

Российские нанотехнологии, 2020, T. 15, № 6, стр. 699-724
АПКОНВЕРТИРУЮЩИЕ КРИСТАЛЛИЧЕСКИЕ НАНОМАТЕРИАЛЫ: СИНТЕЗ, ФОТОЛЮМИНЕСЦЕНТНЫЕ СВОЙСТВА И ПРИМЕНЕНИЕ
Д. Н. Каримов 1, П. А. Демина 1, 2, А. В. Кошелев 1, В. В. Рочева 1, А. В. Соковиков 1, А. Н. Генералова 1, 2, В. П. Зубов 2, Е. В. Хайдуков 1, 3, *, М. В. Ковальчук 1, 3, В. Я. Панченко 1, 3
1 ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
3 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: khaydukov@mail.ru
Поступила в редакцию 02.07.2020
После доработки 02.07.2020
Принята к публикации 17.07.2020
Аннотация
В конце 1990-х годов были продемонстрированы уникальные возможности применения неорганических нанокристаллов, обладающих антистоксовой фотолюминесценцией. Благодаря разработке новых, модификации существующих методов синтеза этого класса наноматериалов и расширению понимания фотофизических процессов, происходящих в нанокристаллах, за достаточно короткий период в данной области был достигнут существенный прорыв. Интерес научного сообщества вызван исключительными люминесцентными свойствами апконвертирующих наноматериалов, которые могут преобразовывать фотоны ближнего ИК-спектра в излучение видимого и УФ-диапазонов. Этот многоквантовый процесс реализуется при низкоинтенсивном возбуждении, что во многом определяет применение наноматериалов данного класса в высокотехнологичных областях, включая биотехнологию, фотохимию, медицину, солнечную энергетику, наносенсорику и др. Целями данного обзора являются рассмотрение механизмов антистоксовой люминесценции, анализ методов синтеза и демонстрация сфер применения фторидных апконвертирующих наноматериалов, в которых они сформировали устойчивую научную и технологическую нишу.
ОГЛАВЛЕНИЕ
Введение
1. Фотофизика процесса апконверсии
2. Активные ионные центры
3. Эффективные пары сенсибилизатор-активатор
4. Кристаллическая матрица наночастицы
5. Методы синтеза наночастиц
5.1. Синтез в среде растворителей
5.2. Синтез наночастиц из расплавов
5.3. Синтез с использованием подхода “сверху-вниз”
6. Гибридные наноконструкции
7. Перспективные направления применения
7.1. Биотехнологии и медицина
7.2. Наносенсоры
7.3. Солнечные элементы
7.4. ИК-индуцируемая фотополимеризация
7.5. Фотокатализ
7.6. Антиконтрафактные технологии
Заключение
ВВЕДЕНИЕ
Явление апконверсии представляет собой нелинейный оптический процесс преобразования света с повышением частоты испускаемых квантов. Процесс должен удовлетворять принципу сохранения энергии и поэтому каждый фотон с высокой энергией излучения требует участия двух или более фотонов возбуждения с более низкой энергией. Механизм апконверсии заметно отличается от других известных нелинейных оптических процессов, таких как многофотонная люминесценция и генерация гармоник. Ключевым отличием является участие в процессе апконверсии реальных энергетических состояний. Из-за этого различия двухфотонная люминесценция и генерация гармоник требуют значительно более высоких интенсивностей возбуждения по сравнению с апконверсией. Этот процесс выражен в материалах, где электронные состояния имеют относительно длительные времена жизни, что позволяет увеличить вероятности взаимодействия с фотонами возбуждения. Механизм может быть эффективно реализован в квантовых системах с частично запрещенными оптическими переходами, поэтому кристаллические материалы, содержащие ионы редкоземельных и переходных элементов, чаще всего изучаются в контексте преобразования с повышением частоты, привлекая внимание с момента открытия данного явления.
Впервые механизм апконверсии был предложен Н. Бломбергеном в 1959 г. [1] и экспериментально обнаружен в кристаллах независимо В.В. Овсянкиным с П.П. Феофиловым [2] и Ф. Аузелем [3] в 1966 г. Из-за низкой эффективности процесса преобразования излучения с повышением частоты эффект апконверсии являлся лишь объектом научных изысканий. Позже, с развитием лазерной техники, этот эффект рассматривался с двух разных позиций. С одной стороны, он являлся паразитным процессом, конкурирующим с лазерной генерацией, например, в кристаллических средах, активированных ионами Er3+; с другой стороны, рассматривался как основа для нелинейной схемы возбуждения лазерной генерации в кристаллах [4]. Однако взрывной интерес к материалам, способным эффективно преобразовывать излучение ближнего ИК-диапазона спектра с повышением частоты, проявился лишь после их успешного синтеза в наноразмерном состоянии [5, 6]. Это обеспечило последовательную конвергенцию эффекта апконверсии в сферу нанотехнологий и привело к созданию нового класса функциональных наноматериалов.
1. ФОТОФИЗИКА ПРОЦЕССА АПКОНВЕРСИИ
Апконверсия является нелинейным оптическим процессом, который реализуется благодаря следующим фотофизическим механизмам, протекающим в активных ионных центрах, помещенных в кристаллическую матрицу: поглощение кванта из основного состояния, поглощение из возбужденного состояния и передача энергии. Иногда в рамках этого процесса для увеличения эффективности конверсии могут применяться схемы, в которых происходит последовательная аккумуляция и миграция энергии между активными центрами (рис. 1).
Рис. 1.
Ключевые фотофизические процессы, вовлеченные в процесс апконверсии; GSA – поглощение из основного состояния, ESA – поглощение из возбужденного состояния, ET – передача энергии, UCL – антистоксовая фотолюминесценция (апконверсия); А – ион-активатор, С – ион-сенсибилизатор, Ак – ион, накапливающий энергию, М – ион, передающий энергию. Схема (а) реализуется в одном активном центре, в схемы (б) и (в) вовлечены два иона одного или разных типов соответственно, в схеме (г) возможно участие трех и более ионов, обеспечивающих поглощение, аккумуляцию, миграцию и испускание энергии [7].
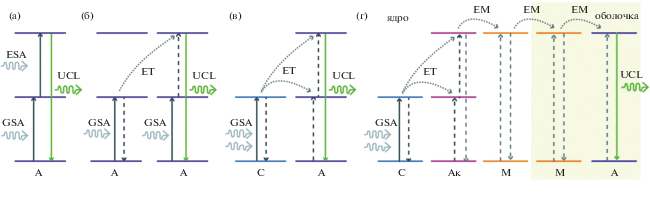
Активные ионы, входящие в состав кристаллической матрицы, принято классифицировать как активаторы и сенсибилизаторы согласно выполняемым ролям в фотопроцессе. Самым простым является механизм апковерсии, в котором участвует один активный центр или ион-активатор, способный поглотить несколько квантов возбуждения через реальное энергетическое состояние с последующим испусканием фотона с повышением частоты (рис. 1а). Более сложным и чаще реализуемым на практике процессом является механизм с привлечением двух активных центров. В этом процессе один ион обеспечивает поглощение кванта света и переходит в возбужденное состояние. Далее при участии процесса переноса энергии возбуждение иона может быть передано на второй ион для его возбуждения в более высоколежащее энергетическое состояние. При этом процессе первый ион переходит в основное состояние. Существуют два типа схожих механизмов. Первый обеспечивается парой идентичных активных ионов, в данном случае взаимодействие вида “активатор–активатор” (рис. 1б), и именно этот процесс наблюдался в первых экспериментах в кристаллах BaF2, легированных ионами Er3+ [2]. Второй подразумевает участие двух различных ионов, т.е. взаимодействие вида “сенсибилизатор–активатор” (рис. 1в), что впервые было продемонстрировано в [3] на системе ионов Yb3+ и Tm3+. В результате передачи энергии от возбужденного иона происходит последовательное возбуждение второго иона через промежуточное до более высоколежащего энергетического состояния с последующим испусканием кванта с большей частотой. Наконец, механизм, представленный на рис. 1г, дополнительно включает в себя процесс аккумуляции и миграции энергии и поэтому требует участия в процессе накапливающих и передающих энергию ионов [7]. Для реализации последнего механизма необходимо применение технологии синтеза структур типа “ядро/оболочка” для обеспечения пространственного разделения активных ионов. Включение в механизм активных центров, обеспечивающих миграцию энергии, облегчает одноступенчатую передачу энергии на ионы-активаторы, значительно снижая требования на время жизни и положение энергетических состояний. Реализация этого механизма позволила впервые включить в механизм апконверсии ионы Eu3+, Tb3+ и Sm3+, промежуточные состояния которых не могут напрямую участвовать в процессе апконверсии, т.е. возбуждаться излучением ближнего ИК-диапазона.
С микроскопической точки зрения процесс апконверсии за счет переноса энергии реализуется благодаря безызлучательной передаче энергии между двумя активными центрами. Как видно из рис. 1, в процесс апконверсии вовлечены ионы, отдающие и принимающие энергию. В этом контексте их еще принято называть соответственно донором и акцептором. Отметим, что передача энергии возможна не только между ионами в возбужденном и основном состоянии, но и между двумя возбужденными ионами, в том числе ионами разного типа. Механизм безызлучательного переноса электронного возбуждения был впервые рассмотрен T. Ферстером для молекул [8]. Ему удалось описать такие взаимодействия теоретически с помощью квантово-механической теории и учетом диполь-дипольного взаимодействия. Позже эта теория была дополнена Д. Декстером для учета мультипольных обменных взаимодействий высших порядков в неорганических матрицах [9].
Вероятность безызлучательного переноса энергии может быть выражена как
где τs – время жизни возбужденного состояния сенсибилизатора; L – расстояние между двумя ионами; L0 – критическое расстояние, на котором вероятность передачи энергии равна вероятности спонтанной безызлучательной релаксации иона-сенсибилизатора; s – целое положительное число, определяющее мультипольность взаимодействия: s = 6, 8 и 10 для диполь-дипольных, диполь-квадрупольных и квадруполь-квадрупольных взаимодействий соответственно. Из этого уравнения вытекает требование к концентрации ионов-сенсибилизаторов и активаторов. При случайном распределении сенсибилизатора и активатора в матрице расстояние L может быть найдено как среднее расстояние между ионами. Этот параметр является крайне важным для оценки эффективности процесса апконверсии при заданных концентрациях легирующих ионов. Расстояние L может быть определено исходя из параметров решетки кристаллической матрицы и заданной концентрации легирующих ионов. Например, для одной из наиболее эффективных апконвертирующих матриц β-Na1.5Y1.5F6, имеющей пр. гр. P63/m, при типичном содержании легирующих ионов-сенсибилизаторов Yb3+ (20%) и ионов-активаторов Er3+ (2%) расстояние между сенсибилизатором и активатором L ~ 0.65 нм [10]. Для расчета оптимальной концентрации легирующих ионов при прогнозировании эффективности апконверсии необходимо использовать более сложные модели, учитывающие также процессы передачи энергии на уровне отдельных активных центров [11].На практике процесс передачи энергии в ходе мультипольного взаимодействия не является резонансным, поэтому для его реализации необходимо привлечение фононов кристаллической решетки, которые способны компенсировать несоответствие в энергетических уровнях. Действительно, если рассматривать реальные энергетические уровни ионов редкоземельных элементов (РЗЭ), можно заметить, что разница энергий между взаимодействующими уровнями может достигать нескольких тысяч см–1 (рис. 2). Для компенсации энергетического зазора может потребоваться более одного фонона [12, 13]. Вероятность переноса энергии с участием фононов экспоненциально зависит от величины несоответствия энергетических уровней ΔЕ [14]:
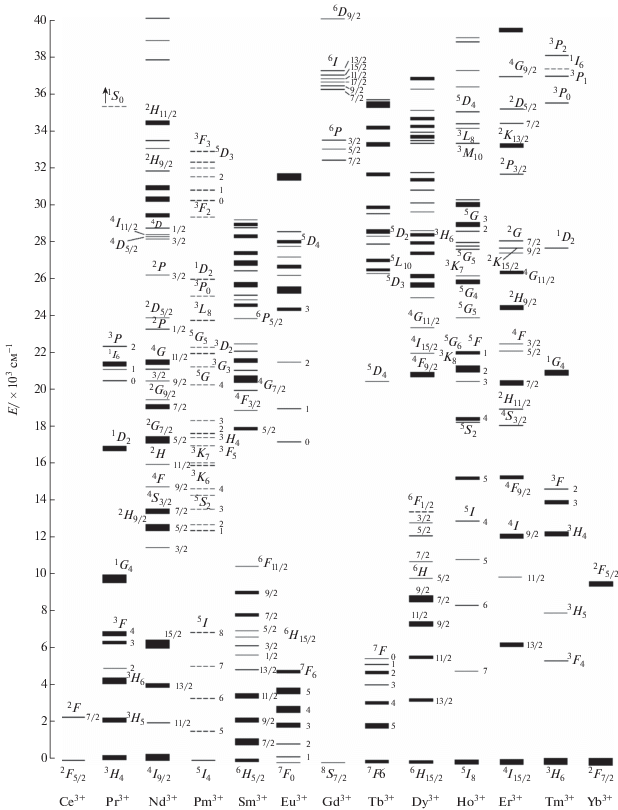
где pph(0) – вероятность переноса энергии при перекрытии линий, β – постоянная, определяемая характеристиками кристаллической матрицы и электрон-фононным взаимодействием.Рис. 2.
Схема энергетических уровней 4f электронной конфигурации для трехвалентных ионов РЗЭ в кристаллической матрице LaF3 [17].
Таким образом, эффективность процесса апконверсии определяется комбинацией легирующих ионов, их концентрацией и фононным спектром кристаллической матрицы.
2. ИОННЫЕ АКТИВНЫЕ ЦЕНТРЫ
В качестве активных центров в апконвертирующих кристаллических материалах обычно используются трехвалентные ионы РЗЭ (R3+), которые имеют электронную конфигурацию 4f n5s25p6 с n = 0–14 для R3+ = La–Lu. Частично заполненная 4f-оболочка отвечает за уникальные спектроскопические свойства и обусловливает наличие большого количества метастабильных энергетических уровней в ИК-, видимой и УФ-областях спектра. Благодаря экранированию внутренних 4f-электронов внешними заполненными 5s- и 5p-оболочками положение энергетических уровней ионов РЗЭ практически не подвержено влиянию кристаллического поля матрицы [15]. Схема энергетических уровней ионов РЗЭ, называемая диаграммой Дике, показана на рис. 2. Она впервые была составлена для этих ионов в кристалле LaCl3 [16]. Позже диаграмма была дополнена для ионов РЗЭ в матрице LaF3 [17] и уточнена в [18] для высоколежащих 4f–4f-переходов всех ионов R3+.
В свободных ионах R3+ переходы между 4f-состояниями запрещены правилами отбора по четности. При помещении этих ионов в несимметричное кристаллическое поле запрет частично снимается. Это обеспечивает наличие метастабильных состояний со временем жизни от сотен микросекунд до миллисекунд, но при этом достаточно низкое сечение таких переходов по сравнению с разрешенными по четности межконфигурационными 4f–5d переходами в ионах R3+ [19].
В нанокристаллах, легированных только ионами-активаторами, двумя основными параметрами, влияющими на квантовый выход апконверсии, являются расстояние между активными ионами и их сечение поглощения. Казалось бы, эффективность поглощения возбуждающего излучения можно повысить благодаря увеличению концентрации ионов активатора в наночастице (НЧ), однако этому препятствует процесс кросc-релаксации, который приводит к потерям энергии [20]. Именно по этой причине концентрация ионов-активаторов в НЧ не может превышать единиц процентов. Так, максимальная концентрация иона эрбия Er3+ в матрице обычно не превышает 3 ат. %, а ионов Tm3+ – 1 ат. %. Кроме того, ионы-активаторы имеют малые значения сечения поглощения ближнего ИК-излучения, что определяет низкую квантовую эффективность апконверсии в НЧ, легированных только одним активным ионом. Это обстоятельство может быть преодолено благодаря использованию пары активных ионов.
Выбор пары сенсибилизатор–активатор требует большого сечения поглощения у иона-сенсибилизатора, наличия “лестничной” схемы метастабильных энергетических состояний у иона-активатора и близости к резонансу энергетических уровней в активной паре. Удивительно, но оптимальная комбинация активных ионов была установлена и продемонстрирована с самого начала, в пионерских работах по апконверсии. Как отметил Ф. Аузель в одной из своих поздних работ: “…Как это часто происходит в науке, наиболее эффективные системы обнаруживаются первыми, здесь это Yb–Er и Yb–Tm” [21]. В действительности одним из наиболее эффективных сенсибилизаторов является ион Yb3+ [22, 23]. Ионы Yb3+, входящие в состав апконвертирующих наночастиц (АНЧ), имеют только два энергетических уровня. В приближении LS-связи эти уровни Yb3+ обозначаются как 2F5/2 и 2F7/2 (рис. 2). Кратность вырождения для уровней 2F5/2 и 2F7/2 равна 6 и 8 соответственно. Электрическое поле матрицы кристалла частично снимает вырождение, но поскольку ион Yb3+ содержит нечетное число электронов, уровни остаются двукратно вырожденными. Систему уровней ионов Yb3+ можно рассматривать как квазидвухуровневую, поскольку в каждом мультиплете устанавливается термодинамически равновесное расщепление заселенностей по подуровням. Это приводит к тому, что сечение поглощения и эффективность люминесценции в значительной мере зависят от симметрии кристаллической матрицы, и, очевидно, чем ниже симметрия матрицы, тем выше эффективность поглощения возбуждающего излучения. Зачастую в качестве “эталонного” сечения поглощения ионов Yb3+ при изучении апконверсии принимается величина σabs ~ 10–20 см2, которая была измерена для матрицы германосиликатного стекла [24]. Вероятно, это является ошибочным, поскольку σabs для конкретной длины волны может варьироваться в зависимости от типа кристаллической матрицы вследствие штарковского расщепления энергетических уровней. Известно, что сечение поглощения для ионов Nd3+, которые также используются в качестве сенсибилизаторов в процессе апконверсии, может существенно изменяться в зависимости от симметрии кристаллического поля матрицы [25–27]. Было показано, что низкосимметричные фторидные соединения, например кристаллы типа LiRF4 и BaR2F8, увеличивают сечение поглощения для ионов R3+ на 2 порядка по сравнению с матрицами, имеющими кубическую кристаллическую решетку [28–31]. Проведенные эксперименты позволили оценить σabs перехода 2F5/2 → 2F7/2 для ионов Yb3+ в НЧ β-NaYF4 на длине волны 975 нм как 2 × × 10–19 см2 [32]. Эти данные указывают на то, что вопрос о сечении поглощения активных ионов в нанокристаллах весьма сложен, поскольку симметрия кристалла на наноразмерном уровне может нарушаться из-за геометрических факторов. Таким образом, значение сечения поглощения ионов R3+ является открытым вопросом, требует детального анализа и проведения дополнительных экспериментов.
3. ЭФФЕКТИВНЫЕ ПАРЫ СЕНСИБИЛИЗАТОР–АКТИВАТОР
Как отмечалось выше, наиболее эффективными ионными парами для реализации процесса апконверсии считаются комбинации Yb3+–Er3+ и Yb3+–Tm3+, которые рассмотрим более детально. На рис. 3 представлена энергетическая диаграмма, поясняющая процесс преобразования излучения на длине волны 980 нм в фотоны ближнего ИК-, видимого и УФ-диапазонов спектра.
Рис. 3.
Схема энергетических уровней для пар сенсибилизатор–активатор, объясняющая механизм апконверсии (а). Характерные спектры фотолюминесценции АНЧ NaYF4:Yb3+/Er3+ и NaYF4:Yb3+/Tm3+ (б). Фотография диспергированных в растворителе АНЧ, люминесцирующих при прохождении через образцы лазерного излучения на длине волны 975 нм (в).
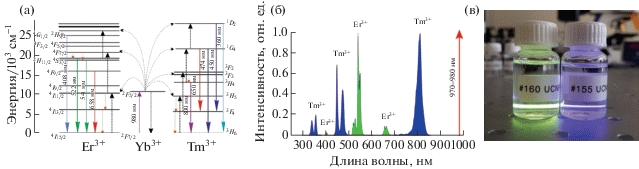
Сенсибилизатор, ион Yb3+, поглощает энергию ИК-излучения, переходя из основного 2F7/2 в возбужденное 2F5/2 состояние. Далее он безызлучательно передает ее близко расположенным соседним ионам Yb3+, тем самым образуя сетку возбужденных Yb3+, до тех пор, пока энергия не захватывается активатором, ионами Er3+ или Tm3+. Активатор, например ион Er3+, переходит из основного 4I15/2 в метастабильное возбужденное состояние 4I11/2, где он может участвовать в следующем синхронном энергообмене с соседним возбужденным ионом Yb3+, в результате чего Er3+ переходит на более высокий энергетический уровень 4F7/2 за счет девозбуждения иона Yb3+. Затем ион Er3+ безызлучательно релаксирует до состояний 2H11/2 и 4S3/2 с последующим испусканием излучения в окрестности 541 нм при переходе 2H11/2 → 4I15/2 и 522 нм при переходе 4S3/2 → 4I15/2. Альтернативно ион Er3+ может безызлучательно девозбуждаться до состояния 4F9/2 с последующим испусканием излучения в окрестности 658 нм при переходе 4F9/2 → 4I15/2. Также к фотолюминесценции (ФЛ) с длиной волны 658 нм (4F9/2 → 4I15/2) может приводить релаксация иона Er3+ из состояния 4I11/2 в состояние 4I13/2 с последующим возбуждением в состояние 4F9/2 при энергообмене с ионом Yb3+ в состоянии 2F5/2. Второй механизм эмиссии с длиной волны в окрестности 658 нм, как правило, наблюдается при интенсивностях возбуждающего излучения ниже 2 Вт/см2 [33]. При достаточной интенсивности возбуждения реализуется трехфотонный процесс апконверсии с длиной волны излучения 408 нм, соответствующий переходу 2H9/2 → 4I15/2. Отметим, что существуют альтернативные схемы, объясняющие механизм ФЛ на длине волны 658 нм из более высоколежащих состояний, с которыми можно ознакомиться, например, в [34].
Из схемы энергетических уровней видно, процессами какой степени являются те или иные переходы в спектре антистоксовой ФЛ. Так, для пары Yb3+ и Tm3+ интенсивность излучения на длине волны 474 нм, соответствующей переходу 1G4 → 3H6 иона тулия, пропорциональна третьей степени от интенсивности накачки в отсутствие насыщения, поскольку для возбуждения этого перехода требуется передача энергии с трех возбужденных ионов Yb3+. По этой причине интенсивность процесса апконверсии имеет нелинейный характер:
где Iex – интенсивность возбуждения, n – требуемое для возбуждения конкретного перехода количество квантов накачки.Теоретическое описание и моделирование процесса апконверсии обычно сводятся к решению скоростных уравнений, описывающих заселенности уровней связанной системы ионов сенсибилизатора и активатора. Такой подход дает довольно хорошее согласование эксперимента с теорией. Однако теоретические модели, описывающие энергетический транспорт в АНЧ, не учитывают время взаимодействия [11], считая перенос энергии бесконечно быстрым. Совсем недавно была предложена феноменологическая модель и показано, что при импульсном возбуждении существенную роль играет миграция энергии по сетке ионов Yb3+ в апконвертирующих нанокристаллах [35]. Также было показано, что наличие этого процесса существенно осложняет анализ времени жизни конкретных уровней и, что более существенно, приводит к неверным оценкам эффективности безызлучательного резонансного переноса энергии в гибридных парах вида АНЧ–органическая молекула. По этой причине необходимы дополнительные исследования, позволяющие глубже изучить механизм апконверсии, в части детального исследования процесса безызлучательной миграции энергии по сетке ионов-сенсибилизаторов в нанокристалле.
4. КРИСТАЛЛИЧЕСКАЯ МАТРИЦА НАНОЧАСТИЦЫ
Определяющую роль в эффективности процесса апконверсии играет выбор материала для кристаллической матрицы, который должен удовлетворять следующим требованиям [10, 22, 36–39]:
– высокий коэффициент пропускания для излучения ИК-возбуждения и широкого диапазона (вплоть до УФ-диапазона) антистоксовой ФЛ, т.е. в качестве основы должны выступать широкозонные диэлектрические материалы, для которых 4f–4f-переходы в ионах РЗЭ попадают в запрещенную зону матрицы;
– высокая изоморфная емкость кристаллической матрицы по отношению к ионам-активаторам, в первую очередь ионам R3+, для обеспечения необходимого уровня легирования;
– требуемая энергия фононов кристаллической решетки матрицы;
– низкая симметрия решетки матрицы и обеспечение необходимой локальной симметрии активных центров в ней, их относительное пространственное положение в кристаллической структуре;
– высокая химическая и термическая стабильность.
Сравнение известных классов широкозонных кислородсодержащих и галогенидных кристаллических соединений показывает, что только материалы на основе неорганических фторидов наиболее полно удовлетворяют приведенным выше требованиям. Значимость класса фторидных материалов для реализации эффективных процессов апконверсии была показана еще в пионерских работах [2], именно фторидные объемные кристаллы в настоящее время являются основой для применения в оптических конверторах и лазерах с накачкой по апконверсионным схемам [39].
Фторидные кристаллы имеют широкую область прозрачности от вакуумного УФ- до среднего ИК-диапазона, узкий фононный спектр (предельная частота фононов во фторидах составляет ~ 350–560 см–1) и высокую химическую стабильность. Самыми низкими значениями энергии фононов (менее 300 см–1) обладают тяжелые галогениды, такие как хлориды, бромиды и йодиды, но их высокая гигроскопичность существенно ограничивает круг применения. Оксидные материалы являются химически стабильными соединениями, но их энергии фононов относительно высоки и обычно сильно превышают значение 500 см–1.
Узкий фононный спектр матрицы является ключевым критерием для процесса апконверсии, так как обеспечивается большее время жизни в “лестничной” схеме метастабильных состояний благодаря снижению вероятности безызлучательной многофононной релаксации возбужденных состояний ионов R3+.
Для увеличения вероятности 4f–4f-переходов ионов R3+ важна как симметрия самой кристаллической матрицы, так и локальная симметрия образующихся активных центров [10]. Так, для гексагональных кристаллов β-NaYF4 (пр. гр. P63/m) с локальной симметрией оптических центров C3h эффективность ФЛ оказывается на порядок выше, чем для кубического аналога α-NaYF4 (пр. гр. $Fm\bar {3}m$) c локальной симметрией центров C4ν [40].
В настоящее время исследовано большое количество фторидных и оксофторидных матриц для создания на их основе АНЧ, а именно: LiRF4, NaRF4, BaRF5, KR3F10, RF3, ROF (R = Y, Gd, Lu, La), CaF2, SrF2, BaF2 [41–56]. Основные усилия направлены на изучение апконверсии в низкосимметричных кристаллических матрицах. Отметим, что исследование апконверсии в кубических фторидных матрицах, например на основе щелочноземельных фторидов [38], несмотря на простоту их синтеза, являются нецелесообразными. В настоящее время наиболее эффективной для задач апконверсии, методически отработанной и практически применяемой кристаллической матрицей для синтеза НЧ, является соединение NaYF4 в гексагональной β-модификации [44, 57].
При поиске новых фторидных кристаллических матриц необходимо учитывать не только приведенные выше требования, но и технологические особенности синтеза АНЧ, такие как морфология, монодисперсность и воспроизводимость спектроскопических характеристик. Совокупность этих факторов задает тенденцию в направлении разработки и совершенствования методов синтеза нанокристаллов именно в области фторидного материаловедения.
5. МЕТОДЫ СИНТЕЗА НАНОЧАСТИЦ
Решающее значение для применения АНЧ имеет разработка стратегий их синтеза с контролируемым составом, размером, кристаллической фазой, спектральными характеристиками и квантовым выходом [58].
Подходы к синтезу могут быть традиционно разделены на две категории: “сверху-вниз” (англ. “top-down”) и “снизу-вверх” (англ. “bottom-up”). В первом случае объемный материал подвергается обработке с использованием механических (например, измельчение) или лазерных (например, абляция) технологий. Подход “снизу-вверх” основан на свойстве молекул формировать супрамолекулярные структуры, которые могут быть получены в процессе гидро/сольвотермального синтеза, термического разложения прекурсоров, микроэмульсионного метода, метода соосаждения, “золь-гель”-метода, микроволнового синтеза. Каждый из этих подходов имеет свои достоинства и недостатки, однако подход “снизу-вверх” позволяет получать монодисперсные НЧ меньшего размера, в то время как подход “сверху-вниз” позволяет эффективнее контролировать химический состав и кристаллическую фазу. По-видимому, перспективным направлением является комбинация двух подходов. Так, в случае синтеза НЧ со структурой типа “ядро/оболочка” для получения ядра можно применять подходы “сверху-вниз”, далее для тонкой настройки фотофизических характеристик формировать оболочки нанообъекта с использованием методик, присущих подходу “снизу-вверх”.
5.1. Синтез в среде растворителей
Методом термического разложения получают НЧ из металлоорганических соединений, выступающих в качестве прекурсоров, которые разлагаются в высококипящих растворителях в присутствии поверхностно-активных веществ (ПАВ). Наиболее широкое применение в качестве прекурсоров нашли трифторацетаты лантаноидов, образующиеся при растворении соответствующих оксидов лантаноидов в водном растворе трифторуксусной кислоты. Термическое разложение обычно проходит в среде 1-октадецена (ОД) в присутствии стабилизаторов, таких как олеиновая кислота (ОК), олеиламин (ОА) или триоктилфосфин оксид (ТОФО), которые содержат функциональную группу для координации с ионами РЗЭ на поверхности образующихся нанокристаллов и длинную углеводородную цепь для предотвращения их агрегации. В процессе синтеза зарождение НЧ происходит мгновенно благодаря высокой скорости разложения прекурсоров, что определяет их узкое распределение по размерам. Природа растворителя, концентрации прекурсоров и стабилизатора, температура и время реакции позволяют синтезировать НЧ с контролируемой морфологией, формой и размером [59–62]. Отметим, что в случае синтеза АНЧ данным методом при участии ТОФО частицы имеют малый диаметр с узким распределением по размерам (d = 9.2 ± 0.73 нм), а использование ОА приводит к полидисперсности в размерном диапазоне от 15 до 40 нм [59]. Вероятно, ТОФО в отличие от ОА более эффективно пассивирует поверхность НЧ, что замедляет их рост и обеспечивает узкое распределение по размерам. Аппаратурное оформление метода термического разложения и изображения частиц, полученные методом просвечивающей электронной микроскопии (ПЭМ), показаны на рис. 4. Метод термического разложения применим для получения НЧ различного химического состава: RF3 [63], ROF [52], NaRF4 [44], BaRF5 [64], KRF4 [65], LiRF4 [66] (где R = Y, Gd, Lu, La). Отметим, что, несмотря на перспективность, метод термического разложения требует проведения реакции в технологически сложных условиях: инертная атмосфера, продолжительное время, высокая температура реакции. Кроме того, при разложении металлсодержащих трифторацетатов образуются токсичные фтор- и оксофторсодержащие соединения [67].
Рис. 4.
Схематическое представление методики термического разложения: 1 – термометр, 2 – реакционная колба, 3 – смесь прекурсоров в среде высококипящих растворителей, 4 – нагревательный элемент, 5 – подвод инертного газа (а); ПЭМ-изображения сферических (б) и гексагональных (в) НЧ NaYF4:Yb/Er, полученных данным методом.

Метод соосаждения на начальном этапе включает в себя получение гомогенной смеси осадителя, в качестве которого могут выступать NaF [68], NH4F [69] или HF [70], и солей РЗЭ, таких как R(NO3)3 и RCl3, в водных растворах или в среде органических высококипящих растворителей (например, ОД, ОК). В случае водных растворов нуклеация и рост частиц протекают в присутствии стабилизирующих агентов (например, ди-N-октадецилдитиофосфат аммония, этилендиаминтетрауксусная кислота (ЭДТА), поливинилпирролидон (ПВП), полиэтиленгликоль (ПЭГ) и др.) при низких температурах с последующим осаждением при достижении НЧ критического размера и прокаливанием конечного продукта. В случае использования высококипящих растворителей применяют олеаты РЗЭ и проводят осаждение при температурах ~200–300°С.
Для успешного проведения реакции соосаждения необходимо, чтобы продукты реакции имели низкую растворимость для быстрого получения пересыщенного раствора, скорость зародышеобразования была выше скорости роста НЧ, а также чтобы вторичные процессы, такие как созревание и агрегация по Оствальду, были относительно медленными и не оказывали существенного влияния на размер и морфологию нанокристаллов. К настоящему времени методом соосаждения могут быть получены следующие НЧ: LaF3 [71], NaYF4 [68, 72], NaGdF4 [73], NaYbF4 [74], KGdF4 [75], NaxScF3 +x [76].
Метод соосаждения является простым и наиболее удобным подходом для синтеза АНЧ благодаря низким затратам на необходимое оборудование, мягким условиям синтеза, простым протоколам и быстропротекающим реакциям, отсутствию токсичных продуктов. Однако в данном процессе трудно контролировать морфологию НЧ и их распределение по размерам. Отметим, что при реакции в сложных многокомпонентных системах одновременно в осадок могут выпадать несколько видов НЧ, что приводит к дополнительным трудностям контроля структуры, морфологии и фазового состава.
В настоящее время активно разрабатывают подходы для управления свойствами нанокристаллов in situ. Например, исследование влияния соотношения стабилизатор/растворитель на размер и форму НЧ показало, что увеличение соотношения ОК:ОД от 3:17 до 3:7 инициирует фазовый переход от моноклинной фазы Na3ScF6 к гексагональной NaScF4 [76], а при ОК:ОД 6:15 отмечаются уменьшение диаметра сферических частиц и их трансформация в микростержни [72, 77]. Замена в реакционной смеси ОК на ОА при синтезе NaYbF4 приводит к уменьшению размера частиц и снижению температуры процесса за счет более эффективного покрытия молекулами ОА поверхности НЧ и уменьшению энергии фазового α–β-перехода в NaYbF4 [78].
Гидро/сольвотермальный метод основан на реакциях, протекающих в закрытых системах при высоких давлении и температуре [79]. При проведении синтеза смешивают прекурсоры, растворители и ПАВ, содержащие функциональные группы, а затем нагревают в специальном реакционном сосуде. Такие ПАВ, как полиэтиленимин, ЭДТА, ОК, цетилтриметиламмоний бромид, выступают в качестве хелатирующих агентов, регулирующих концентрацию катионов, что необходимо для контроля размера, морфологии и формирования кристаллической фазы [80]. В качестве прекурсоров могут выступать хлориды, нитриды или сульфаты РЗЭ. Управление такими параметрами, как температура и концентрация реагентов, позволяет поддерживать высокую скорость зародышеобразования и кристаллизации [81]. В основе гидро/сольвотермального синтеза монодисперсных НЧ лежит процесс массопереноса и выделения продукта на границе раздела фаз, продемонстрированный в [82].
Использование этого метода позволяет получать НЧ различного размера, формы и кристаллической фазы [83–85]. Например, этим методом были синтезированы нано-, микроразмерные гексагональные структуры NaRF4 различной морфологии (рис. 5). В качестве прекурсоров использовали NaF и R(NO3)3 при участии стабилизирующего агента ОК [83]. Изменяя рН реакционной среды, время реакции и температуру, а также концентрацию реагентов, удается контролируемо управлять кристаллической фазой, морфологией частиц и обеспечивать самоорганизацию НЧ в микрокристаллы со сложной архитектурой.
Рис. 5.
ПЭМ-изображения нанокристаллов NaYF4:R3+ различной морфологии, полученных гидротермальным методом в [83] при различных параметрах синтеза: соотношение Y3+/R3+ = 2.03:1, 180°C, 8 ч (а); Y3+/R3+ = 1:2.5, 180°C, 48 ч (б); Y3+/R3+ = 19:1, 195°C, 24 ч (в); Y3+/R3+ = 1:2.3, 195°C, 24 ч (г).

Преимуществами гидро/сольвотермального метода являются высокая реакционная способность реагентов, низкая концентрация образующихся токсичных продуктов, возможность реализации различных реакций в процессе синтеза, а также более низкие температуры реакции по сравнению с методом термического разложения [86]. К недостаткам метода относятся необходимость использования специальных реакционных сосудов и отсутствие возможности визуального наблюдения за процессом синтеза.
Микроэмульсионный метод представляет собой процесс, при котором две несмешивающиеся жидкости при участии амфифильных молекул образуют эмульсию, в микрокаплях которой происходит процесс нуклеации и синтеза НЧ. Для получения НЧ этим методом необходимы две микроэмульсии, одна из которых содержит ионы РЗЭ, а другая – ионы фтора, при смешивании которых инициируется реакция [87]. В микроэмульсионном методе возможно применение недорогого оборудования, а изменение концентрации ПАВ, дозировки растворителя и времени синтеза позволяет контролировать морфологию и размер НЧ, что проиллюстрировано на рис. 6 на примере частиц состава (H3O)Y3F10 · xH2O [88, 89].

Метод имеет большой потенциал для получения комплексных наноструктур из предварительно синтезированных НЧ [90–93]. Так, были получены наноструктуры, содержащие квантовые точки CdSe и NaYF4:Yb3+/Er3+ [92], и АНЧ, импрегнированные НЧ оксида железа, при участии амфифильного блок-сополимера стирола с аллиловым спиртом [93]. Эти мультифункциональные наноструктуры оказались перспективными для флуоресцентного анализа и мультимодальной визуализации. Несмотря на ряд преимуществ, метод имеет ограничения при масштабировании и сложности при выделении целевого конечного продукта.
“Золь-гель”-метод представляет собой синтез НЧ в процессе химической реакции соответствующих водных растворов прекурсоров в присутствии фторирующего агента при низкой (обычно комнатной) температуре с последующим переходом от раствора к коллоидному золю. Золь переходит в гель по мере своего “созревания”, во время которого происходит агрегация частиц, что приводит к образованию металлооксополимерных цепей, формирующих трехмерную структуру геля. Гель разрушается при термообработке с образованием НЧ. Наиболее распространенными прекурсорами для данного метода являются алкоксиды (алкоголяты) металлов. На первом этапе синтеза происходит гидролиз алкоксидов соответствующих металлов. За счет своей электродефицитности алкоксиды металлов имеют высокую реакционную способность к гидролизу. Большое влияние на протекание данной стадии оказывает рН среды, причем чем выше кислотность среды, тем легче происходит перенос протона и тем интенсивнее протекает гидролиз. Далее реализуется сополиконденсация гидролизованных молекул алкоголятов c образованием геля, при прокаливании которого в диапазоне температур 500–1000°С и формируются НЧ.
Этот метод впервые был применен для синтеза НЧ ZrO2, легированных ионами Еr3+ [94]. В [95] для синтеза LiGdF4:Eu3+ и LiYF4:Er3+ использовали алкоголяты соответствующих элементов, полученные взаимодействием безводных хлоридов с изопропиловым спиртом. В качестве фторирующего агента выступал 1,1,1-трифторо-5-метил-2,4-гексадион. Пиролиз геля проводили во фторирующей атмосфере при температуре 550°C. Аналогичная процедура описана в [96] при получении частиц NaYF4:Yb3+/Er3+ из прекурсоров на основе ацетатов в присутствии трифторуксусной кислоты. НЧ BaTiO3:Yb3+/Er3+ были синтезированы путем отжига геля при 800–1000°C, полученного из смеси Ti(OC4H9)4 и ацетатов соответствующих металлов в спиртовой среде [97, 98].
Данный метод синтеза НЧ имеет существенные недостатки. Большинство алкоксидов металлов, за исключением алкоксида кремния, токсичны и имеют высокую стоимость. Высокая реакционная способность алкоксидов затрудняет управление размерами НЧ, которые агрегируют при диспергировании в водной среде [99].
Существует альтернативный “золь-гель”-метод, известный как метод Печини или “полимеризующегося комплекса”, который включает в себя комбинированный процесс комплексообразования металлов и in situ-полимеризацию полученных органических веществ. В качестве прекурсоров выступают неорганические соли металлов (хлориды, нитраты, сульфаты). Для создания стабильных комплексов металлов обычно используют многоосновные органические кислоты, например лимонную кислоту, так как она образует очень стабильные хелатные комплексы с ионами различных металлов. При образовании полимерного геля протекает реакция полиэтерификации данных комплексов в присутствии спиртов (этанол, этиленгликоль, ПЭГ и т.д). В процессе этой реакции формируется жесткая органическая полимерная сетка, которая уменьшает сегрегацию отдельных ионов металлов, обеспечивая композиционную однородность. Отжиг геля при температурах 500–1000°С приводит к образованию чистой фазы многокомпонентных оксидов металлов.
Преимущества метода Печини заключаются в том, что путем изменения соотношения хелатирующий агент/спирт и температуры синтеза можно получать НЧ с узким распределением по размерам, а прекурсоры для синтеза имеют гораздо меньшую стоимость по сравнению с алкоксидами. Этот метод используют для синтеза НЧ Y2O3 [100], Y2Ti2O7 [101], YAlO3 [102] и др.
Для получения фторидных и оксофторидных НЧ “золь-гель”-методом в систему необходимо вводить фторирующие агенты. Так, в [103, 104] для получения YOF и GdOF в качестве источника фтора использовали NH4F, а для получения НЧ BaMgF4:Eu2+ использовали трифторуксусную кислоту [105].
Несмотря на то что при помощи “золь-гель”-процесса можно синтезировать НЧ массово с высокой степенью кристалличности, они имеют широкое распределение по размерам и нерегулярную морфологию, что является существенным ограничением данного метода [106].
Микроволновый синтез НЧ является новым и перспективным методом, поскольку позволяет сократить время химической реакции, увеличивает выход целевого продукта, минимизирует побочные реакции и повышает воспроизводимость. Данный метод схож с гидро/сольвотермальным с той разницей, что для нагрева реакционной смеси используют микроволновое излучение. Энергия кванта микроволнового диапазона в области частот от 0.3 до 300 ГГц слишком мала, чтобы инициировать разрыв химических связей, но при этом позволяет эффективно нагревать объем реакционной смеси по сравнению с внешними источниками тепла [107, 108].
В настоящее время микроволновый нагрев широко применяют в гидротермальном синтезе для получения фторидных НЧ [109–116]. В [111–113] представлены результаты применения микроволнового нагрева реакционной смеси для сольвотермального синтеза НЧ NaYF4 и LaF3 в среде этиленгликоля при температурах 160–200°С. В качестве прекурсоров использовали ацетаты и нитраты соответствующих РЗЭ для синтеза НЧ со средним размером ~10 нм [112]. Сравнительной анализ результатов, полученных при резистивном и микроволновом нагреве, показал, что микроволновый нагрев значительно увеличивает скорость процесса синтеза за счет более эффективного прогревания реакционной смеси [114]. Одним из существенных недостатков микроволнового синтеза является высокая стоимость оборудования.
5.2. Синтез наночастиц из расплавов
Получение НЧ из расплавов солей (из-под флюса). Флюс – высокотемпературный растворитель, используемый при выращивании кристаллов из раствора в расплаве, применяют для материалов с инконгруэнтным характером плавления [117]. Рост нанокристаллов этим методом происходит при температуре значительно ниже температуры плавления растворенного вещества. При синтезе фторидных НЧ происходит взаимодействие ионов металлов с фторирующем агентом, который необходимо вводить в исходный флюс до его расплава. Наиболее подходящим материалом для флюса является NaF за счет высокой растворимости в воде, относительно низкой температуры плавления по сравнению с остальными фторидами и устойчивости к гидролизу на воздухе. В [118] НЧ NaYF4:Yb/Er синтезировали из порошков NaF и YF3 в флюсовом расплаве NaF при температуре 1100°С в течение 10 ч. В результате были получены частицы размером ~600 нм. Недостатком использования флюса NaF являются его частичная растворимость в NaYF4 и образование нецелевых фторидов [119].
Альтернативой NaF являются флюсы на основе водорастворимых солей хлоридов, нитратов и сульфатов. Они обладают более низкой температурой плавления по сравнению с фторидами, образуя меньшее количество побочных продуктов. В качестве фторирующего агента используют NaF, NH4F или NH4НF2. В [120, 121] для синтеза кристаллов β-NaYF4:Yb3+, Er3+/Tm3+ в качестве флюса использовали эквимолярную смесь NaCl:KCl, а в качестве фторирующего агента выступал NaF. Для создания нитратных флюсов применяют соли NH4NO3, KNO3, NaNO3, LiNO3 или их смеси в различных молярных соотношениях [122–124]. В [125] описано получение частиц SrF2 и CaF2 из сульфатных флюсов. Синтез проводился в расплавах на основе Li2SO4–Na2SO4–Sr(Ca)F2 в определенных мольных соотношениях при температуре 700°С. Метод получения НЧ из-под флюса ограничен размером синтезируемых частиц, как правило, в субмикронном и микронном диапазоне.
Синтез с использованием ионных жидкостей. Ионными жидкостями (ИЖ) называется класс органических солей, температура плавления которых ниже температуры кипения воды, а их расплавы находятся в жидком состоянии в широком температурном интервале (300–400°С). Эти соединения состоят из большого асимметричного органического катиона и органического или неорганического аниона, благодаря своей структуре они могут быть использованы как растворители для органических и неорганических веществ. В качестве катиона обычно выступают производные имидазола, пиридина или тетраалкиламмония, а аниона – Cl–, Br–, N(FSO2)$_{2}^{ - }$, C(CF3SO2)$_{3}^{ - }$, BF$_{4}^{ - }$, PF$_{6}^{ - }$, CuCl$_{2}^{ - }$, SnCl$_{3}^{ - }$ и т.д. Выбор аниона определяет гидрофильно-липофильный баланс ИЖ [126].
В процессе синтеза фторидных НЧ ИЖ могут выступать в роли растворителя, матрицы, реагента или ПАВ. Обычно ИЖ используют в качестве среды, в которой ионы фтора взаимодействуют с ионами металлов. Для повышения растворимости реагентов используют бинарные смеси ИЖ со спиртами (этанол, метанол). Однако и сами ИЖ могут служить источником фтора, но только в присутствии воды, обеспечивающей протекание гидролиза. Синтез наночастиц в ИЖ обычно проходит в двухфазной системе, в которой химические реакции образования нерастворимой фторидной фазы протекают на границе раздела фаз. Примером такой системы является этиленгликоль/гексафторфосфат 1-октил-3-метилимидазолия ([C8mim][PF6]) или тетрафторборат 1-бутил-3-метилимидазолия [bmim][BF4]. В процессе получения фторидных НЧ прекурсоры, содержащие ионы РЗЭ, растворяются в полярном растворителе (вода, этиловый спирт, этиленгликоль) и смешиваются с ИЖ. В роли фторирующего агента могут выступать NaF, NH4F и NH4HF2. После тщательного перемешивания реакционную смесь нагревают до требуемой температуры и проводят реакцию в течение длительного (от 12 до 24 ч) времени [127–129]. При использовании микроволнового излучения температура кипения ИЖ достигается в течение очень короткого времени [130].
Механизм образования НЧ в двухфазной системе вода/[bmim][BF4] рассмотрен в [131]. Источником фторид-анионов служили анионы BF$_{4}^{ - }$ ИЖ, которые при нагревании подвергаются быстрому гидролизу в воде с образованием ионов F–. На границе раздела фаз вода/ИЖ происходит взаимодействие фторид-иона с катионами R3+ и формирование фторидных НЧ. В [132] представлен синтез НЧ NaYF4:Yb3+, Er3+/Tm3+ из смеси нитратов РЗЭ и NaCl, растворенных в воде, в присутствии [bmim][BF4] с образованием гексагональных частиц размером до 70 нм.
Важно отметить, что синтез в ИЖ позволяет получать НЧ не только с гидрофобными, но и гидрофильными свойствами поверхности [133]. Недостатком данного метода синтеза является частичный гидролиз как целевого продукта, так и самой ИЖ. Примером может служить образование оксофторида Er4O3F6 при синтезе частиц KErF4 [134].
5.3. Синтез с использованием подхода “сверху-вниз”
Метод лазерной абляции объемных материалов в жидкостях, включая лазерную фрагментацию, представляет уникальные возможности для генерации НЧ [135]. Кроме того, этот бесконтактный одностадийный метод может эффективно использоваться для модификации поверхностей НЧ и, например, их биофункционализации [136]. Этот подход применим для синтеза и управления формой НЧ металлов [137], полупроводников [138, 139], диэлектриков [140, 141] и органических материалов [142].
На сегодня существуют несколько работ, в которых сообщается об абляции объемной мишени в жидкостях короткими лазерными импульсами, приводящей к генерации АНЧ. С использованием этого подхода были получены АНЧ Y2O3:Yb3+/Er3+ и NaYF4:Yb3+/Er3+ [143–145]. Несмотря на демонстрацию эффекта апконверсии в целевом продукте, НЧ обладали несовершенной кристалличностью и полидисперсностью по размерам от 10 до 200 нм. В [146] удалось подобрать условия для получения НЧ YOF с относительно узкой дисперсией по размеру, а в [140] в результате воздействия на НЧ лазерного излучения пикосекундной длительности гексагональные НЧ были перекристаллизованы в одномерные апконвертирующие стержни. Несмотря на прорывные достижения этого подхода при синтезе НЧ широкого класса материалов, метод лазерной абляции пока не находит широкого распространения для генерации фторидных НЧ. Вероятно, это связано с высокой стоимостью оборудования, а также технологическими сложностями в контроле кристалличности, морфологии и размеров частиц.
Механическое измельчение объемного материала позволяет получать НЧ с использованием высокоэнергетических шаровых мельниц планетарного типа. При использовании этого подхода происходит сверхтонкое измельчение микропорошка до наноразмерного состояния за счет комбинации ударного воздействия и трения [147].
В [148] описана возможность получения НЧ FeF3 и GaF3 размером ~15 нм с помощью механического помола в инертной атмосфере. В [149] использовали “сухой” механический помол для получения НЧ La0.95Ba0.05F2.95 из исходного поликристаллического образца, полученного твердофазным синтезом. Размер частиц после помола в течение 3 ч составил порядка 20 нм. Частицы NaYF4:Yb3+/Er3+ размером ~120 нм были синтезированы при помоле микроразмерных частиц, получаемых методом соосаждения [150]. При помоле микронных частиц, полученных гидротермальным методом, формировались НЧ NaYF4:Yb3+/Er3+ размером в диапазоне 100–260 нм [151]. В [152, 153] продемонстрировали синтез НЧ BaY2F8 в диапазоне размеров от 7.5 до 56 нм из объемных монокристаллов, выращенных методом Чохральского.
Получение НЧ механическим дроблением объемных материалов является экологически чистым подходом, не требующим использования агрессивных растворителей. Однако этот метод достаточно сложный из-за длительности процесса и его большой энергоемкости, кроме того, возможно загрязнение конечного продукта компонентами размольной гарнитуры [147].
Механохимический синтез основан на механической активации твердофазных химических реакций, протекающих, как правило, в шаровых мельницах. В процессе помола столкновение мелющих тел характеризуется высокими значениями энергии на единицу площади. Частицам порошка, которые попадают между шарами при их столкновении, передается эта энергия, что приводит к локальному сильному разогреву и спеканию компонентов смеси, за счет чего происходит так называемое деформационное смешивание или механосплавление. Этот процесс протекает при относительно низких температурах, когда все релаксационные процессы заторможены и формирование совершенной кристаллической структуры затруднено, что позволяет синтезировать различные метастабильные промежуточные продукты, в том числе в наносостоянии.
В [149, 154] описано получение НЧ состава Ba1 –xLaxF2 +x путем помола смеси фторидов бария и лантана. НЧ M1 –xEuxF2 +x (M = Ca, Sr, Ba, Pb) получали помолом ацетатов, гидроксидов или карбонатов соответствующих металлов в присутствии NH4F [155]. Получение частиц Ca1 –xLaxF2 +x (x > 0.1) и La1 –yCayFy– 3 (y = 0.15, 0.2) со средним размером 10–30 нм из монокристаллических компонентов CaF2 и LaF3 продемонстрировано в [156, 157]. Отметим, что в [157] впервые наблюдался “механогидролиз” фазы La1 –yCayFy– 3 с образованием оксофторидных соединений. Метод механосплавления использовали для получения НЧ BaFCl:Sm3+, NaYF4:Gd3+/Yb3+/Tm3+, SrFCl:Yb3+/Er3+ NaYF4:Yb3+/Tm3+ [158–161]. Эксперименты проводили в инертной атмосфере, а в качестве прекурсоров использовали стехиометрические количества хлоридов, фторидов или нитратов соответствующих металлов. Время помола варьировалось от 2 до 6 ч. Размеры полученных НЧ находились в диапазоне от 30 до 200 нм.
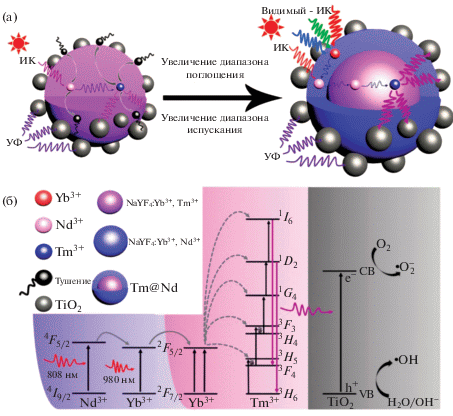
6. ГИБРИДНЫЕ НАНОКОНСТРУКЦИИ
Несмотря на многообещающие достижения в области синтеза АНЧ, одним из существенных ограничений, сдерживающих их применение, остается относительно низкая интенсивность антистоксовой ФЛ, которую принято характеризовать коэффициентом конверсии (КК), т.е. отношением поглощенной к излученной мощности. В настоящий момент разрабатываются различные способы решения этой проблемы, в частности путем создания структур типа “ядро/инертная оболочка” или “ядро/активная оболочка”, оптимизацией концентрации ионов РЗЭ, подбором кристаллической матрицы [162], введением ионов-сенсибилизаторов, например ионов Nd3+, позволяющих изменить длину волны возбуждения НЧ с 980 на 800 нм [163] и др. Однако для специалистов в данной области очевидно, что эти подходы достигли своего предела. Так, максимально достигнутый КК составляет 10–12% для частиц размером ~100–150 нм [164], в то время как КК для частиц размером менее 50 нм, как правило, не превышает 1–3%. При этом важно понимать, что поскольку процесс апконверсии является нелинейным, указанные значения КК достигаются в режиме насыщения, т.е. при интенсивностях выше 30–50 Вт/cм2, в то время как при интенсивностях возбуждающего излучения до 1 Вт/cм2 КК ~ 0.01–0.5%.
Одним из подходов увеличения эффективности фотолюминесценции АНЧ является использование органических красителей, обладающих большим сечением поглощения и высоким квантовым выходом. Органические красители могут усилить ФЛ наночастиц со структурой “ядро/оболочка” в 20–100 раз для частиц размером 30–50 нм и ~3300 раз для частиц размером 20 нм даже без инертной оболочки [165]. Наряду с описанным ранее механизмом передачи энергии между активными ионами (4f–4f-электронные взаимодействия) возбуждение ИК-светом органического красителя, сорбированного на поверхности АНЧ, открывает дополнительный канал для безызлучательной передачи энергии за счет взаимодействия между синглетными или триплетными состояниями органических красителей и 4f-электронными состояниями ионов РЗЭ в НЧ. Безызлучательная передача энергии от красителей к ионам РЗЭ и между активными ионами (например, между сенсибилизатором и активатором) в упрощенном виде может рассматриваться как передача энергии от донора к акцептору по механизму Ферстера, т.е. с использованием диполь-дипольного механизма переноса энергии, в котором донор и акцептор могут быть представлены в виде точечных диполей [21].
Под действием ИК-света краситель переходит в возбужденное синглетное электронное состояние, которое сопровождается рядом процессов девозбуждения. Процесс переноса энергии включает в себя релаксацию молекул красителя в основное состояние при возбуждении ионов РЗЭ. Кроме того, краситель может перейти в триплетное состояние, в этом случае безызлучательная передача энергии осуществляется от красителя к иону РЗЭ посредством спин-запрещенного электронного переход красителя и одновременного возбуждения активного иона. Для усиления эффективности фотолюминесценции АНЧ в качестве органических красителей наиболее широко применяются молекулы, возбуждаемые ИК-светом, с пиками поглощения в окрестности 780–810 нм и длиной волны излучения в диапазоне от 750 до 1150 нм, такие как IR 783, IR 806, IR 808, IR 820, IR 845, ICG и Cyto 840 [166].
Локальное усиление электромагнитного поля вблизи НЧ благородных металлов и связанное с ним усиление процессов излучения и поглощения света в пленках или наноструктурах активно используются в устройствах нанофотоники и плазмоники [167, 168], а также в наносенсорах [169]. Используя структуру спектров плазмонных НЧ, можно усиливать как поглощение, так и испускание света и таким образом создавать эффективные наноразмерные источники света [170] и даже нанолазеры, так называемые спазеры [168, 171]. Эффективность процесса апконверсии в НЧ может быть увеличена за счет создания гибридных структур с плазмонными агентами. Плазмонный резонанс может влиять на три различных физических процесса в апконвертирующих материалах: поглощение света сенсибилизатором, передачу энергии от сенсибилизатора к активатору и эмиссию света активатором [172]. Ранее было показано, что плазмонные гибридные структуры на основе АНЧ демонстрируют усиление ФЛ в ~5−310 раз в зависимости от экспериментальных условий и геометрии создаваемых структур. Так, в [173] было продемонстрировано усиление интенсивности флуоресценции в структурах АНЧ/Au в 22.5 раза при длине спэйсера между слоем золотых НЧ и апконвертирующей пленкой в 8 нм. При изменении толщины неактивной пленки на несколько нанометров эффективность конверсии ИК-излучения в ФЛ видимого диапазона значительно снижалась.
Следовательно, органические красители и плазмонные НЧ имеют большой потенциал для разработки перспективных подходов увеличения эффективности апконверсии путем повышения эффективности “аккумуляции” энергии возбуждения и эмиссии света активными ионами. Однако, несмотря на впечатляющие достижения последних лет, и сам процесс апконверсии, и процесс миграции энергии в гибридных наноконструкциях остаются малоизученными. Главным образом это связано со сложностью синтеза как самих АНЧ, так и функционализацией их поверхности для конъюгации с люминесцентными красителями и плазмонными НЧ. Разброс НЧ по размерам, тушащие дефекты кристаллической структуры и коллективные эффекты, плохо контролируемая концентрация красителей на поверхности и т.д. существенно затрудняют анализ кинетики ФЛ гибридных наноструктур и, как следствие, препятствуют пониманию их фотофизики и внедрению в практику.
7. ПЕРСПЕКТИВНЫЕ НАПРАВЛЕНИЯ ПРИМЕНЕНИЯ
7.1. Биотехнологии и медицина
Успешная интеграция диагностики и терапии в одном агенте с использованием многофункциональных НЧ привела к созданию нового междисциплинарного исследовательского поля под названием “тераностика”, которое является основой инновационных стратегий, позволяющих персонализировать медицину для диагностики и лечения пациентов [174]. В последнее время АНЧ считаются перспективными кандидатами для решения задач тераностики. Они открывают возможности для инструментальных методик оценки in vivo и in vitro накопления в целевых клетках, для их минимально инвазивной визуализации in vivo с целью прогнозирования и мониторинга терапевтического результата в реальном времени [175].
Длина волны возбуждения (~980 нм) и испускаемая АНЧ люминесценция попадают в так называемое “окно прозрачности” биологической ткани (диапазон спектра от 650 до 1300 нм), где проникновение света в ткань происходит с минимальным поглощением и рассеянием. Свет ИК-диапазона может проникать в биоткани глубже, чем УФ- и видимый свет, обеспечивая оптическую визуализацию с большей глубины. Явление апконверсии приводит к уникальным оптическим свойствам НЧ, включая минимальное фотовоздействие на живые организмы и высокое отношение сигнала к шуму при регистрации ФЛ [176] по сравнению с обычными флуоресцентными метками. Кроме того, этот класс НЧ обладает низкой цитотоксичностью и химической инертностью к окружающей среде. Фотолюминесценция АНЧ очень стабильна, они не подвержены фотовыцветанию и фотомерцанию, что позволяет проводить непрерывный мониторинг при визуализации [177].
В современной медицине анализ заболеваний на ранней стадии является определяющей технологией. Несмотря на преимущества АНЧ, невозможно добиться требуемой эффективности диагностики из-за ограничений оптической биовизуализации. Однако неорганическая платформа АНЧ позволяет расширить модальности их применения в визуализации in vivo для интеграции в существующую медицинскую практику, а именно в магнитно-резонансную, рентгеновскую компьютерную, позитронно-эмиссионную томографии [178]. На основе АНЧ были разработаны мультимодальные НЧ с комбинированными функциями визуализации, объединенными в единую наноконструкцию [179].
Метод фотодинамической терапии (ФДТ) является неинвазивной медицинской технологией, включающей в себя три ключевые компоненты: фотосенсибилизатор, свет и кислород, растворенный в биоткани [180]. При возбуждении светом подходящей длины волны молекулы фотосенсибилизатора способны передать энергию поглощенного кванта света окружающим молекулам кислорода, генерируя цитотоксичные активные формы кислорода (АФК), что приводит к гибели клеток. Однако большинство фотосенсибилизаторов возбуждается под действием видимого или УФ-излучения, что существенно ограничивает глубину проникновения света из-за его поглощения и рассеяния биологическими тканями, приводя к слабому терапевтическому эффекту в случае глубоко расположенных опухолей. Оптические свойства АНЧ позволяют реализовать инновационный метод глубокой ФДТ. Апконвертирующие НЧ обладают способностью конвертировать ближнее ИК-излучение в фотоны видимого и УФ-спектрального диапазона, которые, в свою очередь, переводят в возбужденное состояние фотосенсибилизатор, сорбированный на НЧ, через процесс резонансного безызлучательного переноса энергии. Так, в ряде работ была продемонстрирована ИК-ФДТ с использованием наноконструкций: NaYF4:Yb, Er/Хлорин e6 [181], LiYF4:Yb, Er/β-карбоксифталоцианин цинка [182] и NaYbF4:Nd@NaGdF4:Yb, Er@NaGdF4/Хлорин e6 [183]. Кроме того, был создан нанокомплекс на основе нетоксичного эндогенного фотосенсибилизатора – рибофлавина (рис. 7) [184] и продемонстрирована собственная УФ-фототоксичность АНЧ в отношении опухолевых клеток [185].
Рис. 7.
Фотография иммунодефицитной мыши с подкожно трансплантированной опухолью SK-BR-3 через 15 дней после имплантации. Область опухоли отмечена прямоугольником. На изображениях продемонстрировано преимущество визуализации с использованием АНЧ по сравнению с рибофлавином (контраст 30 и 3 соответственно) (а). График роста опухоли SK-BR-3, показывающий стабильное увеличение объема контрольных опухолей (необлученных) и регрессию опухоли после ФДТ (черная стрелка, день 15) с использованием нанокомплекса АНЧ–рибофлавин (б). Серия фотографий опухоли SK-BR-3, сделанных до ФДТ на длине волны 975 нм, через 25 и 50 дней после ФДТ (снизу) и серия фотографий соответствующих контролей (сверху) (в). Гистологические срезы тканей опухоли, окрашенных гематоксилином и эозином, полученных через 1 день после ФДТ. При использовании нанокомплексов АНЧ–рибофлавин наблюдаются характерные для ФДТ повреждения (геморрагии), тогда как контроль не обнаруживает никаких отклонений (г) [184].

Для более детального ознакомления с возможными биомедицинскими применениями АНЧ можем порекомендовать читателям обзор [186].
7.2. Наносенсоры
Люминесцирующие НЧ являются удобной платформой для создания наносенсоров и разработки новых методов диагностики. Ранее сообщалось о создании и применении оптических сенсоров температуры на основе НЧ с возможностью измерения температуры в нанообъеме. Например, была продемонстрирована температурная зависимость линий ФЛ ионов Er3+ [187–190], входящих в состав АНЧ. В работах было показано, что зависимость соотношения интенсивности линий ФЛ ионов Er3+ на длинах волн 520 и 550 нм не зависит от изменения интенсивности возбуждающего лазерного излучения, что делает АНЧ идеальными люминесцентными сенсорами для измерения температуры. Впервые применение “нанотермометра” для измерения температуры в живой клетке было продемонстрировано в [191]. Для решения задач термометрии использовались гидрофильные НЧ NaYF4:Er3+/Yb3+ с полимерным покрытием. После инкубации НЧ с HeLa-клетками нанотермометр был использован для измерения температуры живой клетки от 25°С до ее термически индуцированной гибели при 45°С. В [192] было показано, что применение НЧ со структурой “ядро/оболочка” позволило контролировать температуру с точностью до 0.5°C в физиологическом диапазоне температур. В [193] изучена зависимость спектров флуоресценции АНЧ Y2O3: Tm3+, Yb3+ в температурном диапазоне от комнатной до 480°С и показана их температурная чувствительность.
Наноконструкция типа “нанофосфор/плазмонная НЧ” использовалась для высокочувствительного детектирования за счет специфичного связывания, например, олигонуклеотидов [194] и иммуноглобулинов [195].
Перспективность применения нанокомплекса, в котором реализуется ферстеровский обмен энергией между НЧ NaYF4:Yb3+/Er3+/Tm3+ и органической Ru-содержащей молекулой для детектирования ионов Hg2+ в сверхмалых концентрациях, продемонстрирована в [196].
7.3. Солнечные элементы
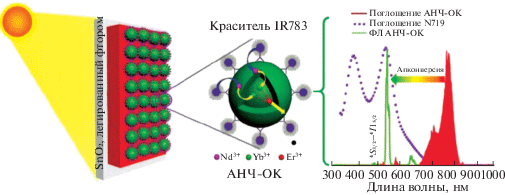
Процесс апконверсии позволяет обойти ограничения, связанные с пропусканием света в полупроводниках. Эта технология позволяет преодолеть предел Шокли–Кейссера для фотоэлементов с одним p–n-переходом путем преобразования солнечного спектра [197]. В [198] показано, что модификация солнечного спектра с помощью преобразователя, повышающего частоту падающего света, может поднять верхний теоретический предел эффективности фотоэлемента из кристаллического кремния с одним p–n-переходом до 40.2% при неконцентрированном облучении солнечным светом. Это значение превышает предел Шокли–Кейссера для кристаллических кремниевых солнечных элементов с шириной запрещенной зоны ~1.1 эВ, составляющий приблизительно 30%. Было предпринято несколько попыток использовать фторидные нанокристаллы, легированные ионами РЗЭ, для увеличения эффективности преобразования солнечного света. АНЧ размещались в качестве заднего отражающего слоя [199], использовались нанокомпозиты “АНЧ–TiO2” [200, 201] или апконвертирующие слои размещались внутри солнечных элементов [202, 203]. Отметим, что эти работы продемонстрировали убедительный отклик на ближнюю ИК-компоненту света, хотя увеличение эффективности таких солнечных элементов остается ограниченным (менее 1%). Главным образом это связано с тем, что используемые апконвертирующие материалы способны эффективно преобразовывать свет ближнего ИК-диапазона с узкой шириной поглощения (10–20 нм) в окрестности 980 нм в случае применения в качестве сенсибилизатора ионов Yb3+ и 800 нм для ионов Nd3+. Однако недавно была представлена прорывная работа [204], в которой добились повышения эффективности преобразования на 13% при использовании гибридных наноструктур типа “АНЧ–органический краситель”, поскольку такие структуры расширяют диапазон поглощаемого света и значительно увеличивают эффективное сечение поглощения наноструктуры (рис. 8).
Рис. 8.
Солнечная ячейка, модифицированная слоем мезопористого оксида титана, сенсибилизированного красителем N719, и гибридными наноструктурами АНЧ–органический краситель IR783 (ОК). Модификация солнечной ячейки АНЧ–ОК расширяет спектральный диапазон поглощения, что способствует эффективному переносу энергии на краситель N719 [204].
7.4. ИК-индуцируемая фотополимеризация
Лишь в нескольких пионерских работах демонстрировалась фотополимеризация, осуществляемая через процесс апконверсии. ИК-свет, преобразованный в зеленую ФЛ наночастиц NaYF4: Yb3+, Er3+, был успешно использован для индуцирования роста тонкой полимерной оболочки (несколько нанометров) на поверхности АНЧ для их гидрофилизации [205] и загрузки лекарственных препаратов [206]. Апконвертирующие микрочастицы были использованы для индуцированного ИК-светом отверждения фотокомпозиций (ФК) [207, 208]. В [209] продемонстрирована “глубокая” фотополимеризация на глубину до 10 см в стеклянной трубке при вертикальном экспонировании лазером на длине волны 980 нм. В [164] впервые в мире были показаны перспективы данной технологии для быстрого трехмерного прототипирования объектов заданной конфигурации (рис. 9а–9д). Уникальные люминесцентные свойства АНЧ позволили продемонстрировать процесс фотополимеризации с использованием стандартных ФК, индуцированный ближним ИК-светом, как на макро-, так и на микромасштабе. Дальнейшее исследование процессов полимеризации в нанокомпозитных материалах показало возможность формирования упорядоченных ансамблей из АНЧ в первоначально гомогенной реакционной смеси (рис. 9е, 9ж) [210]. ИК-индуцируемая фотополимеризация при использовании АНЧ имеет большие перспективы для разработки новых функциональных полимерных композитов и высокоскоростного создания трехмерных полимерных структур со сложной архитектурой, а также как универсальный инструмент для применения в промышленности и биомедицине.
Рис. 9.
Экспериментальная установка для изготовления трехмерных полимерных структур в ФК, содержащей АНЧ, под воздействием ИК-излучения (а) и ее схема (б); структура, полученная с помощью ИК-инициируемой трехмерной фотополимеризации (вид сверху) (в); трехмерная структура, полученная с помощью ИК-инициируемой фотополимеризации (г), и ее антистоксовая ФЛ при возбуждении 975 нм (д) [164]. Изображение трехмерной полимерной микроструктуры, полученной с помощью ИК-инициируемой фотополимеризации (е); Электронно-микроскопическое изображение упорядоченных структур наночастиц, образованных в ФК в процессе ИК-фотополимеризации: сферы и сверхрешетки (ж) [210].

7.5. Фотокатализ
Фотокатализ вызывает все больший интерес благодаря его потенциальному применению в решении энергетических и экологических проблем, например для разложения органических загрязнителей с использованием солнечной энергии или в водородной энергетике [211–213]. Фотокаталитическая активность зависит от способности катализатора генерировать электронно-дырочные пары, что приводит к образованию АФК. При взаимодействии с загрязнителями эти реакционноспособные радикалы могут эффективно окислять загрязняющие вещества и разрушать их до CO2 и H2O [214]. К сожалению, большинство фотокатализаторов может работать только под действием УФ-излучения, на долю которого приходится менее 5% солнечного спектра [215]. Для расширения диапазона поглощения света были разработаны многочисленные стратегии, среди которых особое место занимают подходы с использованием рассматриваемых в обзоре НЧ [215, 216].
Оксид титана (TiO2) является наиболее часто используемым фотокатализатором, что связано с его стоимостью, биосовместимостью, фото- и химической стабильностью [217–219]. Но его применение ограничено узким спектром поглощения и шириной запрещенной зоны в 3.2 эВ, что не позволяет использовать для его активации излучение видимого и ИК-диапазона [220]. Комбинация TiO2 и АНЧ позволяет расширить спектральный диапазон активации этого фотокатализатора (рис. 10). Первый фотокатализатор на основе комбинации НЧ CdF2/BaF2/Er2O3 с TiO2 был разработан в 2005 г. [221] и активировался излучением видимого диапазона. В дальнейшем применение фотокатализатора на основе частиц YF3: Yb3+/Tm3+, покрытых TiO2, позволило использовать свет ИК-диапазона для фотокатализа органических соединений [222]. В [223] представлен эффективный подход к очисткe воды от микотоксина дезоксиниваленола при использовании частиц NaYF4: Yb3+, Tm3+/TiO2 под действием ИК-облучения без образования промежуточных токсических продуктов.
Рис. 10.
Схематическая демонстрация увеличения фотокаталитической активности при переходе от NaYF4:Yb3+/Tm3+@TiO2 к наноконструкции NaYF4:Yb3+/Tm3+@NaYF4:Yb3+/Nd3+@TiO2 (а) и энергетическая диаграмма, описывающая механизм генерации АФК при возбуждении излучением 808 и 980 нм посредством передачи энергии от ионов Tm3+ к частицам TiO2 (б) [215].
Расщепление воды с использованием солнечной энергии – многообещающий подход для возобновляемого производства водорода [224]. Фотоэлектрохимическое расщепление воды с использованием наностержней ZnO, покрытых квантовыми точками CdTe и АНЧ NaYF4: Yb3+/Er3+, применялось для синтеза водорода при активации светом ближнего ИК-диапазона [225]. Использование трехмерного композитного фотоанода на основе АНЧ, нанозолота и TiO2 в качестве одного из трех электродов в фотоэлектрокаталитической системе, работающей под действием солнечного света, позволило увеличить выход водорода в 2 раза [224].
7.6. Антиконтрафактные технологии
Антиконтрафактные технологии находят широкое применение в областях защиты денежных знаков, бланков ценных бумаг, банковских карт и важных документов. Для практического внедрения таких технологий важно выполнение ряда условий: высокая степень защиты, относительная дешевизна, биобезопасность и простота нанесения. Таким требованиям удовлетворяет технология защиты, основанная на нанесении люминесцирующих материалов. Для создания антиконтрафактных меток применяются органические и неорганические люминесцентные материалы, обладающие стоксовой ФЛ [226], как правило, переизлучающие УФ-возбуждение в видимую область спектра. Однако материалы, обладающие широкополосной стоксовой ФЛ, не обеспечивают должного уровня защиты вследствие их доступности. В последние несколько лет для целей антиконтрафактной защиты были предложены НЧ, легированные ионами РЗЭ [227, 228]. Такие НЧ обладают набором узких линий ФЛ, что позволяет значительно повысить степень защиты и создать неподверженные фотовыцветанию метки, которые обнаруживаются только с применением специального оборудования. Перспективными для повышения уровня защищенности наносимых меток являются АНЧ [229, 230]. Одним из очевидных преимуществ использования АНЧ является высокий контраст их детектирования при нанесении на флуоресцирующие под действием УФ поверхности, например бумагу. Вариация оптических свойств АНЧ дает возможность для большого числа комбинаций кодирования. Двумерные изображения могут дополняться рядом модальностей. Например, линиями ФЛ, которые определяются составом НЧ и интенсивностью возбуждения ИК-излучения [231], распределением интенсивности в спектрах антистоксовой ФЛ [232], перестраиваемым временем жизни возбужденных состояний [233]. Высокий уровень защищенности обеспечивается сложностью технологий изготовления НЧ и визуализации наносимых меток. В [234] впервые в мире были продемонстрированы скрытое нанесение и считывание масштабного изображения. Для печати были разработаны чернила, подходящие для стандартных печатающих устройств. В их основе использованы НЧ со структурой “ядро/оболочка” NaYF4:Yb3+, Tm3+/NaYF4, модифицированные амфифильными полимерами. Созданные чернила нетоксичны и биологически безопасны, а скрытые изображения, напечатанные на бумаге (рис. 11), невидимы в условиях естественного освещения, но визуализируются при подсветке лазерным излучением с длиной волны 975 нм.
Рис. 11.
Графическое изображение (а) и QR-код (б), напечатанные АНЧ и визуализируемые при облучении ИК-лазером, и аналогичный QR-код (в), напечатанный с использованием обычных чернил [234].

ЗАКЛЮЧЕНИЕ
Проведенный анализ показывает, что апконвертирующие нанокристаллы выделились в отдельное междисциплинарное научное поле и рассматриваются как отдельный класс функциональных материалов нового поколения, обеспечивающих единую наноплатформу для решения задач в различных областях науки и техники. Благодаря применению АНЧ достигнуты впечатляющие результаты в биотехнологиях и медицине, солнечной энергетике, антиконтрафактных технологиях, 3D-печати, оптической микроскопии сверхвысокого разрешения и т.д. Однако, несмотря на разработанные стратегии синтеза для получения АНЧ высокого качества с контролируемым составом, формой и размером, а также с требуемой кристаллической фазой, очевидно, что существующие методы их получения далеки от совершенства в силу сложности и ресурсоемкости. Более того, контроль размера, формы, поверхностных свойств АНЧ, а также разработка воспроизводимого метода синтеза нанокристаллов остаются ключевыми проблемами на пути их широкого применения в науке и технике. Несмотря на значительный прогресс в теоретических аспектах, процессы, лежащие в основе явления апконверсии, остаются малоизученными. Оптимизация фотофизических параметров АНЧ с целью повышения их квантового выхода остается важной задачей, решение которой откроет новые возможности для их применения.
Поэтому поиск и разработка новых экономически целесообразных подходов к синтезу и развитие теоретических моделей, описывающих процесс апконверсии, остаются актуальными на пути к получению высококачественных нанокристаллов. Именно междисциплинарный комплексный подход к изучению явления апконверсии, направляемый конкретными приложениями, в полной мере реализуется в Российской Федерации консорциумом на базе ФНИЦ “Кристаллография и фотоника” РАН, ИБХ РАН и НИЦ “Курчатовский институт”, обладающим компетенциями в области полного цикла, включающего в себя синтез, модификацию нанокристаллов, исследование их свойств и инженерию на апконвертирующей наноплатформе.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (проект № 19-02-00877 в части анализа методов синтеза наночастиц, проект № 18-29-20064 в части анализа методов синтеза и свойств гибридных наноструктур) и Министерства науки и высшего образования в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН в части анализа фотофизики процесса апконверсии.
Список литературы
Bloembergen N. // Phys. Rev. Lett. 1959. V. 2. № 3. P. 84. https://doi.org/10.1103/PhysRevLett.2.84
Овсянкин В.В., Феофилов П.П. // Письма в ЖЭТФ. 1966. Т. 3. С. 494.
Auzel F. // Compt. Rend. Acad. Sci. Paris B. 1966. V. 263. P. 819.
Каминский А.А., Антипенко Б.М. Многоуровневые функциональные схемы кристаллических лазеров. М.: Наука, 1989. 269 с.
Heer S., Kömpe K., Güdel H.-U., Haase M. // Adv. Mater. 2004. V. 16. № 23–24. P. 2102. https://doi.org/10.1002/adma.200400772
Yi G., Lu H., Zhao S. et al. // Nano Lett. 2004. V. 4. № 11. P. 2191. https://doi.org/10.1021/nl048680h
Wang F., Deng R., Wang J. et al. // Nat. Mater. 2011. V. 10. № 12. P. 968. https://doi.org/10.1038/nmat3149
Förster T. // Ann. Phys. 1948. V. 437. № 1–2. P. 55. https://doi.org/10.1002/andp.19484370105
Dexter D.L. // J. Chem. Phys. 1953. V. 21. № 5. P. 836. https://doi.org/10.1063/1.1699044
Nadort A., Zhao J., Goldys E.M. // Nanoscale. 2016. V. 8. № 27. P. 13099. https://doi.org/10.1039/C5NR08477F
Villanueva-Delgado P., Krämer K.W., Valiente R. // J. Phys. Chem. C. 2015. V. 119. № 41. P. 23648. https://doi.org/10.1021/acs.jpcc.5b06770
Wright J.C. // Top. Appl. Phys. 1976. V. 15. P. 239. https://doi.org/10.1007/BFb0111143
Auzel F. // Phys. Rev. B. 1976. V. 13. № 7. P. 2809. https://doi.org/10.1103/PhysRevB.13.2809
Miyakawa T., Dexter D.L. // Phys. Rev. B. 1970. V. 1. № 1. P. 70. https://doi.org/10.1103/PhysRevB.1.70
Liu G. // Spectroscopic properties of rare earths in optical materials / Eds. Hull R. et al. Springer series in materials science. 2005. V. 83. P. 1. Berlin; Heidelberg: Springer, https://doi.org/10.1007/3-540-28209-2_1
Dieke G.H., Crosswhite H.M. Spectra and energy levels of rare earth ions in crystals. New York: Wiley, 1968. 401 p.
Carnall W.T., Goodman G.L., Rajnak K., Rana R.S. // J. Chem. Phys. 1989. V. 90. P. 3443. https://doi.org/10.1063/1.455853
Peijzel P.S., Meijerink A., Wegh R.T. et al. // J. Solid State Chem. 2005. V. 178. № 2. P. 448. https://doi.org/10.1016/j.jssc.2004.07.046
Знаменский Н.В., Малюкин Ю.В. Спектры и динамика оптических переходов редкоземельных ионов в кристаллах. М.: Физматлит, 2008. 187 с.
Wen S., Zhou J., Zheng K. et al. // Nat. Commun. 2018. V. 9. P. 2415. https://doi.org/10.1038/s41467-018-04813-5
Auzel F. // Chem. Rev. 2004. V. 104. P. 139. https://doi.org/10.1021/cr020357g
Wang F., Liu X. // Chem. Soc. Rev. 2009. V. 38. № 4. P. 976. https://doi.org/10.1039/B809132N
Ehlert O., Thomann R., Darbandi M., Nann T. // ACS Nano. 2008. V. 2. № 1. P. 120 https://doi.org/10.1021/nn7002458
Paschotta R., Nilsson J., Tropper A.C., Hanna D. // IEEE J. Quantum Electronics. 1997. V. 33. P. 1049. https://doi.org/10.1109/3.594865
Kaminskii A.A., Agamalyan N.R., Deniseneo G.A. et al. // Phys. Status Solidi A. 1982. V. 70 P. 397. https://doi.org/10.1002/pssa.2210700206
Kaminskii A.A., Ngoc T., Sarkisov S.E. et al. // Phys. Status Solidi A. 1980. V. 59. P. 121. https://doi.org/10.1002/pssa.2210590117
Kaminskii A.A., Sarkisov S.E., Kürsten H.D., Schultze D. // Phys. Status Solidi A. 1982. V. 72. P. 207. https://doi.org/10.1002/pssa.2210720121
Brown M.R., Roots K.G., Shand W.A. // J. Phys. C: Solid State Phys. 1969. V. 2. P. 593. https://doi.org/10.1088/0022-3719/2/4/304
Caspers H.H., Rast H.E. // J. Lumin. 1975. V. 10. P. 347. https://doi.org/10.1016/0022-2313(75)90001-0
Esterowitz L., Bartoli F.J., Allen R.E. et al. // Phys. Rev. B. 1979. V. 19. P. 6442. https://doi.org/10.1103/PhysRevB.19.6442
Da Gama A.A.S., De Sá G.F., Porcher P., Caro P. // J. Chem. Phys. 1981. V. 75. P. 2583. https://doi.org/10.1063/1.442410
Krylov I.V., Akasov R.A., Rocheva V.V. et al. // Front. Chem. 2020. V. 8. № 295. P. 295. https://doi.org/10.3389/fchem.2020.00295
Mai H.X., Zhang Y.W., Sun L.D., Yan C.H. // J. Phys. Chem. C. 2007. V. 111. № 37. P. 13721. https://doi.org/10.1021/jp073920d
Anderson R.B., Smith S.J., May P.S., Berry M.T. // J. Phys. Chem. Lett. 2013. V. 5. № 1. P. 36. https://doi.org/10.1021/jz402366r
Alyatkin S., Asharchuk I., Khaydukov K. et al. // Nanotechnology. 2017. V. 28. № 3. P. 35401. https://doi.org/10.1088/1361-6528/28/3/035401
Thirumalai J. Luminescence: An Outlook on the Phenomena and their Applications. Rijeka: InTech, 2016. 378 p. https://doi.org/10.5772/62517
Ong L.C., Gnanasammandhan M.K., Nagarajan S., Zhang Y. // Luminescence. 2010. V. 25. № 4. P. 290. https://doi.org/10.1002/bio.1229
Кузнецов С.В., Осико В.В., Ткаченко Е.А., Федоров П.П. // Успехи химии. 2006. Т. 75. № 12. С. 1193.
Kaminskii A.A. // Laser Photonics Rev. 2007. V. 1. № 2. P. 93. https://doi.org/10.1002/lpor.200710008
Aebischer A., Hostettler M., Hauser J. et al. // Angew. Chem. Int. Ed. 2006. V. 45. № 17. P. 2802. https://doi.org/10.1002/anie.200503966
Mahalingam V., Naccache R., Vetrone F., Capobianco J.A. // Chem. Eur. J. 2009. V. 15. № 38. P. 96 https://doi.org/10.1002/chem.200901371
Chen G., Ohulchanskyy T.Y., Kachynski A. et al. // ACS Nano. 2011. V. 5. № 6. P. 4981. https://doi.org/10.1021/nn201083j
Zhang B.F., Frigoli M., Angiuli F. et al. // Chem. Commun. 2012. V. 48. № 58. P. 7244. https://doi.org/10.1039/C2CC33052K
Boyer J.C., Cuccia L.A., Capobianco J.A. // Nano Lett. 2007. V. 7. № 3. P. 847. https://doi.org/10.1021/nl070235
Vetrone F., Naccache R., Mahalingam V. et al. // Adv. Funct. Mater. 2009. V. 19. № 18. P. 2924. https://doi.org/10.1002/adfm.200900234
Naccache R., Vetrone F., Mahalingam V. et al. // Chem. Mater. 2009. V. 21. № 4. P. 717. https://doi.org/10.1021/cm803151y
Liu Q., Sun Y., Yang T. et al. // J. Am. Chem. Soc. 2011. V. 133. № 43. P. 17122. https://doi.org/10.1021/ja207078s
Vetrone F., Mahalingam V., Capobianco J.A. // Chem. Mater. 2009. V. 21. № 9. P. 1847. https://doi.org/10.1021/cm900313s
Zhang C., Li C., Li G. et al. // J. Mater. Chem. 2011. V. 21. № 3. P. 717. https://doi.org/10.1039/C0JM02948C
Mahalingam V., Vetrone F., Capobianco J.A. et al. // J. Mater. Chem. 2009. V. 19. № 20. P. 3149. https://doi.org/10.1039/B900300B
Yang D., Li C., Li G. et al. // J. Mater. Chem. 2011. V. 21. № 16. P. 5923. https://doi.org/10.1039/C0JM04179C
Yi G., Peng Y., Gao Z. // Chem. Mater. 2011. V. 23. № 11. P. 2729. https://doi.org/10.1021/cm103175s
Sun X., Zhang Y.W., Du Y.P. et al. // Chem. Eur. J. 2007. V. 13. № 8. P. 2320. https://doi.org/10.1002/chem.200601072
Du Y.P., Zhang Y.W., Sun L.D., Yan C.H. // J. Phys. Chem. C. 2008. V. 112. № 2. P. 405. https://doi.org/10.1021/jp076717r
Quan Z., Yang D., Yang P. et al. // Inorgan. Chem. 2008. V. 47. № 20. P. 9509. https://doi.org/10.1021/ic8014207
Du Y.P., Sun X., Zhang Y.W. et al. // Cryst. Growth Des. 2009. V. 9. № 4. P. 2013. https://doi.org/10.1021/cg801371r
Yi G.S., Chow G.M. // Chem. Mater. 2007. V. 19. № 3. P. 341. https://doi.org/10.1021/cm062447y
Shan J., Ju Y. // Nanotechnology. 2009. V. 20. № 27. P. 27560. https://doi.org/10.1088/0957-4484/20/27/275603
Shan J., Qin X., Yao N., Ju Y. // Nanotechnology. 2007. V 18. № 44. P. 445607. https://doi.org/10.1088/0957-4484/18/44/445607
Yan C., Zhao H., Perepichka D.F., Rosei F. // Small. 2016. V. 12. № 29. P. 3888. https://doi.org/10.1002/smll.201601565
Lu S., Tu D., Li X. et al. // Nano Research. 2016. V. 9. № 1. P. 187. https://doi.org/10.1007/s12274-015-0979-4
Cheng T., Marin R., Skripka A. et al. // J. Am. Chem. Soc. 2018. V. 140. № 40. P. 12890. https://doi.org/10.1021/jacs.8b07086
Chen G., Qiu H., Fan R. et al. // J. Mater. Chem. 2012. V. 22. № 38. P. 20190. https://doi.org/10.1039/C2JM32298F
Yi G.S., Lee W.B., Chow G.M. // J. Nanosci. Nanotechnol. 2007. V. 7. № 8. P. 2790. https://doi.org/10.1166/jnn.2007.638
Zhang D., De G., Zi L. et al. // J. Colloid Interface Sci. 2018. V. 512. P. 141. https://doi.org/10.1016/j.jcis.2017.10.012
Zhou B., Xu B., He H. et al. // Nanoscale. 2018. V. 10. № 6. P. 2834. https://doi.org/10.1039/C7NR07709B
Boyer J.C., Vetrone F., Cuccia L.A., Capobianco J.A. // J. Am. Chem. Soc. 2006. V. 128. № 23. P. 7444. https://doi.org/10.1021/ja061848b
Shao B., Zhao Q., Jia Y. et al. // Chem. Commun. 2014. V. 50. № 84. P. 12706. https://doi.org/10.1039/C4CC05191B
Du P., Luo L., Huang X., Yu J.S. // J. Colloid Interface Sci. 2018. V. 514. P. 172. https://doi.org/10.1016/j.jcis.2017.12.027
Kumar A., Tiwari S.P., Swart H.C., da Silva J.C.G.E. // Opt. Mater. 2019. V. 92. P. 347. https://doi.org/10.1016/j.optmat.2019.04.050
Yi G.S., Chow G.M. // J. Mater. Chem. 2005. V. 15. № 41. P. 4460. https://doi.org/10.1039/B508240D
Na H., Woo K., Lim K., Jang H.S. // Nanoscale. 2013. V. 5. № 10. P. 4242. https://doi.org/10.1039/C3NR00080J
Wang F., Han Y., Lim C. et al. // Nature. 2010. V. 463. № 7284. P. 1061. https://doi.org/10.1038/nature08777
Damasco J.A., Chen G., Shao W. et al. // ACS Appl. Mater. Interfaces. 2014. V. 6. № 16. P. 13884. https://doi.org/10.1021/am503288d
Wong H.T., Vetrone F., Naccache R. et al. // J. Mater. Chem. 2011. V. 21. № 41. P. 16589. https://doi.org/10.1039/C1JM12796A
Capobianco J.A., Teng X., Zhu Y. et al. // J. Am. Chem. Soc. 2012. V. 134. № 20. P. 8340. https://doi.org/10.1021/ja3016236
Liu D., Xu X., Du Y. et al. // Nat. Commun. 2016. V. 7. P. 10254. https://doi.org/10.1038/ncomms10254
Chen B., Kong W., Wang N. et al. // Chem. Mater. 2019. V. 31. № 13. P. 4779. https://doi.org/10.1021/acs.chemmater.9b01050
Feng S., Xu R. // Acc. Chem. Res. 2001. V. 34. № 3. P. 239. https://doi.org/10.1021/ar0000105
Li C., Lin J. // J. Mater. Chem. 2010. V. 20. № 33. P. 6831. https://doi.org/10.1039/C0JM00031K
Muhr V., Wilhelm S., Hirsch T., Wolfbeis O.S. // Acc. Chem. Res. 2014. V. 47. № 12. P. 3481. https://doi.org/10.1021/ar500253g
Wang X., Zhuang J., Peng Q. et al. // Nature. 2005. V. 437. № 7055. P. 121. https://doi.org/10.1038/nature03968
Wang L., Li Y. // Chem. Mater. 2007. V. 19. № 4. P. 727. https://doi.org/10.1021/cm061887m
Zhang F., Wan Y., Yu T. et al. // Angew. Chem. Int. Ed. 2007. V. 46. № 42. P. 7976. https://doi.org/10.1002/anie.200702519
Qi W., Wu Q., Shapter J.G. et al. // ACS Omega. 2018. V. 3. № 12. P. 18730. https://doi.org/10.1021/acsomega.8b02919
Hu H., Chen Z., Cao T. et al. // Nanotechnology. 2008. V. 19. № 37. P. 375702. https://doi.org/10.1088/0957-4484/19/37/375702
Gunaseelan M., Yamini S., Kumar G.A., Senthilselvan J. // Opt. Mater. 2018. V. 75. P. 174. https://doi.org/10.1016/j.optmat.2017.10.012
Lucier B.E., Johnston K.E., Arnold D.C. et al. // J. Phys. Chem. C. 2014. V. 118. № 2. P. 1213. https://doi.org/10.1021/jp408148b
Richard B., Lemyre J.L., Ritcey A.M. // Langmuir. 2017. V. 33. № 19. P. 4748. https://doi.org/10.1021/acs.langmuir.7b00773
Cai H., Shen T., Kirillov A.M. et al. // Inorgan. Chem. 2017. V. 56. № 9. P. 5295. https://doi.org/10.1021/acs.inorgchem.7b00380
Wu W., Wang L., WangY. et al. // J. Colloid Interface Sci. 2020. V. 563. P. 308. https://doi.org/10.1016/j.jcis.2019.12.084
Liu L., Jiao J., Wei W. et al. // Scripta Mater. 2019. V. 169. P. 61. https://doi.org/10.1016/j.scriptamat.2019.04.038
Xu H., Cheng L., Wang C. et al. // Biomaterials. 2011. V. 32. № 35. P. 9364. https://doi.org/10.1016/j.biomaterials.2011.08.053
Patra A., Friend C.S., Kapoor R., Prasad P.N. // J. Phys. Chem. B. 2002. V. 106. № 8. P. 1909. https://doi.org/10.1021/jp013576z
Lepoutre S., Boyer D., Mahiou R. // Opt. Mater. 2006. V. 28. № 6–7. P. 592. https://doi.org/10.1016/j.optmat.2005.09.053
Cao B.S., He Y.Y., Zhang L., Dong B. // J. Lumin. 2013. V. 135. P. 128. https://doi.org/10.1016/j.jlumin.2012.10.031
Chen X., Liu Z., Sun Q. et al. // Opt. Commun. 2011. V. 284. № 7. P. 2046. https://doi.org/10.1016/j.optcom.2010.12.007
Meneses-Franco A., Campos-Vallette M., Vásquez S.O., Soto-Bustamante E.A. // Materials. 2018. V. 11. № 10. P. 1950. https://doi.org/10.3390/ma11101950
Lin J., Yu M., Lin C., Liu X // J. Phys. Chem. C. 2007. V. 111. № 16. P. 5835. https://doi.org/10.1021/jp070062c
Lü Q., Li A., Guo F. et al. // Nanotechnology. 2008. V. 19. № 14. P. 145701. https://doi.org/10.1088/0957-4484/19/14/145701
Chen Z.S., Gong W.P., Chen T.F., Li S.L. // Bull. Mater. Sci. 2011. V. 34. № 3. P. 429. https://doi.org/10.1007/s12034-011-0116-2
Hernández-Rodriguez M.A., Lozano-Gorrín A.D., Lavín V. et al. // Opt. Express. 2017. V. 25. № 22. P. 27845. https://doi.org/10.1364/OE.25.027845
Grzyb T., Węcławiak M., Rozowska J., Lis S. // J. Alloys Compd. 2013. V. 576. P. 345. https://doi.org/10.1016/j.jallcom.2013.05.207
Grzyb T., Tymiński A. // J. Alloys Compd. 2016. V. 660. P. 235. https://doi.org/10.1016/j.jallcom.2015.11.122
Fujihara S., Kishiki Y., Kimura T. // J. Alloys Compd. 2002. V. 333. № 1–2. P. 76. https://doi.org/10.1016/S0925-8388(01)01696-6
Chang H., Xie J., Zhao B. et al. // Nanomaterials. 2015. V. 5. № 1. P. 1. https://doi.org/10.3390/nano5010001
Kappe C.O // Angew. Chem. Int. Ed. 2004. V. 43. № 46. P. 6250. https://doi.org/10.1002/anie.200400655
Stuerga D., Delmotte M. Microwaves in organic synthesis. Weinheim: Wiley-VCH Verlag GmbH, 2006. 1033 p.
Tong L., Li X., Hua R. et al. // J. Nanosci. Nanotechnol. 2016. V. 16. № 1. P. 816. https://doi.org/10.1166/jnn.2016.10889
Reddy K.L., Prabhakar N., Arppe R. et al. // J. Mater. Sci. 2017. V. 52. № 10. P. 5738. https://doi.org/10.1007/s10853-017-0809-z
Mi C., Tian Z., Cao C. et al. // Langmuir. 2011. V. 27. № 23. P. 14632. https://doi.org/10.1021/la204015m
Li F., Li C., Liu X. et al. // Dalton Trans. 2013. V. 42. № 6. P. 2015. https://doi.org/10.1039/C2DT32295A
Suzuki Y., Yin S., Sato T. // Mater. Focus. 2015. V. 4. № 1. P. 58. https://doi.org/10.1166/mat.2015.1208
Wang D., Ren L., Zhou X. et al. // Nanotechnology. 2012. V. 23. № 22. P. 225705. https://doi.org/10.1088/0957-4484/23/22/225705
Wang H.Q., Nann T. // Acs Nano. 2009. V. 3. № 11. P. 3804. https://doi.org/10.1021/nn9012093
Wang H.Q., Tilley R.D., Nann T. // Cryst. Eng. Commun. 2010. V. 12. № 7. P. 1993. https://doi.org/10.1039/B927225A
Fedorov P.P., Alexandrov A.A. // J. Fluorine Chem. 2019. P. 109374. https://doi.org/10.1016/j.jfluchem.2019.109374
Teshima K., Lee S., Shikine N. et al. // Cryst. Growth Des. 2011. V. 11. № 4. P. 995. https://doi.org/10.1021/cg100932k
Федоров П.П., Маякова М.Н., Кузнецов С.В. и др. // Журн. неорган. химии. 2016. Т. 61. № 11. С. 1529.
Ding M., Huang W., Cao L. et al. // Mater. Lett. 2012. V. 86. P. 58. https://doi.org/10.1016/j.matlet.2012.07.031
Ding M., Chen D., Zhong J. et al. // Sci. Adv. Mater. 2017. V. 9. № 3–4. P. 688. https://doi.org/10.1166/sam.2017.2680
Zhang X., Yang P., Li C. et al. // Chem. Commun. 2011. V. 47. № 44. P. 12143. https://doi.org/10.1039/C1CC15194K
Ding M., Lu C., Cao L. et al. // Cryst. Eng. Commun. 2013. V. 15. № 30. P. 6015. https://doi.org/10.1039/C3CE40477C
Huang X., Hu G., Xu Q. et al. // J. Alloys Compd. 2014. V. 616. P. 652. https://doi.org/10.1016/j.jallcom.2014.07.067
Пройдакова В.Ю., Александров А.А., Воронов В.В., Федоров П.П. // Журн. неорган. химии. 2020. Т. 65. № 6. С. 764. https://doi.org/10.31857/S0044457X20060161
Welton T., Wasserscheid P. Ionic liquids in synthesis. Weinheim: Wiley-VSH, 2008. 720 p.
Zhang C., Chen J. // Chem. Commun. 2010. V. 46. № 4. P. 592. https://doi.org/10.1039/B919044A
Zhou N., Qiu P., Wang K. et al. // Nanoscale Res. Lett. 2013. V. 8. № 1. P. 1. https://doi.org/10.1186/1556-276X-8-518
Song Y., Li Y., Zhao T. et al. // J. Colloid Interface Sci. 2017. V. 487. P. 281. https://doi.org/10.1016/j.jcis.2016.10.044
Guricová M., Pinc J., Malinčik J. et al. // Rev. Inorgan. Chem. 2019. V. 39. № 2. P. 77. https://doi.org/10.1515/revic-2018-0016
Li C., Yang P., Xu Z. et al. // Cryst. Eng. Commun. 2011. V. 13. № 3. P. 1003. https://doi.org/10.1039/C0CE00186D
Liu X., Zhao J., Sun Y. et al. // Chem. Commun. 2009. № 43. P. 6628. https://doi.og/10.1039/B915517A
He M., Huang P., Zhang C. et al. // Adv. Funct. Mater. 2011. V. 21. № 23. P. 4470. https://doi.org/10.1002/adfm.201101040
González-Mancebo D., Becerro A.I., Cantelar E. et al. // Dalton Trans. 2017. V. 46. № 20. P. 6580. https://doi.org/10.1039/C7DT00453B
Yang G.W. // Prog. Mater. Sci. 2007. V. 52. № 4. P. 648. https://doi.org/10.1016/j.pmatsci.2006.10.016
Barchanski A., Funk D., Wittich O. et al. // J. Phys. Chem. C. 2015. V. 119. № 17. P. 9524. https://doi.org/10.1021/jp511162n
Mafuné F., Kohno J., Takeda Y. et al. // J. Phys. Chem. B. 2000. V. 104. № 35. P. 8333. https://doi.org/10.1021/jp001803b
Katsuki D., Sato T., Suzuki R. et al. // Appl. Phys. A. 2012. V. 108. № 2. P. 321. https://doi.org/10.1007/s00339-012-6962-y
Sasaki T., Liang C., Nichols W.T. et al. // Appl. Phys. A. 2004. V. 79. № 4–6. P. 1489. https://doi.org/10.1007/s00339-004-2827-3
Sajti L., Karimov D.N., Rocheva V.V. et al. // Nano Res. 2020. https://doi.org/10.1007/s12274-020-3163-4
Edmonds A.M., Sobhan M.A., Sreenivasan V.K. et al. // Part. Part. Syst. Char. 2013. V. 30. № 6. P. 506. https://doi.org/10.1002/ppsc.201200112
Maurer E., Barcikowski S., Gökce B. // Chem. Eng. Technol. 2017. V. 40. № 9. P. 1535. https://doi.org/10.1002/ceat.201600506
Onodera Y., Nunokawa T., Odawara O., Wada H. // J. Lumin. 2013. V. 137. P. 220. https://doi.org/10.1016/j.jlumin.2012.12.033
Ikehata T., Onodera Y., Nunokawa T. et al. // Appl. Surface Sci. 2015. V. 348. P. 54. https://doi.org/10.1016/j.apsusc.2014.12.097
Gemini L., Schmitz T., Kling R. et al. // Chem. Phys. Chem. 2017. V. 18. № 9. P. 1210 https://doi.org/10.1002/cphc.201601266
Anjana R., Kurias K.M., Jayaraj M.K. // Opt. Mater. 2017. V. 72. P. 730. https://doi.org/10.1016/j.optmat.2017.07.021
Avvakumov E.G., Senna M., Kosova N.V. Soft mechanochemical synthesis: a basis for new chemical technologies. London: Kluwer Academic Publishers, 2002. 206 p.
Guérault H., Greneche J.M. // J. Phys: Condens. Matter. 2000. V. 12. № 22. P. 4791. https://doi.org/10.1088/0953-8984/12/22/311
Chable J., Martin A.G., Bourdin A. et al. // J. Alloys Compd. 2017. V. 692. P. 980. https://doi.org/10.1016/j.jallcom.2016.09.135
Patel D.N., Sarkisov S.S., Darwish A.M., Ballato J. // Opt. Express. 2016. V. 24. № 18. P. 21147. https://doi.org/10.1364/OE.24.021147
Yuan D., Yi G.S., Chow G.M. // J. Mater. Res. 2009. V. 24. № 6. P. 2042. https://doi.org/10.1557/jmr.2009.0258
Toncelli A., Ahmadi B. // 2011 International Workshop on Biophotonics. IEEE, 2011. P. 1. https://doi.org/10.1109/IWBP.2011.5954826
Hakim R., Damak K., Gemmi M. et al. // J. Phys. Chem. C. 2015. V. 119. № 5. P. 2844. https://doi.org/10.1021/jp510851w
Duvel A., Bednarcik J., Sepelak V., Heitjans P. // J. Phys. Chem. C. 2014. V. 118. № 13. P. 7117. https://doi.org/10.1021/jp410018t
Heise M., Schol G., Krahl T., Kemnitz E. // Solid State Sci. 2019. V. 91. P. 113. https://doi.org/10.1016/j.solidstatesciences.2019.03.014
Соболев Б.П., Фадеева В.И., Свиридов И.А. и др. // Кристаллография. 2005. Т. 50. № 3. С. 524.
Соболев Б.П., Свиридов И.А., Фадеева В.И. и др. // Кристаллография. 2008. Т. 53. № 5. С. 919.
Liu Z., Stevens-Kalceff M.A., Wang X., Riesen H. // Chem. Phys. Lett. 2013. V. 588. P. 193. https://doi.org/10.1016/j.cplett.2013.10.024
Wang X.L., Liu Z.Q., Stevens-Kalceff M.A., Riesen H. // Inorgan. Chem. 2014. V. 53. № 17. P. 8839. https://doi.org/10.1021/ic500712b
Zhang J., Riesen N., Riesen H. // Nanoscale. 2017. V. 9. № 41. P. 15958. https://doi.org/10.1039/C7NR05108E
Zhang J., Riesen H. // Chem. Phys. Lett. 2015. V. 641. P. 1. https://doi.org/10.1016/j.cplett.2015.10.040
Chen G., Agren H., Ohulchanskyy T.Y., Prasad P.N. // Chem. Soc. Rev. 2015. V. 44. № 6. P. 1680. https://doi.org/10.1039/C4CS00170B
Chen G., Qiu H., Prasad P.N., Chen X. // Chem. Rev. 2014. V. 114. P. 5161. https://doi.org/10.1021/cr400425h
Rocheva V.V., Koroleva A.V., Savelyev A.G. et al. // Sci. Reports. 2018. V. 8. № 1. P. 3663. https://doi.org/10.1038/s41598-018-21793-0
Wang X., Valiev R.R., Ohulchanskyy T.Y. et al. // Chem. Soc. Rev. 2017. V. 46. № 14. P. 4150. https://doi.org/10.1039/C7CS00053G
Shao Q., Li X., Hua P. et al. // J. Colloid Interface Sci. 2017. V. 486. P. 121. https://doi.org/10.1016/j.jcis.2016.09.067
Okamoto K., Niki I., Shvartser A. et al. // Nat. Mater. 2004. V. 3. № 9. P. 601. https://doi.org/10.1038/nmat1198
Berini P., De Leon I. // Nat. Photonics. 2012. V. 6. № 1. P. 16. https://doi.org/10.1038/nphoton.2011.285
David C., Guillot N., Shen H. et al. // Nanotechnology. 2010. V. 21. № 47. P. 475501. https://doi.org/10.1088/0957-4484/21/47/475501
Liu M., Chen R., Adamo G. et al. // Nanophotonics. 2013. V. 2. № 2. P. 153. https://doi.org/10.1515/nanoph-2012-0040
Noginov M.A., Zhu G., Belgrave A.M. et al. // Nat. Lett. 2009. V. 460. № 7259. P. 1110. https://doi.org/10.1038/nature08318
Lu D., Mao C., Cho S.K. et al. // Sci. Reports. 2016. V. 6. P. 18894. https://doi.org/10.1038/srep18894
Feng A.L., You M.L., Tian L. et al. // Sci. Reports. 2015. V. 5. P. 7779. https://doi.org/10.1038/srep07779
Dai Z. Advances in Nanotheranostics II. Cancer Theranostic Nanomedicine. Singapore: Springer, 2016. V. 7. 381 p. https://doi.org/10.1007/978-981-10-0063-8
Chen M., Yin M. // Prog. Polym. Sci. 2014. V. 39. № 2. P. 365. https://doi.org/10.1016/j.progpolymsci.2013.11.001
Yang D., Ma P., Hou Z. et al. // Chem. Soc. Rev. V. 44. № 6. P. 1416. https://doi.org/10.1039/c4cs00155a
Zhou J., Liu Q., Feng W. et al. // Chem. Rev. 2015. V. 115. № 1. P. 395. https://doi.org/10.1021/cr400478f
Key J., Leary J.F. // Int. J. Nanomed. 2014. V. 9. P. 711. https://doi.org/10.2147/IJN.S53717
Generalova A.N., Chichkov B.N., Khaydukov E.V. // Adv. Colloid Interface Sci. 2017. V. 245. P. 1. https://doi.org/10.1016/j.cis.2017.05.006
Dolmans D.E., Fukumura D., Jain R.K. // Nat. Rev. Cancer. 2003. V. 3. № 5. P. 380. https://doi.org/10.1038/nrc1071
Wang C., Tao H., Cheng L., Liu Z. // Biomaterials. 2011. V. 32. № 26. P. 6145. https://doi.org/10.1016/j.biomaterials.2011.05.007
Wang M., Chen Z., Zheng W. et al. // Nanoscale. 2014. V. 6. № 14. P. 8274. https://doi.org/10.1039/C4NR01826E
Ai F., Ju Q., Zhang X. et al. // Sci. Reports. 2015. V. 5. P. 10785. https://doi.org/10.1038/srep10785
Khaydukov E.V., Mironova K.E., Semchishen V.A. et al. // Sci. Reports. 2016. V. 6. № 1. P. 35103. https://doi.org/10.1038/srep35103
Mironova K.E., Khochenkov D.A., Generalova A.N. // Nanoscale. 2017. V. 9. № 39. P. 14921. https://doi.org/10.1039/C7NR04092J
Гребеник Е.А., Костюк А.Б., Деев С.М. // Успехи химии. 2016. Т. 85. № 12. С. 1277.
Wang X., Kong X., Yu Y. et al. // J. Phys. Chem. C. 2007. V. 111. № 41. P. 15119. https://doi.org/10.1021/jp0686689
Lei Y., Song H., Yang L. et al. // J. Chem. Phys. 2005. V. 123. № 17. P. 174710. https://doi.org/10.1063/1.2087487
Wang Y., Tu L., Zhao J. et al. // J. Phys. Chem. C. 2009. V. 113. № 17. P. 7164. https://doi.org/10.1021/jp9003399
Bai X., Song H., Pan G. et al. // J. Phys. Chem C. 2007. V. 111. № 36. P. 13611. https://doi.org/10.1021/jp070122e
Vetrone F., Naccache R., Zamarrón A. et al. // ACS Nano. 2010. V. 4. № 6. P. 3254. https://doi.org/10.1021/nn100244a
Sedlmeier A., Achatz D.E., Fischer L.H. et al. // Nanoscale. 2012. V. 4. № 22. P. 7090. https://doi.org/10.1039/C2NR32314A
Li D., Wang Y., Zhang X. et al. // Opt. Commun. 2012. V. 285. № 7. P. 1925. https://doi.org/10.1016/j.optcom.2011.12.075
Van De Rijke F., Zijlmans H., Li S. et al. // Nat. Biotechnol. 2001. V. 19. № 3. P. 273. https://doi.org/10.1038/85734
Wang M., Hou W., Mi C.C. et al. // Anal. Chem. 2009. V. 81. № 21. C. 8783. https://doi.org/10.1021/ac901808q
Liu Q., Peng J., Sun L., Li F. // ACS Nano. 2011. V. 5. № 10. P. 8040. https://doi.org/10.1021/nn202620u
Shockley W., Queisser H.J. // J. Appl. Phys. 1961. V. 32. № 3. P. 510. https://doi.org/10.1063/1.1736034
Trupke T., Shalav A., Richards B.S. et al. // Sol. Energy Mater. Sol. Cells. 2006. V. 90. № 18–19. P. 3327. https://doi.org/10.1016/j.solmat.2005.09.021
Shan G.B., Assaaoudi H., Demopoulos G.P. // ACS Appl. Mater. Interfaces. 2011. V. 3. № 9. P. 3239. https://doi.org/10.1021/am200537e
Shan G.B., Demopoulos G.P. // Adv. Mater. 2010. V. 22. № 39. P. 4373. https://doi.org/10.1002/adma.201001816
Yu J., Yang Y., Fan R. et al. // Nanoscale. 2016. V. 8. № 7. P. 4173. https://doi.org/10.1039/C5NR08319B
Liang L., Liu Y., Zhao X.Z. // Chem. Commun. 2013. V. 49. № 38. P. 3958. https://doi.org/10.1039/C3CC41252K
Liang L., Liu Y., Bu C. et al. // Adv. Mater. 2013. V. 25. № 15. P. 2174. https://doi.org/10.1002/adma.201204847
Hao S., Shang Y., Li D. et al. // Nanoscale. 2017. V. 9. № 20. P. 6711. https://doi.org/10.1039/C7NR01008G
Beyazit S., Ambrosini S., Marchyk N. et al. // Angew. Chem. Int. Ed. 2014. V. 53. № 34. P. 8919. https://doi.org/10.1002/anie.201403576
Xiao Q., Ji Y., Xiao Z. et al. // Chem. Commun. 2013. V. 49. № 15. P. 1527. https://doi.org/10.1039/C2CC37620B
Méndez-Ramos J., Ruiz-Morales J.C., Acosta-Mora P., Khaidukov N.M. // J. Mater. Chem. C. 2016. V. 4. № 4. P. 801. https://doi.org/10.1039/C5TC03315B
Darani M.K., Bastani S., Ghahari M. et al. // Prog. Org. Coat. 2017. V. 104. P. 97. https://doi.org/10.1016/j.porgcoat.2016.11.005
Liu R., Chen H., Li Z. et al. // Polym. Chem. 2016. V. 7. № 14. P. 2457. https://doi.org/110.1039/C6PY00184J
Demina P.A., Arkharova N.A., Asharchuk I.M. et al. // Molecules. 2019. V. 24. № 13. P. 2476. https://doi.org/10.3390/molecules24132476
Pan Y., Feng P., Yin M. et al. // ChemistrySelect. 2019. V. 4. № 38. P. 11346. https://doi.org/10.1002/slct.201902646
Wang Y., Suzuki H., Xie J. et al. // Chem. Rev. 2018. V. 118. № 10. P. 5201. https://doi.org/10.1021/acs.chemrev.7b00286
Balaji R., Kumar S., Reddy K. L. et al. // J. Alloys Compd. 2017. V. 724. P. 481. https://doi.org/10.1016/j.jallcom.2017.07.050
Fagnoni M., Dondi D., Ravelli D., Albini A. // Chem. Rev. 2007. V. 107. № 6. P. 2725. https://doi.org/10.1021/cr068352x
Huang H., Li H., Wang Z. et al. // Chem. Eng. J. 2019. V. 361. P. 1089. https://doi.org/10.1016/j.cej.2018.12.174
Challagulla S., Payra S., Bajaj M., Roy S. // Bull. Mater. Sci. 2019. V. 42. № 3. P. 102. https://doi.org/10.1007/s12034-019-1804-6
Yang L., Huang J., Ji W., Mao M. // Powder Technol. 2020. V. 360. P. 956. https://doi.org/10.1016/j.powtec.2019.10.053
Fujishima A., Zhang X., Tryk D.A. // Surf. Sci. Rep. 2008. V. 63. № 12. P. 515. https://doi.org/10.1016/j.surfrep.2008.10.001
Linsebigler A.L., Lu G., Yates J.T. // Chem. Rev. 1995. V. 95. № 3. P. 735. https://doi.org/10.1021/cr00035a013
Xu X., Sun Y., Zhang Q. et al. // Opt. Mater. 2019. V. 94. P. 444. https://doi.org/10.1016/j.optmat.2019.05.038
Wang J., Wen F., Zhang Z. et al. // J. Environ. Sci. China. 2005. V. 17. № 5. P. 727.
Qin W., Zhang D., Zhao D. et al. // Chem. Commun. 2010. V. 46. № 13. P. 2304. https://doi.org/10.1039/b924052g
Zhou Y., Wu S., Wang F. et al. // Chemosphere. 2020. V. 238. P. 124648. https://doi.org/10.1016/j.chemosphere.2019.124648
Boppella R., Marques Mota F., Lim J.W. et al. // ACS Appl. Energy Mater. 2019. V. 2. № 5. P. 3780. https://doi.org/10.1021/acsaem.9b00469
Chen C.K., Chen H.M., Chen C.-J., Liu R.-S. // Chem. Commun. 2013. V. 49. № 72. P. 7917. https://doi.org/10.1039/C3CC42567C
Yoon B., Lee J., Park I.S. et al. // J. Mater. Chem. C. 2013. V. 1. № 13. P. 2388. https://doi.org/10.1039/C3TC00818E
Gupta B.K., Haranath D., Saini S. et al. // Nanotechnology. 2010. V. 21. № 5. P. 055607. https://doi.org/10.1088/0957-4484/21/5/055607
Anh T.K., Loc D.X., Huong T.T. et al. // Int. J. Nanotechnol. 2011. V. 8. № 3–5. P. 335. https://doi.org/10.1504/IJNT.2011.03821
Meruga J.M., Baride A., Cross W. et al. // J. Mater. Chem. C. 2014. V. 2. № 12. P. 2221. https://doi.org/10.1039/C3TC32233E
You M., Zhong J., Hong Y. et al. // Nanoscale. 2015. V. 7. № 10. P. 4423. https://doi.org/10.1039/C4NR06944G
Khaydukov E.V., Semchishen V.A., Zvyagin A.V. // Opt. Lett. 2015. V. 40. № 7. P. 1169. https://doi.org/10.1364/OL.40.001169
Zhao J., Jin D., Schartner E.P. et al. // Nat. Nanotechnol. 2013. V. 8. № 10. P. 729. https://doi.org/10.1038/nnano.2013.171
u Y., Zhao J., Zhang R. et al. // Nat. Photonics. 2014. V. 8. № 1. P. 32. 10.1038/nphoton.2013.32
Хайдуков Е.В., Рочева В.В., Миронова К.Е. и др. // Российские нанотехнологии. 2015. Т. 10. № 11–12. С. 87.
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии