

Нефтехимия, 2019, T. 59, № 5, стр. 601-606
Гидродеоксигенация подсолнечного масла на катализаторах NiMoS/B2O3–Al2O3
А. А. Непомнящий 1, Е. А. Булучевский 1, *, А. В. Лавренов 1
1 Институт проблем переработки углеводородов СО РАН
644024 Омск, Россия
* E-mail: bulu@ihcp.ru
Поступила в редакцию 05.03.2019
После доработки 05.03.2019
Принята к публикации 13.05.2019
Аннотация
Показана возможность использования боратсодержащего оксида алюминия в качестве носителя для катализатора гидродеоксигенации подсолнечного масла при 380°С, 4.0 МПа и массовой скорости подачи сырья 1 ч–1 с выходом жидких продуктов 81–85 мас. %. Установлено, что с повышением содержания оксида бора в катализаторе, за счет увеличения его кислотности, возрастает доля изоалканов в продуктах до 77–78%, а также увеличивается вклад реакций декарбоксилирования/декарбонилирования в образование продуктов.
Рост доли низкосортной нефти в общем объеме переработки с одновременным ужесточением экологических норм заставляют задуматься о поиске новых сырьевых источников для производства моторных топлив. В качестве одного из перспективных источников сырья рассматривается биомасса, главное достоинство которой – быстрая возобновляемость. Считается, что использование биомассы для получения топлив существенно снижает воздействие парниковых газов, образующихся при сгорании топлива, на климатические изменения [1].
Жиры, как компонент биомассы, содержащие в своем составе углеводородные фрагменты С15–С18, наиболее перспективны для производства дизельных топлив (ДТ). Биодизельные топлива второго поколения получают путем гидропереработки масложирового сырья. Отсутствие кислород-, азот- и серосодержащих соединений, непредельных и полиароматических углеводородов (УВ), а также высокие значения цетановых чисел обуславливают высокие эксплуатационные и экологические характеристики таких топлив не только по сравнению с традиционным ДТ, но и по сравнению с биодизельными топливами первого поколения – метиловыми эфирами жирных кислот.
На сегодняшний день промышленные технологии производства ДТ из масложирового сырья разработаны рядом зарубежных фирм, среди которых Neste Oil (Финляндия) [2], UOP/Eni (США, Италия) [3], Syntroleum (США) [4], Conoc-oPhillips (США, Ирландия) [5], PETROBRAS (Бразилия) [6], Haldor Topsøe (Дания) [7], Nippon Oil (Япония) [8], SK Energy (Южная Корея) [9]. В большей части таких процессов используются модифицированные схемы и традиционные катализаторы гидроочистки, в некоторых случаях сырье подвергается предва-рительному гидрированию [10]. В качестве продуктов получают смеси УВ С15–С18 с высоким содержанием н-алканов, главный недостаток которых – высокие температуры застывания, что делает невозможным их использование в качестве компонентов зимних ДТ [11]. Для улучшения низкотемпературных характеристик такие компоненты дополнительно подвергают изомеризационной депарафинизации на цеолитсодержащих катализаторах [2, 10, 12, 13]. Использование двухстадийной схемы переработки существенно удорожает процесс, а также приводит к снижению выхода жидких продуктов.
Попыткам реализации стадий гидродеоксигенации масложирового сырья и изомеризации образующихся н-алканов на одном бифункциональном катализаторе посвящено большое количество исследований. Такие катализаторы содержат гидрирующие металлические, оксидные, или сульфидные компоненты, нанесенные на кислотные подложки, свойства которых определяют изомерный состав, а, следовательно, и температуру застывания получаемых продуктов. Наиболее часто в качестве кислотных компонентов катализаторов исследуются материалы, содержащие цеолиты различных типов – SAPO-11 [14–16], SAPO-31 [17], МСМ-41 [18], SBA-15 [19], HY [20], Beta [21], ZSM-22 [16] и др. В некоторых работах описано использование анион-модифицированных оксидов металлов WOx/Al2O3 [22, 23], или Ce0.6Zr0.4O2 [24].
В настоящей работе в качестве катализатора гидродеоксигенации подсолнечного масла и изомеризации образующихся при этом алканов исследован никель-молибден-сульфидный катализатор на основе кислотного носителя B2O3–Al2O3 различного состава. Основная цель работы – исследование влияния содержания оксида бора в катализаторе на показатели процесса гидродеоксигенации и состав получаемых продуктов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление и исследование катализаторов. Боратсодержащие носители B2O3–Al2O3 (BA) готовили путем смешения псевдобемита промышленного производства (ЗАО “Промышленные катализаторы”, г. Рязань) с водными растворами борной кислоты (ч. д. а.) с последующими сушкой (120°С, 12 ч) и прокаливанием (550°С, 16 ч) [25]. Номинальное содержание оксида бора в катализаторах варьировалось от 5 до 30 мас. % (образцы BA-5–BA-30), посредством использования растворов борной кислоты с различной концентрацией.
Введение никеля и молибдена в катализатор осуществляли методом пропитки готового носителя (фракция 0.2–0.5 мм) растворами биметаллических цитратных комплексов никеля и молибдена под вакуумом. Пропиточные растворы готовили с использованием лимонной кислоты (ГОСТ 908-04, РЕАХИМ), (NH4)6Mo7O24 ⋅ 4H2O (ООО “ЦТ Лантан”) и Ni(CH3COO)2 ⋅ 4H2O (х. ч., РЕАХИМ). По окончанию пропитки образцы высушивали при 220°С в течение 2 ч. Мольное соотношение металлов Ni/(Ni + Mo) в активном компоненте катализатора составляло 0.33.
Содержание никеля, молибдена и оксида бора в катализаторах определяли методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой на спектрометре Varian 710-ES (Agilent Technologies).
Испытания катализаторов. В качестве сырья для процесса гидродеоксигенации использовали рафинированное подсолнечное масло производства ООО “МЭЗ Юг Руси”; содержание кислот С16 и С18 составляло 6.4 и 92.5 мас. % соответственно, суммарное содержание непредельных кислот составляло 89.4 мас. % [23].
Испытания катализаторов проводили на лабораторной установке проточного типа со стационарным слоем катализатора. Перед испытанием катализаторы сушили в токе водорода (300 мл/мин) при температуре 120°С в течение 5 ч. Сульфидирование катализаторов осуществляли в две ступени при 230 и 340°С при массовой скорости подачи сырья 2 ч–1 и соотношении H2 : сырье = 300 нм3 : м3. В качестве осерняющего агента использовали раствор диметилдисульфида в прямогонном бензине (0.6 мас. % в пересчете на S).
Процесс гидродеоксигенации подсолнечного масла проводили при температуре 380°С и давлении 4.0 МПа, массовой скорости подачи сырья 1 ч–1, соотношение H2 : масло = 2500 нм3 : м3. В реакционную смесь дополнительно дозировали сероводород в количестве 0.4% от объема подаваемого водорода.
Жидкие и газообразные продукты процесса разделяли в сепараторе при атмосферном давлении и 20°С. Состав газообразных продуктов определяли в режиме on line с использованием двухканального газового хроматографа Хромос ГХ-1000. Неорганические компоненты газовой фазы (H2, H2S, CO, CO2 и Н2О) анализировали с применением двух насадочных колонок (длина 3 м, диаметр 4 мм), заполненных сорбентом Porapak R и активированным углем, в комбинации с детектором по теплопроводности. Углеводородные компоненты газовой фазы (С1–С4, С5+) анализировали с применением капиллярной колонки (J & W DB-1, длина 60 м) и пламенно-ионизационного детектора. Углеводородный состав стабильного жидкого продукта определяли с использованием газового хроматографа Хромос ГХ-1000, снабженного капиллярной колонкой (Restek Rtx-1, длина 100 м) и пламенно-ионизационным детектором. С целью определения полноты удаления кислорода и оценки группового состава получаемых продуктов жидкий углеводородный продукт также анализировали методом ЯМР на ядрах 1H и 13C (Bruker Avance 400, 400 МГц).
Все образцы катализаторов испытывали в течение 150 ч. Отбор проб осуществляли каждые четыре часа в течение первых суток, далее – через каждые 24 ч.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В табл. 1 представлены данные по фактическому содержанию никеля, молибдена и оксида бора (мас. %) в составе катализаторов. Как видно из представленных данных, фактическое содержание никеля и молибдена близко к расчетному. Содержание оксида бора несколько меньше расчетного для образца ВА-30, что может быть связано с возгонкой B2O3 при прокаливании носителя [25].
Таблица 1.
Химический состав катализаторов
| Образец | Содержание элементов, мас. % | |||||
|---|---|---|---|---|---|---|
| номинальное | фактическое | |||||
| Ni | Mo | B2O3 | Ni | Mo | B2O3 | |
| NiMo/BA-0 | 3.6 | 12.0 | 0.0 | 3.7 ± 0.2 | 12.1 ± 0.6 | 0.0 |
| NiMo/BA-5 | 3.6 | 12.0 | 5.0 | 3.8 ± 0.1 | 12.2 ± 0.4 | 5.1 ± 0.1 |
| NiMo/BA-10 | 3.6 | 12.0 | 10.0 | 3.7 ± 0.1 | 12.1 ± 0.1 | 9.9 ± 0.4 |
| NiMo/BA-15 | 3.6 | 12.0 | 15.0 | 3.6 ± 0.1 | 11.9 ± 0.2 | 15.3 ± 0.7 |
| NiMo/BA-20 | 3.6 | 12.0 | 20.0 | 3.7 ± 0.1 | 12.2 ± 0.1 | 19.8 ± 0.3 |
| NiMo/BA-25 | 3.6 | 12.0 | 25.0 | 3.7 ± 0.2 | 12.2 ± 0.2 | 25.0 ± 0.2 |
| NiMo/BA-30 | 3.6 | 12.0 | 30.0 | 3.9 ± 0.2 | 12.6 ± 0.5 | 28.7 ± 0.9 |
Анализ жидких продуктов процесса методом ЯМР-спектроскопии на ядрах 1H показал, что в спектрах всех продуктов присутствуют исключительно пики с химическими сдвигами, характерными для алифатических протонов. Пики с химическими сдвигами, характерными для протонов, входящих в состав кислородсодержащих молекул, непредельных, или ароматических УВ в спектрах не обнаружены. Таким образом, можно констатировать, что на всех испытанных катализаторах протекает полная деоксигенация сырья, а также исчерпывающее гидрирование двойных связей в жирнокислотных остатках непредельных кислот, содержащихся в масле.
Основные продукты превращения подсолнечного масла – жидкие УВ C5+, газообразные вещества и вода. Выход жидких УВ на поданое масло на всех катализаторах (рис. 1а) находится в пределах интервала значений теоретического выхода, который, с учетом состава исходного масла, составляет 81% при удалении всего кислорода в виде СО и СО2 и 86% при удалении всего кислорода в виде воды. При этом катализаторы с содержанием оксида бора 10% и менее обеспечивают более высокие значения выхода жидких продуктов, чем образцы с высоким содержанием модификатора. Особенно сильно это проявляется в первые 24 ч эксперимента, где разница выходов на образцах NiMoS/BA-0 и NiMoS-BA-30 может составлять до 4%. Это объясняется более высокой кислотностью катализаторов с содержанием оксида бора 15–30 мас. % [26]. Рост кислотности катализаторов способствует протеканию, во-первых, реакций декарбоксилирования/декарбонилирования, снижая выход углеводородов С16 и С18 [27, 28], во-вторых – вторичных реакций крекинга образующихся алканов. Так на рис. 1б показано, что выход УВ С10+ на образцах, содержащих 15–30 мас. % оксида бора, в первые часы эксперимента не превышает 72–74%, при этом выход продуктов крекинга – УВ С5–С9 может составлять 10–12%. С увеличением времени работы катализатора, интенсивность протекания реакций крекинга снижается, и выход УВ С10+ выходит на стационарный уровень 78–82% после первых суток испытаний. Такой “период приработки” катализатора, вероятнее всего, связан с дезактивацией наиболее сильных кислотных центров катализатора, на которых протекает крекинг алканов.
Рис. 1.
Зависимости выхода (Y) жидких продуктов (а) и углеводородов С10+ (б) от времени работы катализатора для образцов с содержанием B2O3 0–30 мас. %.
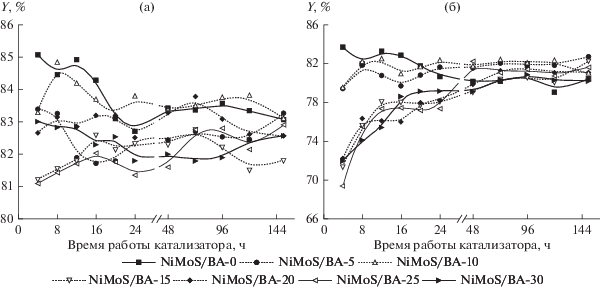
Следует отметить, что в составе продуктов, полученных на всех катализаторах, присутствуют УВ С21+, хотя в сырье не содержится жирных кислот с числом атомов углерода более 18. Максимальный выход УВ С21+ наблюдается на образце NiMoS/Al2O3 и составляет 3–3.5%, практически не изменяясь в течение всего времени испытаний. На образцах с более высокой кислотностью – NiMoS/BA-25 и NiMoS-BA-30 выход С21+ не превышает 0.5% в первые часы эксперимента и увеличивается до 1.5–2% при времени работы катализатора более 100 ч. Вероятнее всего, образование таких УВ связано с изначальным присутствием в сырье олигомеров непредельных жирных кислот, которые подвергаются гидрированию, а затем крекингу на испытываемых образцах катализаторов.
Общий выход газообразных продуктов (рис. 2а, 2б) зависит от типа используемого носителя, и увеличивается с повышением его кислотности. Для немодифицированного образца он не превышает 7–8%, тогда как для образцов с высокой кислотностью, содержащих 25–30 мас. % B2O3, выход газов может достигать 15–16%. С течением времени эксперимента этот показатель практически не изменяется. Усредненный по времени выход компонентов газа – метана, этана, пропана, бутана, CO и CO2 в зависимости от содержания оксида бора представлен на рис. 2б. Как видно из представленных данных, выход пропана составляет 4.7–5.0 мас. % для всех образцов, что соответствует теоретическому значению выхода, рассчитанному для случая полного гидрирования всех глицерольных остатков – 5.0 мас. %. Сопоставления выходов пропана и суммарного выхода жидких увуглеводородов, показало, что их молярное соотношение для всех катализаторов составляет 0.33, что соответствует молярному соотношению глицерина и жирнокислотных остатков в маслах. Таким образом, пропан в ходе процесса образуется исключительно при гидрировании глицерольных остатков.
Рис. 2.
Зависимость выхода газа от времени работы катализатора для образцов с содержанием B2O3 0–30 мас. % (а); зависимость усредненного по времени состава газов от содержания B2O3 в катализаторах (б).
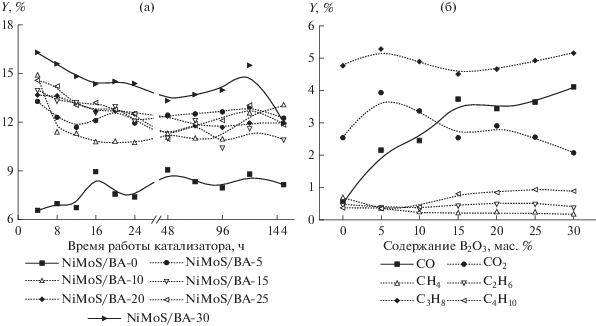
В условиях эксперимента возможно протекание реакций восстановления CO2 до CO водородом, гидрирования CO и СО2 до метана и реакция окисления CО присутствующей в реакционной среде водой, поэтому для оценки интенсивности протекания реакций декарбоксилирования/декарбонилирования наиболее корректным является сопоставление суммарного мольного выхода компонентов C1 и мольного выхода углеводородов с нечетным количеством атомов углерода. Это соотношение находится в пределах 0.95–1.1 и близко к стехиометрическому, из чего можно сделать вывод о минимальном вкладе вторичных реакций гидрогенолиза и крекинга УВ в образование метана в процессе. Что касается соотношения выходов газов C1 в процессе, выход метана не превышает 0.5% и не зависит от кислотности образца. Выход монооксида углерода с ростом содержания бора в образцах возрастает, а выход CO2, напротив, снижается. Вероятнее всего, с изменением кислотности носителя меняется соотношение скоростей реакций декарбоксилирования и декарбонилирования, однако, причины такого явления в литературе, по всей видимости, до сих пор не описаны.
На рис. 3а приведены зависимости содержания изоалканов в продуктах от времени работы катализатора для испытанной серии образцов. Как видно из представленных данных, на немодифицированном катализаторе в начальный период времени массовая доля изоалканов в продуктах не превышает 12%, и в течение 12 ч снижается до 7–8%. Введение оксида бора существенно повышает активность катализатора в реакции изомеризации. Содержание разветвленных алканов в продукте возрастает, и для образцов с максимальной кислотностью носителя (при содержании B2O3 20–30 мас. %) может достигать 77–78% в начальные моменты времени. Однако, как и в случае немодифицированного катализатора, изомеризующая способность образцов постепенно снижается, и после 150 ч работы не превышает 25%. Таким образом, во время работы катализаторов происходит дезактивация кислотных центров.
Рис. 3.
Зависимости содержания изоалканов в продуктах (а) и массовой доли углеводородов с нечетным числом атомов углерода в продуктах C15–18 (б) от времени работы катализатора для образцов с содержанием B2O3 0–30 мас. %.
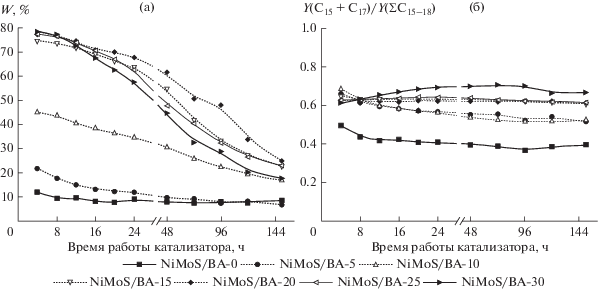
По массовой доле УВ с нечетным числом атомов углерода в продуктах можно оценить соотношение скоростей реакций деоксигенации, в которых кислород удаляется в виде воды и реакций декарбоксилирования/декарбонилирования. Как видно из рис. 3б, данный показатель зависит от кислотности носителя, и возрастает от 0.4 для NiMoS/BA-0 до 0.7 для NiMoS/BA-30. Эти данные хорошо согласуются как с общим снижением выхода жидких продуктов, так и с повышением общего выхода газа и суммарного содержания оксидов углерода в этом же ряду катализаторов. Таким образом, можно утверждать, что увеличение кислотности катализаторов путем модифицирования носителей оксидом бора не только увеличивает содержание изомеров в составе продуктов гидродеоксигенации масел, но и приводит к интенсификации реакций декарбоксилирования/декарбонилирования.
В результате проведенных исследований установлено, что гидродеоксигенация подсолнечного масла при температуре 380°С, давлении 4 МПа и массовой скорости подачи сырья 1 ч–1 на катализаторах NiMoS/B2O3–Al2O3 позволяет получить жидкие УВ дизельной фракции, не имеющие в своем составе серо- и кислородсодержащих соединений, с выходом 81–85%.
Введение 20–30 мас.% оксида бора в катализатор гидродеоксигенации масел позволяет получать смеси УВ дизельной фракции с массовой долей изоалканов в продуктах до 77–78%, однако дезактивация кислотных центров катализатора наступает в течение первых суток его работы. При этом катализаторы NiMoS/B2O3–Al2O3 обладают стабильностью в реакциях гидродеоксигенации 150 ч и более.
Работа выполнена в рамках государственного задания ИППУ СО РАН в соответствии с Программой фундаментальных научных исследований государственных академий наук на 2013-2020 годы по направлению V.46, проект № V.46.4.4 (номер госрегистрации в системе ЕГИСУ НИОКТР AAAA-A17-117021450098-2).
Список литературы
Al-Sabawi M., Chen J. // Energy Fuels. 2012. V. 26. № 9. P. 5373.
Laakkonen M., Myllyoja J., Toukoniitty Bl., Hujanen M., Saastamoinen A., Toivo A. Patent US № 20130090502. 2012.
Petry J.A., Marker T.L. Patent EP № 1728844. 2006.
Abhari R., Tomlinson L., Havlik P., Jannasch N. Patent US № 7968757. 2008.
Ghonasgi D.R., Sughrue L., Yao E.J., Xu X. Patent US № 7955401. 2007.
Gomes J.R., Zotin J. L., Pacheco M.E., Cotta Ferreira Da Silva R.M. Patent US № 20100270207. 2010.
Jonson E. // Energies. 2019. V. 12. № 2. P. 250.
Fujiyama Y., Okuhara T. Patent US № 6045690. 1997.
Ju S.J., Jeon H.J., Park J.M., Jeong I.Y., Kim G.R., Kang S.Y., Ryu J.W., Lee S.I., Kim D.W. Patent EP № 2628781. 2010.
Kalnes T.N., Marker T., Koers K.P., Shonnard D.R. // Biofuels Technology Q4. 2008. P. 7.
Šimácek P., Kubicka D., Šebor G., Pospíšil M. // Fuel. 2010. V. 89. № 3. P. 611.
Jakkula J., Aalto P., Niemi V., Kiiski U., Nikkonen J., Mikkonen S., Piirainen O. // WO 2004/022674 A1. 2004.
Jakkula J., Aalto P., Niemi V., Kiiski U., Nikkonen J., Mikkonen S., Piirainen O. // EP 1396531 B2. 2004.
Herskowitz M., Landau M.V., Reizner Y., Berger D. // Fuel. 2013. V. 111. P. 157.
Liu Q., Zuo H., Zhang Q., Wang T., Ma L. // Chin. J. of Catal. 2014. V. 35. № 5. P. 748.
Wang C., Tian Z., Wang L., Xu R., Liu Q., Qu W., Ma H., Wang B. // ChemSusChem. 2012. V. 5. № 10. P. 1974.
Kikhtyanin O.V., Rubanov A.E., Ayupov A.B., Echevsky G.V. // Fuel. 2010. V. 89. № 10. P. 3085.
Kubicka D., Bejblova M., Vlk J. // Top. Catal. 2010. V. 53. № 3–4. P. 168.
Duan J., Han J., Sun H., Chen P., Lou H., Zheng X. // Cat. Commun. 2012. V. 17. P. 76.
Zarchin R., Rabaev M., Vidruk-Nehemya R., Landau M.V., Herskowitz M. // Fuel. 2015. V. 139. P. 684.
Sousa F. P., Noemí Silva L., B. de Rezende D., A. de Oliveira L.C., Pasa V.M.D. // Fuel. 2018. V. 223. P. 149.
Janampelli S., Darbha S. // Catal. Today. 2018. V. 309. P. 219.
Непомнящий А.А., Булучевский Е.А., Лавренов А.В., Юрпалов В.Л., Гуляева Т.И., Леонтьева Н.Н., Талзи В.П. // Журн. прикладной химии. 2017. Т. 90. № 12. С. 1613.
Jeona K.W., Shima J.O., Choa J.W., Janga W.J., Naa H.S., Kima H.M., Leea Y.L., Jeonb B.H., Baec J.W., Roha H.S. // Fuel. 2019. V. 236. P. 928.
Карпова Т.Р., Булучевский Е.А., Лавренов А.В., Леонтьева Н.Н., Тренихин М.В., Гуляева Т.И., Талзи В.П. // Химия в интересах устойчивого развития. 2013. Т. 21. № 1. С. 61.
Юрпалов В.Л., Дроздов В.А., Карпова Т.Р., Лавренов А.В. // Химия в интересах устойчивого развития. 2017. Т. 25. № 1. С. 109.
Kubička D., Horáček J., Setnička M. Bulánek R., Zukal A., Kubičková I. // Appl. Catal. B: Environmental. 2014. V. 145. P. 101.
Qian E.W., Chen N., Gong S. // J. of Molecular Catalysis A: Chemical. 2014. V. 387. P. 76.
Дополнительные материалы отсутствуют.
Инструменты
Нефтехимия