

Российские нанотехнологии, 2021, T. 16, № 1, стр. 103-110
ПОЛУЧЕНИЕ НАНОВОЛОКНИСТЫХ МАТЕРИАЛОВ НА ОСНОВЕ ВЫСОКОМОЛЕКУЛЯРНОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ МЕТОДОМ ЭЛЕКТРОФОРМОВАНИЯ
Т. Х. Тенчурин 1, *, А. Д. Шепелев 1, С. И. Белоусов 1, А. А. Пучков 1, Е. В. Ястремский 1, С. Н. Чвалун 1
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: tenchurin.timur@mail.ru
Поступила в редакцию 24.11.2020
После доработки 24.11.2020
Принята к публикации 16.12.2020
Аннотация
Методом электроформования получены нановолокнистые каркасы на основе гиалуроновой кислоты, имитирующие естественный внеклеточный матрикс, перспективный для использования в тканевой инженерии. Гиалуроновая кислота входит в состав многих органов и тканей и является биологически активным компонентом, способным принимать участие в пролиферации и миграции клеток. Высокая вязкость растворов высокомолекулярного биополимера существенно затрудняет получение нановолокнистых каркасов на его основе. Проведенные реологические исследования позволили определить состав растворителя, способствующего снижению вязкости прядильного раствора. Наибольшая ньютоновская вязкость 1%-ного раствора гиалуроновой кислоты в воде составила 25 Па с, а при использовании смеси гидрата аммиака (10%) и диметилформамида в соотношении 2 : 1 уменьшилась до 1.147 Па с. С помощью специальной прядильной ячейки с фильерой малого диаметра и использованием высокого давления (до 10 атм) удалось получить нановолокна на основе гиалуроновой кислоты диаметром от 100 до 300 нм.
ВВЕДЕНИЕ
Дальнейший прогресс в области регенеративной медицины связывают с созданием трехмерных материалов, наделенных биоподобными свойствами. Разработанные к настоящему времени синтетические каркасы с морфологическими, структурными и механическими свойствами, необходимыми для прикрепления и пролиферации клеток, не обеспечивают адекватного микроокружения для адгезии, миграции и направленной дифференцировки клеток [1]. В связи с этим продолжается поиск оптимальных путей модификации синтетических материалов компонентами внеклеточного матрикса, в том числе коллагеном, фибронектином, а также гиалуроновой кислотой (ГК) [2–5]. ГК рассматривали ранее в качестве биологически инертного наполнителя, обеспечивающего вязкоупругое поведение живых тканей. Однако в последние время было показано, что молекулы ГК способны выполнять в тканях целый ряд сигнальных функций, в том числе принимать участие в пролиферации и миграции клеток [6].
Молекула ГК, синтезированная в организме человека, состоит из повторяющихся дисахаридных звеньев D-глюкуроновой кислоты и D-N-ацетилглюкозамина, соединяющихся посредством чередующихся β-1,4- и β-1,3-гликозидных связей, и имеет молекулярную массу более >106 г/моль [6]. За счет возникновения многочисленных межмолекулярных водородных связей цепочки ГК способны переплетаться между собой и образовывать трехмерную сетку зацеплений в водных растворах, характеризующихся аномально высокой вязкостью даже при низких концентрациях [7].
Как показали проведенные исследования, биологическая активность ГК обусловлена не только клеточной спецификой, но и в значительной степени определяется размерами самой молекулы. Высокомолекулярная ГК (нативная) обладает противовоспалительным действием и антиангиогенной активностью, участвует в репаративной регенерации тканей и морфогенезе. Напротив, низкомолекулярная ГК (2 × 104–106 г/моль) стимулирует выделение провоспалительных цитокинов, хемокинов и факторов роста. Ее повышенный уровень был обнаружен при различных заболеваниях, в том числе астме, фиброзе легких, ревматоидном артрите, онкологии и т.д. [6, 8].
Вследствие своих уникальных биологических и физико-химических свойств ГК находит широкое применение в медицине. Конъюгирование антидиабетических пептидов, гидрокортизона и преднизолона с ГК улучшает стабильность и терапевтический эффект лекарственных веществ. ГК способна специфически связываться с рецептором CD44, экспрессируемым в различных опухолевых клетках, что позволяет использовать ее для разработки таргетных методов лечения онкологических заболеваний. ГК применяют для создания ранозаживляющих покрытий, так как она содействует пролиферации фибробластов, их миграции и адгезии к раневой поверхности, а также стимулирует образование коллагена. ГК входит в состав стекловидного тела, слезной железы, эпителия роговицы, конъюнктивы и слезной жидкости. Поэтому офтальмологические продукты на ее основе биосовместимы, не вызывают реакцию на инородное тело. Значительный интерес представляет использование ГК для регенерации органов и тканей с помощью тканеинженерных конструкций (ТИК) [8, 9]. Создание ТИК проводят на основе биосовместимых материалов, которые по своей архитектонике, химическим и механическим свойствам способны имитировать естественный внеклеточный матрикс (ВКМ) [10]. Последний представляет собой высокопористый каркас, состоящий в основном из волокон коллагена, протеогликанов и гликопротеинов. ГК присутствует во всем объеме ВКМ и связывает протеогликаны за счет электростатических связей.
Одним из эффективных способов получения клеточных каркасов является метод электроформования, позволяющий получать нановолокнистые материалы с высокой пористостью и удельной поверхностью [11, 12]. Среди множества работ, описывающих получение волокон в электрическом поле, к сожалению, крайне мало исследований посвящено формованию водных растворов ГК [13–15] в силу их высокой вязкости. Оптимальным с реологической точки зрения растворителем для ГК считается смесь гидрата аммиака и апротонных растворителей типа N,N-диметилфорамида (ДМФА) и N-метилпирролидона (НМП). Щелочная среда, создаваемая аммиаком, способствует разрушению водородных и увеличению гибкости гликозидных связей, что при pH > 11 существенно снижает вязкость растворов ГК [16, 17]. В свою очередь ДМФА облегчает процесс электропрядения, так как способствует снижению сил поверхностного натяжения [14]. Несмотря на то что при использовании растворов низкомолекулярной ГК удалось получить нановолокнистые материалы [15], процесс электроформования высокомолекулярной ГК требует дальнейшего изучения и оптимизации. В результате электроформования были получены единичные образцы нановолокон ГК, объемный расход раствора ГК составлял не более 0.01 мкл/мин [13, 14], однако данных об исследовании процесса деградации ГК в этих средах не приводится. Последнее особенно важно, поскольку биологические\сигнальные свойства молекул ГК зависят от их длины [6].
Для успешного получения волокон из растворов высокомолекулярной ГК необходимо исследовать реологические свойства ее растворов и связать их с условиями электропрядения.
МЕТОДЫ И МАТЕРИАЛЫ
Приготовление прядильных растворов. Для изготовления волокнистых материалов использовали гиалуронат натрия (производитель ООО “ПТК Туше Флора”) со средней молекулярной массой Mw = 1.2–2.2 × 106 г/моль. При приготовлении формовочных растворов концентрацией от 1 до 2.5% использовали смесь водного 10–30%-ного раствора гидрата аммиака (производитель Sigma-Aldrich) и N,N-диметилформамида или N-метилпирролидона (производитель ООО “Компонент-Реактив”) в соотношение 2:1, а также воду. Для повышения волокнообразующих свойств растворов на основе ГК в их состав вводили полиэтиленоксид (ПЭО, Mw = 2000 кДа, Sigma-Aldrich) концентрацией от 1.1 до 1.2%.
Реологические свойства растворов. Реологические свойства изучали на реометре Антон Паар МСР 501 (Physica MSR 501 Anton Paar) с измерительной системой “конус–плоскость” 50 мм (СР – 50) и CC27/E/TI.
Электропроводность растворов. Для измерения электропроводности растворов использовали кондуктометр Эксперт-002.
Определение pH растворов. Для определения значения pH растворов использовали pH-метр S40 (Меттлер Толедо).
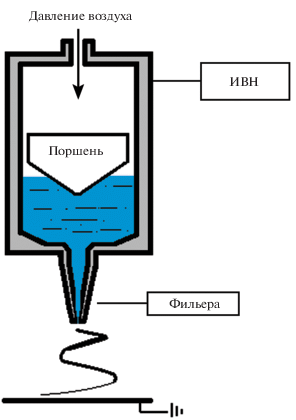
Процесс электроформования проводили при параметрах, обеспечивающих устойчивый процесс формования волокон, комнатной температуре и относительной влажности воздуха от 60 до 80%. В качестве приемного электрода использовали заземленный металлический диск диаметром 70 мм. При получении волокон соблюдали следующие условия формования в геометрии игла–п лоскость: напряжение, подаваемое на фильеру, составляло 29–47 кВ, межэлектродное расстояние 18–20 см и объемный расход 0.6–1.2 мл/ч. В качестве фильеры использовали стеклянные капилляры с диаметром канала от 150 до 200 мкм и сопротивлением от 130 до 150 мм.вд.ст. (при расходе воздуха 1 см3/с). Для проведения электроформования использовали специальную установку, работающую под давлением от 1 до 10 атм. (рис. 1).
Рис. 1.
Схема установки для электроформования высоковязких растворов. ИВН – источник высокого напряжения.
Оптическая микроскопия. Для первичной оценки морфологии получаемых волокон использовали оптический микроскоп ZEISS Axio Imager.
Сканирующая электронная микроскопия. Морфологию образцов исследовали с помощью сканирующего электронного микроскопа FEI Versa 3D DualBeam в режиме высокого вакуума. Изображения получены с помощью детектора вторичных электронов при ультранизком ускоряющем напряжении 1 кВ.
Гель-проникающая хроматография (ГПХ). Для контроля молекулярно-массовых характеристик раствора ГК в NH3 ⋅ H2O (10%) и ДМФА была проведена ГПХ на хроматографической системе «Knauer» с использованием рефрактометрического детектора и колонки PL aquagel-OH, подходящей для исследования образцов с молекулярной массой от 6000 до 10 000 000 Да. Исследования проводили при 40°С и скорости потока элюента 1 мл/мин. Для исследования готовили растворы образцов ГК в воде с концентрацией 3 мг/мл.
РЕЗУЛЬТАТЫ
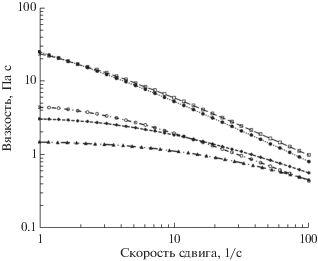
Исследование реологических свойств растворов ГК. Вязкость растворов является важнейшим параметром в методе электроформования, определяющим в конечном итоге производительность процесса, диаметр и морфологию получаемых волокон [18, 19]. Для проведения реологических исследований были приготовлены растворы на основе гиалуроната натрия концентрацией от 1 до 2.5% в различных растворителях. Как известно, вязкость растворов существенно зависит от молекулярной массы полимера [20] и межмолекулярного взаимодействия [7]. Поэтому неудивительно, что вязкость 1%-ного раствора ГК в воде составила ~25 Па с (рис. 2), что вызвано образованием водородных связей между ацетамидогруппой –NH и карбоксилатной группой, а также гидродинамическим размером молекулярного клубка ГК (~ от 210 до 410 нм) [16, 17].
Рис. 2.
Зависимость вязкости 1%-ного раствора гиалуроната натрия от состава растворителя: ● – H2O; □ – NH3 ⋅ H2O (33%) + N-метилпирролидон (2:1); ○ – NH3 ⋅ H2O (33%) + диметилформамид (2:1); ♦ – NH3 ⋅ H2O (10%) + N-метилпирролидон (2:1);▲ – NH3 ⋅ H2O (10%) + диметилформамид (2:1).
Для разрушения водородных связей проводили защелачивание растворов с помощью гидрата аммиака различной концентрации, а для снижения поверхностного натяжения и электропроводности вводили апротонные растворители ДМФА или НМП [21]. Измеренные значения pH и электропроводности приготовленных растворителей представлены в табл. 1.
Таблица 1.
Зависимость pH и электропроводности от состава растворителей
| Состав растворителя | pH | Электро-проводность, мкСм/см |
|---|---|---|
| NH3 ⋅ H2O (33%) + НМП (2: 1) | 13 | 271 |
| NH3 ⋅ H2O (33%) + ДМФА (2: 1) | 12.9 | 783 |
| NH3 ⋅ H2O (33%) | 12.83 | 523 |
| NH3 ⋅ H2O (10%) + НМП (2: 1) | 12.22 | 274 |
| NH3 ⋅ H2O (10%) + ДМФА (2: 1) | 12.15 | 1241 |
| NH3 ⋅ H2O (10%) | 12.13 | 1192 |
| ДМФА | 4.8 | |
| НМП | 1.8 |
Как и следовало ожидать, с увеличением содержания аммиака возрастает pH растворов. Интересно отметить, что электропроводность растворов снижается только при добавлении НМП, а при введении ДМФА она, наоборот, возрастает. После добавления ГК в состав раствора его электропроводность закономерно увеличивается [13]. Из рис. 2 видно, что вязкость щелочных растворов ГК меньше, чем вязкость растворов ГК в воде. Наименьшей вязкостью обладают растворы ГК в NH3 ⋅ H2O (10%) и ДМФА (2:1). При этом показатель pH для этого растворителя меньше, чем для остальных сред (табл. 1). Полученные результаты, по всей видимости, можно объяснить тем, что, с одной стороны, при увеличении pH разрушается сетка водородных связей, а с другой стороны, возрастает гидродинамический размер клубка ГК вследствие накопления на поверхности цепи отрицательного заряда. На основе приведенных результатов для приготовления прядильных растворов с приемлемой вязкостью следует использовать смесь 10–33%-ного гидрата аммиака с ДМФА. Отметим, что полученные выводы отличаются от опубликованных ранее результатов, в которых для приготовления растворов использовали только 33%-ный гидрат аммиака в ДМФА (2:1) [13].
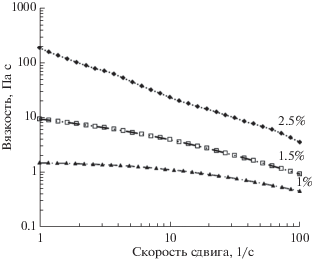
Как видно из рис. 3, при увеличении концентрации ГК с 1 до 2.5% вязкость раствора в 10%-ном гидрате аммиака в ДМФА возрастает более чем в 100 раз, достигая таким образом недостижимых для обычных шприцевых насосов давления для проталкивания поршня. Поэтому для успешного электроформования растворов ГК разработали специальную прядильную ячейку (рис. 1), рассчитанную на высокие давления, что позволило использовать тонкие капилляры (с диаметром выходного отверстия 150–200 мкм), позволявшие, как показывают исследования, изменять реологические свойства прядильного раствора (за счет скорости сдвига) без разрыва цепи ГК [7]. Рассчитать скорость сдвига при движении раствора ГК в капилляре можно по формуле $\dot {\gamma } = 4Q{\text{/}}\pi {{R}^{3}}$, где Q – объемный расход, а R – радиус капилляра. С учетом использованных параметров формования максимальная скорость сдвига у стенки капилляра составляет ~2000–3000 с–1. В результате создается преимущественная ориентация макромолекул, существенным образом влияющая на процесс растяжения струи в электрическом поле, а вязкость 1%-ного раствора снижается до 50–70 мПа с. Кроме того, проведенные ранее исследования показывают, что уменьшение размеров поперечного сечения капилляра благоприятно сказывается на процессе волокнообразования, так как способствует растяжению струи и снижению диаметра получаемых волокон [22, 23].
Рис. 3.
Зависимость вязкости растворов гиалуроната натрия в NH3 ⋅ H2O (10%) + диметилформамид (2:1) от концентрации и скорости сдвига.
Исследование процесса электроформования растворов ГК. Основной трудностью в процессе формования растворов ГК является их высокая вязкость, препятствующая формированию устойчивой заряженной струи в электрическом поле. Высокая электропроводность растворов ГК должна способствовать преодолению вязких сил. При этом существует риск возникновения перезарядки осажденного волокнистого слоя, что приведет к его вспуханию и вытяжению навстречу дрейфующим волокнам.
Проведенные реологические исследования показали, что для приготовления прядильных растворов следует использовать 10–33%-ный гидрат аммиака в ДМФА (рис. 1). В этом случае получим наименее вязкий прядильный раствор. Однако использование концентрированных растворов гидрата аммиака приводит к деградации ГК. Поэтому для дальнейших исследований целесообразно было продолжить работу с 10%-ным гидратом аммиака в ДМФА и НМП, так как эти растворители имеют более низкое поверхностное натяжение по сравнению с водой при приемлемой вязкости растворов на их основе. Кроме того, было проведено исследование процесса электроформования ГК из водных растворов с различной электропроводностью для определения ее влияния на процесс волокнообразования.
Для исследования процесса электроформования из воды приготовили серию растворов с различным содержанием NaCl. Полученные результаты представлены на рис. 4.
Рис. 4.
Микрофотографии волокон, полученных из раствора ГК в воде в зависимости от содержания NaCl: 0 (а), 0.0004 (б), 0.004 г/мл (в).
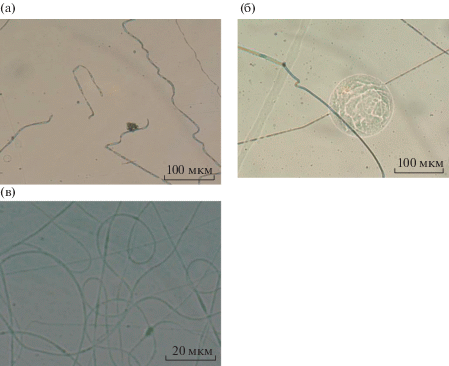
Как видно на рис. 4а, при отсутствии соли в растворе ГК его электропроводящих свойств не хватало для инициирования стабильного процесса электроформования. На приемном электроде можно было наблюдать как волокна с дефектами в виде перетяжек, так и их обрывки. Процесс формования шел нестабильно, периодически прекращаясь. К сожалению, при дальнейшем увеличении электропроводности (рис. 4б, 4в) решить данную проблему не удалось. При таком режиме формования волокнистого материала на его поверхности постоянно присутствовали следы дефектов от капель. Тем не менее отметим снижение диаметра полученных волокон до ~300–200 нм.
Для преодоления факторов, препятствующих проведению электроформования из растворов ГК в 10%-ном гидрате аммиака в ДМФА, потребовалось приложить напряжение выше 29 кВ и сократить межэлектродное расстояние до 20 см. Кроме того, для успешного формования ГК в состав прядильных растворов вводили добавку высокомолекулярного ПЭО. Как показали исследования, в случае введения в состав раствора ПЭО менее 1.1% процесс электроформования превращался в электрораспыление либо сопровождался получением волокон, содержащих дефекты в виде капель из-за недостатка волокнообразующих свойств. Поэтому для получения бездефектных волокон концентрацию ПЭО в растворе повысили до 1.2% (рис. 5).
Рис. 5.
Микрофотографии волокон, полученных из раствора ГК концентрацией 1.5% с добавкой ПЭО 1.1 (а) и 1.2% (б).

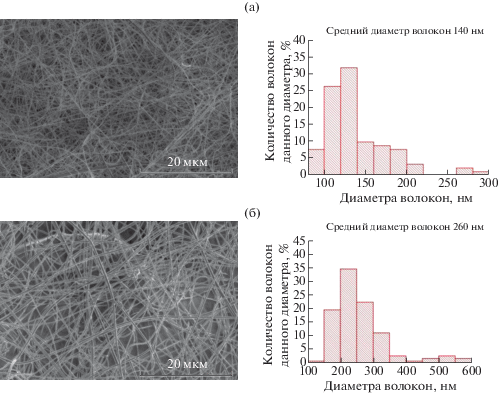
Вязкость прядильных растворов является существенным параметром, определяющим морфологию изготовленных волокнистых материалов. Как видно из рис. 6, с увеличением концентрации ГК в растворе от 1.5 до 2.5% средний диаметр волокон возрастает с 140 до 260 нм.
Рис. 6.
Гистограммы распределения волокон по размерам, рассчитанные из анализа микрофотографий нетканых материалов, полученных из раствора ГК концентрацией 1.5 (а) и 2.5% (б).
Несмотря на стабильный процесс электроформования растворов ГК из смеси гидрата аммиака и НМП, на приемном электроде можно было наблюдать перезарядку волокнистого слоя. Вероятно, вследствие малой интенсивности испарения НМП с поверхности струи (температура кипения НМП 202°С) полученные волокна содержали остаточный растворитель, существенно увеличивающий их проводимость. Результаты представлены на рис. 7.
Рис. 7.
Микрофотографии волокон диаметром от 2 до 8 мкм, полученных из раствора ГК концентрацией 1.5% в 10%-ном гидрате аммиака и НМП (а), в сравнении с волокнами диаметром от 100 до 200 нм, полученными из 10%-ного гидрата аммиака и ДМФА (б).

Диаметр волокон, полученных из растворов на основе НМП (рис. 7а), существенно больше размеров волокон, изготовленных из растворов на основе ДМФА (рис. 7б), что может быть связано с увеличением вязкости прядильного раствора, а также уменьшением электропроводности при замене ДМФА на НМП.
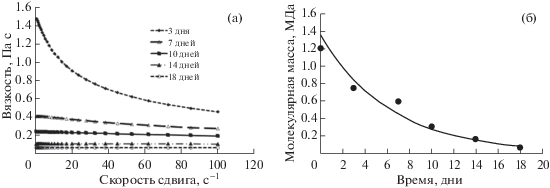
Исследование процесса деградации ГК. Деградация ГК в организме может происходить по двум причинам: ферментативным (гиалуронидазы) и окислительным за счет активных форм кислорода [6]. Химические методы деградации ГК включают в себя кислотный и щелочный гидролиз, а также окислительную деградацию. Разложение ГК в щелочных условиях происходит в две стадии вследствие случайного разрыва гликозидных связей [24]. Как видно из рис. 8, начальная вязкость (1.47 Па с) 1%-ного раствора ГК в среде NH3 ⋅ H2O (10%) и ДМФА (2:1) через 7 дней снижается примерно в 4 раза, а в течение 18 дней уменьшается до ~0.05 Па с, что соответствует значительному падению молекулярной массы – 95%. Таким образом, с биологической точки зрения использовать формовочный раствор для получения нановолокон необходимо в течение первых двух дней после его приготовления.
ЗАКЛЮЧЕНИЕ
Водные растворы ГК обладают чрезвычайно высокой вязкостью, что препятствует проведению электроформования. Реологические исследования позволили определить состав растворителя (NH3 ⋅ H2O (10–33%) + диметилформамид (2:1)), позволяющий снизить начальную ньютоновскую вязкость прядильных растворов. Кроме того, предложена новая схема формования ГК, которая заключается в продавливании ее высоковязких растворов под давлением через коническую фильеру с малым сечением. При прохождении растворов ГК через такую фильеру вследствие неньютоновского поведения снижается их вязкость, что также способствует успешному процессу электроформования. В результате использования реологических данных и предложенной схемы формования удалось получить нановолокнистый материал на основе ГК с диаметром волокна от 100 до 300 нм. Исследование кинетики деградации ГК позволило определить оптимальные сроки использования формовочных растворов ГК после их приготовления.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований и Правительства города Москвы (грант № 19-33-70071 мол_а_мос).
Список литературы
Li Y., Xiao Y., Liu C. // Biomaterials. 2010. V. 31. P. 4639. https://doi.org/10.1021/acs.chemrev.6b00654
Fullana M.J., Wnek G.E. // Drug Deliv. Transl. Res. 2012. V. 2. P. 313. https://doi.org/10.1007/s13346-012-0087-x
Wu T., Zheng H., Chen J. et al. // J. Mater. Chem. B. 2017. V. 5. P. 139. https://doi.org/10.1039/C6TB02484J
Llopis-Hernandez V., Cantini M., Gonzalez-Garcia C., Salmeron-Sanchez M. // Int. Mater. Rev. 2014. V. 60. P. 245. https://doi.org/10.1179/1743280414Y.0000000049
Arnal-Pastor M., Martínez Ramos C., Pérez Garnés M. et al. // Mater. Sci. Eng. C. 2013. V. 33. P. 4086. https://doi.org/10.1016/j.msec.2013.05.058
Cyphert J.M., Trempus C.S., Garantziotis S. // Int. J. Cell Biol. 2015 V. 7. P. 563818. https://doi.org/10.1155/2015/563818
Maleki A., Kjøniksen A.-L., Nyström B. // Polym. Bull. 2007. V. 59. P. 217. https://doi.org/10.1007/s00289-007-0760-2
Fallacara A., Baldini E., Manfredini S., Vertuani S. // Polymers-Basel. 2018. V. 10. P. 701. https://doi.org/10.3390/polym10070701
Gracaa M.F.P., Miguela S.P., Cabral C.S.D., Correia I.J. // Carbohyd. Polym. 2020. V. 241. P. 116364. https://doi.org/10.1016/j.carbpol.2020.116364
Pieri A.De, Byerley A.M., Musumeci C.R. et al. // Spine. 2020. V. 3. P. e1117. https://doi.org/10.1002/jsp2.1117
Abrigo M., McArthur S.L., Kingshott P. // Macromol Biosci. 2014. V. 14. P. 772. https://doi.org/10.1002/mabi.201300561
Liaoa Y., Loha Ch.-H., Tiana M. et al. // Prog. Polym. Sci. 2018. V. 77. P. 69. https://doi.org/10.1016/j.progpolymsci.2017.10.003
Brenner E.K., Schiffman J.D., Thompson E.A. et al. // Carbohyd. Polym. 2012. V. 87. P. 926. https://doi.org/10.1016/j.carbpol.2011.07.033
Pabjańczyk-Wlazło E., Król P., Krucińska I. et al. // Adv. Polym. Technol. 2018. V. 37. P. 1929. https://doi.org/10.1002/adv.21851
Pabjańczyk-Wlazło E., Krucińska I., Chrzanowski M. et al. // Fibres Text. East. Eur. 2017. V. 25. P. 45. https://doi.org/10.5604/12303666.1237225
Cowman M., Schmidt T.A., Raghavan P., Stecco A. // F1000Res . 2015. V. 4. P. 622. https://doi.org/10.12688/f1000research.6885.1
Gatej I., Popa M., Rinaudo M. // Biomacromolecules. 2005. V. 6. P. 61. https://doi.org/10.1021/bm040050m
Demir M.M., Yilgor I., Yilgor E., Erman B. // Polymer. 2002. V. 43. P. 3303. https://doi.org/10.1016/S0032-3861(02)00136-2
Mit-uppatham C., Nithitanakul M., Supaphol P. // Macromol. Chem. Phys. 2004. V. 205. P. 2327. https://doi.org/10.1002/macp.200400225
Gupta P., Elkins C., Long T.E., Wilkes G.L. // Polymer. 2005. V. 46. P. 4799. https://doi.org/10.1016/j.polymer.2005.04.021
Li J., He A., Han Ch.C. et al. // Macromol. Rapid Commun. 2006. V. 27. P. 114. https://doi.org/10.1002/marc.200500726
Esmaeilirad Ah., Ko J., Vrukosuyev M. et al. // Mater. Res. Express. 2017. V. 4 P. 015302. https://doi.org/10.1088/2053-1591/4/1/015302
Kong Ch.S., Lee S.G., Lee S.H. et al. // J. Macromol. Sci. B. 2011. V. 50. P. 528. https://doi.org/10.1080/00222341003784386
Stern R., Kogan G., Jedrzejas M.J., Šoltés L. // Biotechnol. Adv. 2007. V. 25. P. 537. https://doi.org/10.1016/j.biotechadv.2007.07.001
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии