995) Protection against HIV-1 infection in hu-PBL-SCID mice by passive immunization with a neutralizing human monoclonal antibody against the gp120 CD4-binding site. AIDS. 9, F1–F6.
Dexter D.L., Diamond M., Creveling J., Chen S.F. (1993) Chemotherapy of mammary carcinomas arising in ras transgenic mice. Invest. New Drugs. 11, 161–168.
Haddad A.F., Young J.S., Amara D., Berger M.S., Raleigh D.R., Aghi M.K., Butowski N.A. (2021) Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neuro-Oncol. Adv. 3, vdab100.
Nomura T., Tamaoki N., Takakura A., Suemizu H. (2008) Basic concept of development and practical application of animal models for human diseases. Curr. Topics Microbiol. Immunol. 324, 1–24.
Coussens L.M., Hanahan D., Arbeit J.M. (1996) Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am. J. Pathol. 149, 1899–1917.
Winkler E.S., Bailey A.L., Kafai N.M., Nair S., McCune B.T., Yu J., Fox J.M., Chen R.E., Earnest J.T., Keeler S.P., Ritter J.H., Kang L.I., Dort S., Robichaud A., Head R., Holtzman M.J., Diamond M.S. (2020) SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 21, 1327–1335.
Ren R.B., Costantini F., Gorgacz E.J., Lee J.J., Racaniello V.R. (1990) Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis. Cell. 63, 353–362.
Mouse Genome Sequencing C., Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., Antonarakis S.E., Attwood J., Baertsch R., Bailey J., Barlow K., Beck S., Berry E., Birren B., Bloom T., Bork P., Botcherby M., Bray N., Brent M.R., Brown D.G., Brown S.D., Bult C., Burton J., Butler J., Campbell R.D., Carninci P., Cawley S., Chiaromonte F., Chinwalla A.T., Church D.M., Clamp M., Clee C., Collins F.S., Cook L.L., Copley R.R., Coulson A., Couronne O., Cuff J., Curwen V., Cutts T., Daly M., David R., Davies J., Delehaunty K.D., Deri J., Dermitzakis E.T., Dewey C., Dickens N.J., Diekhans M., Dodge S., Dubchak I., Dunn D.M., Eddy S.R., Elnitski L., Emes R.D., Eswara P., Eyras E., Felsenfeld A., Fewell G.A., Flicek P., Foley K., Frankel W.N., Fulton L.A., Fulton R.S., Furey T.S., Gage D., Gibbs R.A., Glusman G., Gnerre S., Goldman N., Goodstadt L., Grafham D., Graves T.A., Green E.D., Gregory S., Guigo R., Guyer M., Hardison R.C., Haussler D., Hayashizaki Y., Hillier L.W., Hinrichs A., Hlavina W., Holzer T., Hsu F., Hua A., Hubbard T., Hunt A., Jackson I., Jaffe D.B., Johnson L.S., Jones M., Jones T.A., Joy A., Kamal M., Karlsson E.K., Karolchik D., Kasprzyk A., Kawai J., Keibler E., Kells C., Kent W.J., Kirby A., Kolbe D.L., Korf I., Kucherlapati R.S., Kulbokas E.J., Kulp D., Landers T., Leger J.P., Leonard S., Letunic I., Levine R., Li J., Li M., Lloyd C., Lucas S., Ma B., Maglott D.R., Mardis E.R., Matthews L., Mauceli E., Mayer J.H., McCarthy M., McCombie W.R., McLaren S., McLay K., McPherson J.D., Meldrim J., Meredith B., Mesirov J.P., Miller W., Miner T.L., Mongin E., Montgomery K.T., Morgan M., Mott R., Mullikin J.C., Muzny D.M., Nash W.E., Nelson J.O., Nhan M.N., Nicol R., Ning Z., Nusbaum C., O’Connor M.J., Okazaki Y., Oliver K., Overton-Larty E., Pachter L., Parra G., Pepin K.H., Peterson J., Pevzner P., Plumb R., Pohl C.S., Poliakov A., Ponce T.C., Ponting C.P., Potter S., Quail M., Reymond A., Roe B.A., Roskin K.M., Rubin E.M., Rust A.G., Santos R., Sapojnikov V., Schultz B., Schultz J., Schwartz M.S., Schwartz S., Scott C., Seaman S., Searle S., Sharpe T., Sheridan A., Shownkeen R., Sims S., Singer J.B., Slater G., Smit A., Smith D.R., Spencer B., Stabenau A., Stange-Thomann N., Sugnet C., Suyama M., Tesler G., Thompson J., Torrents D., Trevaskis E., Tromp J., Ucla C., Ureta-Vidal A., Vinson J.P., Von Niederhausern A.C., Wade C.M., Wall M., Weber R.J., Weiss R.B., Wendl M.C., West A.P., Wetterstrand K., Wheeler R., Whelan S., Wierzbowski J., Willey D., Williams S., Wilson R.K., Winter E., Worley K.C., Wyman D., Yang S., Yang S.P., Zdobnov E.M., Zody M.C., Lander E.S. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature. 420, 520–562.
Dorner M., Horwitz J.A., Robbins J.B., Barry W.T., Feng Q., Mu K., Jones C.T., Schoggins J.W., Catanese M.T., Burton D.R., Law M., Rice C.M., Ploss A. (2011) A genetically humanized mouse model for hepatitis C virus infection. Nature. 474, 208–211.
Burm R., Collignon L., Mesalam A.A., Meuleman P. (2018) Animal models to study hepatitis C virus infection. Front. Immunol. 9, 1032.
Li H., Zhuang Q., Wang Y., Zhang T., Zhao J., Zhang Y., Zhang J., Lin Y., Yuan Q., Xia N., Han J. (2014) HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell Mol. Immunol. 11, 175–183.
Boberg A., Brave A., Johansson S., Wahren B., Hin-kula J., Rollman E. (2008) Murine models for HIV vaccination and challenge. Expert Rev. Vaccines. 7, 117–130.
Mestas J., Hughes C.C. (2004) Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738.
Masemann D., Ludwig S., Boergeling Y. (2020) Advances in transgenic mouse models to study infections by human pathogenic viruses. Internat. J. Mol. Sci. 21(23), 9289.
Inuzuka T., Takahashi K., Chiba T., Marusawa H. (2014) Mouse models of hepatitis B virus infection comprising host–virus immunologic interactions. Pathogens. 3, 377–389.
Li Y.T., Wu H.L., Liu C.J. (2021) Molecular mechanisms and animal models of HBV-related hepatocellular carcinoma: with emphasis on metastatic tumor antigen 1. Internat. J. Mol. Sci. 22(17), 9380.
Moriya K., Nakagawa K., Santa T., Shintani Y., Fujie H., Miyoshi H., Tsutsumi T., Miyazawa T., Ishibashi K., Horie T., Imai K., Todoroki T., Kimura S., Koike K. (2001) Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61, 4365–4370.
Liu Y., Maya S., Ploss A. (2021) Animal models of hepatitis B virus infection-success, challenges, and future directions. Viruses. 13(5), 777
Leonard J.M., Abramczuk J.W., Pezen D.S., Rutledge R., Belcher J.H., Hakim F., Shearer G., Lamperth L., Travis W., Fredrickson T., Notkinsand A.L., Martin M.A. (1988) Development of disease and virus recovery in transgenic mice containing HIV proviral DNA. Science. 242, 1665–1670.
Goudreau G., Carpenter S., Beaulieu N., Jolicoeur P. (1996) Vacuolar myelopathy in transgenic mice expressing human immunodeficiency virus type 1 proteins under the regulation of the myelin basic protein gene promoter. Nat. Med. 2, 655–661.
Vogel J., Hinrichs S.H., Reynolds R.K., Luciw P.A., Jay G. (1988) The HIV tat gene induces dermal lesions resembling Kaposi’s sarcoma in transgenic mice. Nature. 335, 606–611.
Brady H.J., Abraham D.J., Pennington D.J., Miles C.G., Jenkins S., Dzierzak E.A. (1995) Altered cytokine expression in T lymphocytes from human immunodeficiency virus Tat transgenic mice. J. Virol. 69, 7622–7629.
Bravo Cruz A.G., Damania B. (2019) In vivo models of oncoproteins encoded by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 93(11), e01053-18.
Yu M., Chandra J. (2021) Therapeutic DNA vaccine against HPV16-associated cancer. Meth. Mol. Biol. 2197, 241–252.
Pascolo S. (2005) HLA class I transgenic mice: development, utilisation and improvement. Expert Opin. Biol. Therapy. 5, 919–938.
Pascolo S., Bervas N., Ure J.M., Smith A.G., Lemonnier F.A., Perarnau B. (1997) HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J. Exp. Med. 185, 2043–2051.
Boucherma R., Kridane-Miledi H., Bouziat R., Rasmussen M., Gatard T., Langa-Vives F., Lemercier B., Lim A., Berard M., Benmohamed L., Buus S., Rooke R., Lemonnier F.A. (2013) HLA-A*01:03, HLA-A*24:02, HLA-B*08:01, HLA-B*27:05, HLA-B*35:01, HLA-B*44:02, and HLA-C*07:01 monochain transgenic/H-2 class I null mice: novel versatile preclinical models of human T cell responses. J. Iimmunol. 191, 583–593.
Zottnick S., Voss A.L., Riemer A.B. (2020) Inducing immunity where it matters: orthotopic HPV tumor models and therapeutic vaccinations. Front. Immunol. 11, 1750.
Eiben G.L., Velders M.P., Schreiber H., Cassetti M.C., Pullen J.K., Smith L.R., Kast W.M. (2002) Establishment of an HLA-A*0201 human papillomavirus type 16 tumor model to determine the efficacy of vaccination strategies in HLA-A*0201 transgenic mice. Cancer Res. 62, 5792–5799.
Sun S., Li J. (2017) Humanized chimeric mouse models of hepatitis B virus infection. Internat. J. Infect. Dis.: IJID: Official Publ. Internat. Soc. Infect. Dis. 59, 131–136.
Tateno C., Yoshizane Y., Saito N., Kataoka M., Utoh R., Yamasaki C., Tachibana A., Soeno Y., Asahina K., Hino H., Asahara T., Yokoi T., Furukawa T., Yoshizato K. (2004) Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 165, 901–912.
Dandri M., Burda M.R., Torok E., Pollok J.M., Iwanska A., Sommer G., Rogiers X., Rogler C.E., Gupta S., Will H., Greten H., Petersen J. (2001) Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology. 33, 981–988.
Tsuge M., Hiraga N., Takaishi H., Noguchi C., Oga H., Imamura M., Takahashi S., Iwao E., Fujimoto Y., Ochi H., Chayama K., Tateno C., Yoshizato K. (2005) Infection of human hepatocyte chimeric mouse with genetically engineered hepatitis B virus. Hepatology. 42, 1046–1054.
Yuan L., Jiang J., Liu X., Zhang Y., Zhang L., Xin J., Wu K., Li X., Cao J., Guo X., Shi D., Li J., Jiang L., Sun S., Wang T., Hou W., Zhang T., Zhu H., Zhang J., Yuan Q., Cheng T., Li J., Xia N. (2019) HBV infection-induced liver cirrhosis development in dual-humanised mice with human bone mesenchymal stem cell transplantation. Gut. 68, 2044–2056.
Hu J., Lin Y.Y., Chen P.J., Watashi K., Wakita T. (2019) Cell and animal models for studying hepatitis B virus infection and drug development. Gastroenterology. 156, 338–354.
Ilan E., Arazi J., Nussbaum O., Zauberman A., Eren R., Lubin I., Neville L., Ben-Moshe O., Kischitzky A., Litchi A., Margalit I., Gopher J., Mounir S., Cai W., Daudi N., Eid A., Jurim O., Czerniak A., Galun E., Dagan S. (2002) The hepatitis C virus (HCV)-Trimera mouse: a model for evaluation of agents against HCV. J. Infect. Dis. 185, 153–161.
Mercer D.F., Schiller D.E., Elliott J.F., Douglas D.N., Hao C., Rinfret A., Addison W.R., Fischer K.P., Churchill T.A., Lakey J.R., Tyrrell D.L., Kneteman N.M. (2001) Hepatitis C virus replication in mice with chimeric human livers. Nat. Medicine. 7, 927–933.
Kneteman N.M., Weiner A.J., O’Connell J., Collett M., Gao T., Aukerman L., Kovelsky R., Ni Z.J., Zhu Q., Hashash A., Kline J., Hsi B., Schiller D., Douglas D., Tyrrell D.L., Mercer D.F. (2006) Anti-HCV therapies in chimeric scid-Alb/uPA mice parallel outcomes in human clinical application. Hepatology. 43, 1346–1353.
Turrini P., Sasso R., Germoni S., Marcucci I., Celluci A., Di Marco A., Marra E., Paonessa G., Eutropi A., Laufer R., Migliaccio G., Padron J. (2006) Development of humanized mice for the study of hepatitis C virus infection. Transplant. Proc. 38, 1181–1184.
Nolan K., Verzosa G., Cleaver T., Tippimanchai D., DePledge L.N., Wang X.J., Young C., Le A., Doebele R., Li H., Malkoski S.P. (2020) Development of syngeneic murine cell lines for use in immunocompetent orthotopic lung cancer models. Cancer Cell Internat. 20, 417.
Talmadge J.E., Singh R.K., Fidler I.J., Raz A. (2007) Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 170, 793–804.
Takaki H., Oshiumi H., Shingai M., Matsumoto M., Seya T. (2017) Development of mouse models for analysis of human virus infections. Microbiol. Immunol. 61, 107–113.
Zitvogel L., Pitt J.M., Daillere R., Smyth M.J., Kroemer G. (2016) Mouse models in oncoimmunology. Nat. Rev. Cancer. 16, 759–773.
Gunther J.H., Jurczok A., Wulf T., Brandau S., Deinert I., Jocham D., Bohle A. (1999) Optimizing syngeneic orthotopic murine bladder cancer (MB49). Cancer Res. 59, 2834–2837.
Mittal V.K., Bhullar J.S., Jayant K. (2015) Animal models of human colorectal cancer: current status, uses and limitations. W. J. Gastroenterol. 21, 11854–11861.
Tsukiyama-Kohara K. (2012) Role of oxidative stress in hepatocarcinogenesis induced by hepatitis C virus. Internat. J. Mol. Sci. 13, 15271–15278.
Bayurova E., Jansons J., Skrastina D., Smirnova O., Mezale D., Kostyusheva A., Kostyushev D., Petkov S., Podschwadt P., Valuev-Elliston V. (2019) HIV-1 reverse transcriptase promotes tumor growth and metastasis formation via ROS-dependent upregulation of twist. Oxidative Med. Cell. Longevity. 2019, 6016278. https://doi.org/10.1155/2019/601627
Lawson J.S., Salmons B., Glenn W.K. (2018) Oncogenic viruses and breast cancer: mouse mammary tumor virus (MMTV), bovine leukemia virus (BLV), human papilloma virus (HPV), and Epstein–Barr virus (EBV). Fron. Oncol. 8, 1.
Wei T., Buehler D., Ward-Shaw E., Lambert P.F. (2020) An infection-based murine model for papillomavirus-associated head and neck cancer. mBio. 11(3), e00908-20.
Spurgeon M.E., Lambert P.F. (2019) Sexual transmission of murine papillomavirus (MmuPV1) in Mus musculus. eLife. 8, e50056.
Yu L., Majerciak V., Xue X.Y., Uberoi A., Lobanov A., Chen X., Cam M., Hughes S.H., Lambert P.F., Zheng Z.M. (2021) Mouse papillomavirus type 1 (MmuPV1) DNA is frequently integrated in benign tumors by microhomology-mediated end-joining. PLoS Pathog. 17, e1009812.
Liang X., Paden C.R., Morales F.M., Powers R.P., Jacob J., Speck S.H. (2011) Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS Pathog. 7, e1002220.
Butel J.S. (2000) Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 21, 405–426.
Замараев А.В., Животовский Б., Копеина Г.С. (2020) Вирусные инфекции: негативный регулятор апоптоза и фактор онкогенности. Биохимия. 85(10), 1398–1410.
Sun W., Yang J. (2010) Functional mechanisms for human tumor suppressors. J. Cancer. 1, 136–140.
Attardi L.D., Jacks T. (1999) The role of p53 in tumour suppression: lessons from mouse models. Cell. Mol. Life Sci: CMLS. 55, 48–63.
Clarke A.R., Hollstein M. (2003) Mouse models with modified p53 sequences to study cancer and ageing. Cell Death Differ. 10, 443–450.
Levine A.J., Oren M. (2009) The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 9, 749–758.
Ahuja D., Saenz-Robles M.T., Pipas J.M. (2005) SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 24, 7729–7745.
Hudson A.L., Colvin E.K. (2016) Transgenic mouse models of SV40-induced cancer. ILAR J. 57, 44–54.
Engeland K. (2018) Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 25, 114–132.
Lipinski M.M., Jacks T. (1999) The retinoblastoma gene family in differentiation and development. Oncogene. 18, 7873–7882.
Gupta S., Kumar P., Das B.C. (2018) HPV: molecular pathways and targets. Curr. Problems Cancer. 42, 161–174.
Matlashewski G., Schneider J., Banks L., Jones N., Murray A., Crawford L. (1987) Human papillomavirus type 16 DNA cooperates with activated ras in transforming primary cells. EMBO J. 6, 1741–1746.
Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D. (2011) RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 11, 761–774.
Kivinen L., Tsubari M., Haapajarvi T., Datto M.B., Wang X.F., Laiho M. (1999) Ras induces p21Cip1/Waf1 cyclin kinase inhibitor transcriptionally through Sp1-binding sites. Oncogene. 18, 6252–6261.
Feltkamp M.C., Smits H.L., Vierboom M.P., Minnaar R.P., de Jongh B.M., Drijfhout J.W., ter Schegget J., Melief C.J., Kast W.M. (1993) Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol. 23, 2242–2249.
Mermod M., Hiou-Feige A., Bovay E., Roh V., Sponarova J., Bongiovanni M., Vermeer D.W., Lee J.H., Petrova T.V., Rivals J.P., Monnier Y., Tolstonog G.V., Simon C. (2018) Mouse model of postsurgical primary tumor recurrence and regional lymph node metastasis progression in HPV-related head and neck cancer. Intern. J. Cancer. 142, 2518–2528.
Paolini F., Massa S., Manni I., Franconi R., Venuti A. (2013) Immunotherapy in new pre-clinical models of HPV-associated oral cancers. Hum. Vaccin. Immunother. 9, 534–543.
Henkle T.R., Lam B., Kung Y.J., Lin J., Tseng S.H., Ferrall L., Xing D., Hung C.F., Wu T.C. (2021) Development of a novel mouse model of spontaneous high-risk HPVE6/E7-expressing carcinoma in the cervicovaginal tract. Cancer Res. 81, 4560–4569.
Haverkos H.W. (2004) Viruses, chemicals and co-carcinogenesis. Oncogene. 23, 6492–6499.
Isaguliants M., Bayurova E., Avdoshina D., Kondrashova A., Chiodi F., Palefsky J.M. (2021) Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers. 13(2), 305.
Kornek M., Raskopf E., Guetgemann I., Ocker M., Gerceker S., Gonzalez-Carmona M.A., Rabe C., Sauerbruch T., Schmitz V. (2006) Combination of systemic thioacetamide (TAA) injections and ethanol feeding accelerates hepatic fibrosis in C3H/He mice and is associated with intrahepatic up regulation of MMP-2, VEGF and ICAM-1. J. Hepatol. 45, 370–376.
Longnecker D.S., Kuhlmann E.T., Freeman D.H., Jr. (1990) Characterization of the elastase 1-simian virus 40 T-antigen mouse model of pancreatic carcinoma: effects of sex and diet. Cancer Res. 50, 7552–7554.
Lau T.C., Fiebig-Comyn A.A., Shaler C.R., McPhee J.B., Coombes B.K., Schertzer J.D. (2021) Low dietary fiber promotes enteric expansion of a Crohn’s disease-associated pathobiont independent of obesity. Am. J. Physiol. Endocrinol. Metabolism. 321, E338–E350.
Morrison K.E., Jasarevic E., Howard C.D., Bale T.L. (2020) It’s the fiber, not the fat: significant effects of dietary challenge on the gut microbiome. Microbiome. 8, 15.
Wang L., Yi T., Zhang W., Pardoll D.M., Yu H. (2010) IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 70, 10112–10120.
He D., Li H., Yusuf N., Elmets C.A., Athar M., Katiyar S.K., Xu H. (2012) IL-17 mediated inflammation promotes tumor growth and progression in the skin. PLoS One. 7, e32126.
Isaguliants M., Krotova O., Petkov S., Jansons J., Bayurova E., Mezale D., Fridrihsone I., Kilpelainen A., Podschwadt P., Agapkina Y., Smirnova O., Kostic L., Saleem M., Latyshev O., Eliseeva O., Malkova A., Gorodnicheva T., Wahren B., Gordeychuk I., Starodubova E., Latanova A. (2021) Cellular immune response induced by DNA immunization of mice with drug resistant integrases of HIV-1 clade A offers partial protection against growth and metastatic activity of integrase-expressing adenocarcinoma cells. Microorganisms. 9(6), 1219. https://doi.org/10.3390/microorganisms9061219
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология