

Нефтехимия, 2019, T. 59, № 4, стр. 477-482
Использование нуклида 33S для определения дисперсности частиц сульфидной фазы MoS2, нанесенной на мезопористый оксид кремния
А. Г. Попов 1, *, А. И. Никифоров 1, В. Л. Сушкевич 1, И. А. Тябликов 1, И. И. Иванова 1, 2
1 Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: andreygpopov@yahoo.com
Поступила в редакцию 11.09.2018
После доработки 25.02.2019
Принята к публикации 14.03.2019
Аннотация
Предложен новый подход для определения дисперсности частиц сульфидной фазы катализаторов гидроочистки. Метод включает в себя сульфидирование катализатора сульфидом водорода, содержащим нуклид 33S, проведение реакции гидрообессеривания тиофена и на заключительной стадии определение изотопного состава серы на катализаторе комбинацией методов термопрограмированного окисления и масс-спектрометрии (ТПО-МС). Показано, что в ходе гидрообессеривания часть наиболее лабильной серы 33S на гранях гексагональных призм MoS2 замещается на 32S из тиофена, и степень этого замещения можно определить методом ТПО-МС. Обнаружено, что с уменьшением размера частиц сульфидной фазы, которое подтверждается методом просвечивающей электронной микроскопии (ПЭМ), увеличивается доля лабильной серы, способной к замещению в ходе гидрообессеривания.
Гидрообессеривание является одним из наиболее важных процессов нефтепереработки с тоннажем около 4 млрд т в год [1]. Поскольку требования к содержанию серы в топливе постоянно повышаются, возникает необходимость в развитии новых, более эффективных катализаторов и технологий гидрообессеривания [2]. Положение осложняется тем, что в гидрообессеривание вовлекают все более тяжелое сырье.
Одним из путей повышения эффективности катализаторов гидрообессеривания является увеличение дисперсности их сульфидной фазы. Для определения характеристки дисперсности катализатора используют метод ПЭМ, который позволяет напрямую определять размер частиц сульфидной фазы [3–5]. К недостаткам метода можно отнести то, что это локальный метод. Поэтому для определения среднего размера частиц требуется усреднение по большому объему экспериментальных данных. Кроме того, метод требует дорогостоящего оборудования.
В настоящей работе мы предлагаем использовать для определения дисперсности сульфидных систем альтернативный подход, основанный на использовании нуклида серы 33S. Применение изотопов серы для характеристики сульфидных катализаторов в литературе известно [6–8]. Благодаря использованию радиоактивного нуклида 35S при исследовании реакции гидрообессеривания дибензотиофена в системах (Co, Ni)–MoS2 удалось доказать обмен между серой катализатора и сырья в ходе гидрообессеривания [6]. Однако использование радиоактивной серы сопряжено с целым рядом трудностей, среди которых низкая разрешающая способность метода, длительность экспериментов и повышенные требования к оборудованию для работы с радиоактивными веществами.
Таких недостатков лишен метод на основе 33S, который был использован для уточнения механизма гидрообессеривания тиофена [9]. Мы применили аналогичный подход для определения дисперсности сульфидных систем. Для этого была получена серия мезопористых SiO2, которые были использованы в качестве носителей катализаторов состава MoS2/SiO2 с разной дисперсностью сульфидной фазы. Применение оксида кремния вместо традиционно используемого оксида алюминия связано с возможностью варьирования в широких пределах площади его поверхности. Дисперсность модельной серии сульфидных катализаторов была охарактеризована двумя независимыми методами – с помощью ПЭМ и методом изотопного вытеснения 33S–32S. Было показано, что новый метод может быть использован для определения дисперсности сульфидных систем.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В рамках данного исследования использовали каталитическую композицию, состоящую из оксида молибдена MoO2, нанесенного на мезопористый SiO2, с различной удельной площадью поверхности и диаметром пор, посредством синтеза с использованием органических молекул-экспандеров, с последующим сульфидированием катализатора до MoS2/SiO2. MoS2 получали сульфидированием нанесенного оксида молибдена непосредственно в реакторе каталитической установки.
Для синтеза мезопористого SiO2 с экспандером, модифицирующим темплат, в течение суток при комнатной температуре перемешивали смесь 2 г Pluronic P123, 60 г дистиллированной воды, 9.6 мл концентрированной HCl. Далее в смесь добавляли 0.02 г NH4F и продолжали перемешивание в течение нескольких часов, после чего вводили в смесь рассчитанное количество триметилбензола (ТМБ), либо гептана. В полученный раствор медленно прикапывали 4.90 мл тетраэтоксисилана (ТЭОС) при постоянном перемешивании (n ТМБ (гептан)/n (ТЭОС) = 4–9). Далее производили перемешивание смеси в течение 24 ч при комнатной температуре. После этого проводили гидротермальную обработку образца в течение 24 ч при 110°C. Твердую фазу отделяли и промывали горячей дистиллированной H2O. После высушивания образов при 100°C вещество прокаливали при 550°C в течение 6 ч для удаления темплата.
Характеристики пористой структуры образцов полученных мезопористых SiO2 определяли с использованием метода низкотемпературной адсорбции-десорбции азота. Съемку изотерм проводили по стандартной методике на поромере ASAP 2010 (Micromeritics, США). Расчет характеристик пористой структуры проводили с использованием программного обеспечения прибора.
Молибден, в расчете 16 мас. % MoO3, наносили на мезопористый SiO2 путем пропитки материала носителя рассчитанным количеством раствора (NH4)6Mo7O24. Количество молибдена в конечном катализаторе подтверждали методом рентгенофлуоресцентного анализа на приборе Thermo Scientific ARL Perform`X с родиевой трубкой 3.5 кВт. Перед анализом навески образцов массой 0.15–0.20 г прессовали в таблетки с борной кислотой.
Сульфидирование MoO2/SiO2 сероводородом проводили в установке, состоящей из проточного реактора под атмосферным давлением с катализатором и реактора, предназначенного для in situ генерации H2S (рис. 1). Образующийся в реакторе сероводород с помощью помпы циркулировал через слой катализатора. В стандартном эксперименте 50 мг MoO2/SiO2 (фракция 0.25–0.5 мм) загружали в реактор. Смесь 8 мг 32S, либо 33S (атомная доля 33S = 99.18%) и 30 мг парафина помещали в генератор H2S. Перед экспериментом установка продували водородом, катализатор отжигали при 300°C. В процессе сульфидирования включалась помпа, настроенная на скорость 50 мл/мин. В системе был оставлен небольшой поток водорода с целью создания избыточного давления 1.1 бар. В этих условиях оксидный катализатор сульфидировался при 400°C при скорости потока H2 = 5 мл/мин. После окончания сульфидирования каталитическую установку продували водородом при скорости потока = 20 мл/мин в течение 1 ч.

Реакцию гидрообессеривания тиофена на MoS2/SiO2 вели при 360°C и скорости потока водорода = 5 мл/мин, который насыщался тиофеном в сатураторе при 25°C. Продукты реакции гидроочистки анализировали методами in situ масс-спектрометрии с использованием масс-спектрометра RGA200 (Stanford Research Systems) и газовой хроматографии на приборе Кристалл 5000, колонка Al2O3/KCl (50 м). Селективности продуктов реакции десульфуризации рассчитывали по уравнению:
После гидрообессеривания тиофена на катализаторе в течение 1 ч, каталитическую установку охлаждали до комнатной температуры при постоянной продувке водородом. Отработанный катализатор подвергали ТПО смесью 3% O2 в He со скоростью нагрева 10°C/мин и скоростью газового потока 30 мл/мин. Продукты ТПО катализатора анализировались in situ МС.
Массовую долю лабильной серы в образцах ωSлаб определяли по кривым выделения 33SO2 и 32SO2 в ходе ТПО-МС анализа по следующей формуле:
Размер частиц MoS2 исследовали с помощью метода ПЭМ на электронном микроскопе FEI Tecnai Osiris с ускоряющим напряжением 200 кВ.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Путем добавления в ходе синтеза экспандеров (н-гептана или мезитилена) были получены образцы мезопористого SiO2 с различными текстурными свойствами. Характеристики полученных образцов представлены в табл. 1. Можно видеть, что удельная площадью поверхности образцов варьировалась в широких пределах от 130 до 800 м2/г.
Таблица 1.
Свойства образцов мезопористого SiO2
| Образец | Sуд. пов-ти, м2/г | Vмикропор, см3/г | Vпор, см3/г | Dпор, А |
|---|---|---|---|---|
| Mo-SiO2-130 | 130 | 0.002 | 0.46 | 100 |
| Mo-SiO2-200 | 200 | 0.005 | 0.91 | 160 |
| Mo-SiO2-250 | 250 | 0.007 | 1.23 | 200 |
| Mo-SiO2-380 | 380 | 0.014 | 0.91 | 130 |
| Mo-SiO2-510 | 510 | 0.020 | 0.97 | 140 |
| Mo-SiO2-610 | 610 | 0.045 | 1.27 | 100 |
| Mo-SiO2-720 | 720 | 0.031 | 1.06 | 110 |
| Mo-SiO2-800 | 800 | 0.005 | 1.10 | 80 |
Синтезированные образцы были использованы в качестве носителей для катализаторов гидрообессеривания состава MoS2/SiO2. Для приготовления серии катализаторов одинаковое количество прекурсора молибдена наносили на мезопористый SiO2 с различной удельной площадью поверхности. Количество нанесенного молибдена была подтверждено данными РФА. Основные характеристики полученных катализаторов отражены в табл. 2.
Сравнение среднего размера частиц сульфидной фазы по данным ПЭМ позволяет сделать вывод, что, как и предполагалось, дисперсность сульфидной фазы уменьшается с падением удельной площади поверхности носителя. Размер частиц MoS2 изменялся в пределах от 2.9 до 9.5 нм, что выше, чем для сульфидных систем на основе оксида алюминия [10]. Вероятно, это связано с более слабым взаимодействием оксида кремния с активной фазой. Кроме того, катализатор с самой низкой удельной поверхностью носителя Mo-SiO2-130 показал более сложное бимодальное распределение частиц MoS2 по размеру.
Различия в дисперсности сульфидной фазы нашли свое отражение в катализе. На рис. 2 показано, как увеличение удельной площади поверхности исходного носителя влияло на конверсию тиофена. Рост конверсии хорошо согласуется с данными ПЭМ об уменьшении размера частиц активной фазы с увеличением удельной площади поверхности носителя. Выход графика зависимости на плато при большой поверхности носителя, вероятно, связан с тем, что достигается минимальный предел размера частиц MoS2.
Рис. 2.
Зависимость конверсии тиофена от площади удельной поверхности носителя для катализатора гидрообессеривания.

Таким образом, была получена серия образцов MoS2/SiO2 одинакового состава, но с разным размером частиц активной фазы. Эта серия была использована в качестве модельной в экспериментах по изотопному вытеснению 33S–32S в ходе гидрообессеривания тиофена.
Сульфидирование образцов катализаторов проводили непосредственно перед реакцией с помощью H233S, который генерировали из элементной серы 33S и парафина. Методика эксперимента аналогична описанной в работе [9]. После сульфидирования проводили реакцию гидрообессеривания тиофена с контролем продуктов с помощью масс-спектрометра. В ходе реакции наряду с продуктами разложения тиофена (бутаны, бутилены, H2S) наблюдали выделение изотопного H233S, который мог образоваться только за счет 33S сульфидной фазы. Таким образом, можно предположить, что в ходе гидрообессеривания тиофена происходит замещение серы сульфидной фазы на серу, которая высвободилась при разложении тиофена. В данной работе мы не изучали механизм такого замещения, однако на основе литературных данных [11, 12] можно сделать вывод, что процесс идет по пути изотопного вытеснения, а не изотопного обмена. Наличие такого замещения серы было зафиксировано также в работе [9] для сульфидов молибдена, нанесенных на оксид алюминия. В дальнейшем способную к замещению серу мы будем называть лабильной. Предполагается, что лабильная сера расположена на боковых гранях гексагональных призм MoS2. Реакцию прекращали через 60 мин после начала, когда выделение H233S прекращалось, то есть вся способная к обмену 33S заместилась на 32S.
Катализаторы после реакции подвергали ТПО, контролируя выделение оксида серы SO2 с помощью масс-спектрометра. Результаты типичных экспериментов ТПО-МС представлены на рис. 3. Для образца Mo-SiO2-720 на кривой выделения SO2(33) можно видеть основной максимум при температуре 330°С (рис. 3д). Небольшой пик при 430°С, возможно, связан с разложением поверхностных сульфатов, которые могут образовываться на начальной стадии окисления. На кривой выделения SO2 также обнаруживается один основной пик с максимумом при 296°С, то есть ниже, чем для 33SO2. Это указывает на более легкое окисление серы 32S, которая оказалась на катализаторе в результате вытеснения 33S серой 32S тиофена в ходе его гидрообессеривания. Для Mo-SiO2-720 доля лабильной серы составила 29% от всей серы на катализаторе (табл. 2).
Рис. 3.
Сравнение размеров частиц и данных ТПО-МС образцов Mo-SiO2-130 (а, в, д) и Mo-SiO2-720 (б, г, е). Данные ПЭМ (а, б); распределение частиц по размеру на основе ПЭМ (в, г); результаты ТПО-МС (д, е).

На образце Mo-SiO2-130 (рис. 3е) обнаружена более сложная картина окисления. На кривой 33SO2 наряду с пиком при 330°С присутствует также интенсивный пик при 386°С. На кривой SO2 также представлены два пика при 296 и 386°С. Наличие дополнительного высокотемпературного пика, вероятно, связано с наличием крупных частиц сульфидной фазы, которое подтверждается данными ПЭМ (рис. 3б и 3г). Такое бимодальное распределение частиц по размеру наблюдалось только для Mo-SiO2-130 с наименьшей удельной площадью поверхности. Согласно данным ПЭМ более мелкие частицы MoS2 имели размер 5.4 нм, а крупные – около 9.5 нм. Доля лабильной серы составила 20% для мелких частиц и всего 2% для крупных. По-видимому, поскольку лабильная сера располагается на боковых гранях гексагональных призм MoS2, при увеличении размера частиц доля этой поверхностной серы уменьшается.
На образце Mo-SiO2-130 (рис. 3е) обнаружена более сложная картина окисления. На кривой 33SO2 наряду с пиком при 330°С присутствует также интенсивный пик при 386°С. На кривой SO2 также представлены два пика при 296 и 386°С. Наличие дополнительного высокотемпературного пика, вероятно, связано с наличием крупных частиц сульфидной фазы, которое подтверждается данными ПЭМ (рис. 3б и 3г). Такое бимодальное распределение частиц по размеру наблюдалось только для Mo-SiO2-130 с наименьшей удельной площадью поверхности. Согласно данным ПЭМ более мелкие частицы MoS2 имели размер 5.4 нм, а крупные – около 9.5 нм. Доля лабильной серы составила 20% для мелких частиц и всего 2% для крупных. По-видимому, поскольку лабильная сера располагается на боковых гранях гексагональных призм MoS2, при увеличении размера частиц доля этой поверхностной серы уменьшается.
Для всей серии образцов была получена зависимость доли лабильной серы от размера частиц MoS2, которая представлена на рис. 4.
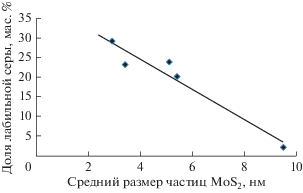
Можно видеть хорошую корреляцию между этими параметрами, что открывает путь к использованию 33S–32S вытеснения для характеристики сульфидных катализаторов. Этот метод наряду с ПЭМ дает представление о дисперсности сульфидной фазы. Однако ПЭМ является локальным методом и для корректного определения размера частиц требуется усреднение по большому количеству индивидуальных частиц. Обмен 33S–32S – метод интегральный и характеризует весь катализатор в целом. Кроме того, к преимуществам метода можно отнести то, что он в сочетании с химическим анализом на серу, напрямую определяет концентрацию лабильной серы в катализаторе, которая, в свою очередь, может влиять на каталитическую активность сульфидных систем.
Список литературы
Srivastava V.C. // RSC Advances. 2012. V. 2. № 3. P. 759.
European Standard for Gasoline EN 228/1999 and for Diesel EN 590/1999, 1999.
Breysse M., Geantet C., Afanasiev P., Blanchard J., Vrinat M. // Catalysis Today. 2008. V. 130. № 1. P. 3.
Hensen E.J.M., Kooyman P.V., Van der Meer Y., Van der Kraan A.M., De Beer V.H.J., Van Veen J.A. R., Van Santen R.A. // J. of Catalysis. 2001. V. 199. № 2. P. 224.
Topsøe H. // Applied Catalysis A: General. 2007. V. 322. P. 3.
Kabe T., Ishihara A., Qian W. // Catalysis Surveys from Asia. 1999. V. 3. № 1. P. 17.
Kogan V.M, Rozhdestvenskaya N.N., Korshevets I.K. // Applied Catalysis A: General. 2002. V. 234. № 1–2. P. 207.
Dobrovolszky M., Paal Z., Tétényi P. // Applied Catalysis A: General. 1996. V. 142. № 1. P. 159.
Sushkevich V.L., Popov A.G., Ivanova I.I. // Angewandte Chemie International Edition. 2017. V. 56. № 36. P. 10 872.
Nikulshin P.A., Ishutenko D.I., Mozhaev A.A., Maslakov K.I., Pimerzin A.A. // J. of Catalysis. 2014. V. 312. P. 152.
Kogan V.M. // Applied Catalysis A: General. 2002. V. 237. P. 161.
Kogan V.M. Isaguliants G.V. // Catalysis Today. 2008. V. 130. P. 243.
Дополнительные материалы отсутствуют.
Инструменты
Нефтехимия