

Нейрохимия, 2019, T. 36, № 1, стр. 14-23
Кофилин: молекулярно-клеточные функции и роль в функционировании нервной системы
Т. Ф. Ковалева 1, Н. С. Максимова 1, И. Ю. Жуков 1, В. И. Першин 1, 2, И. В. Мухина 1, 2, М. Р. Гайнуллин 1, 3
1 Приволжский исследовательский медицинский университет
Нижний Новгород, Россия
2 Национальный исследовательский Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
3 Norwegian PSC Research Center, Oslo University Hospital Rikshospitalet
Oslo, Norway
Поступила в редакцию 26.03.2018
После доработки 13.06.2018
Принята к публикации 08.05.2018
Аннотация
Актин-связывающие белки и, в частности, представители семейства актин-деполимеризующий фактор (ADF)/кофилин, участвуют в регуляции актинового цитоскелета в ответ на различные внутри- и внеклеточные сигналы. Последние исследования указывают на исключительную роль этой группы белков в развитии и функционировании нервной системы. В настоящем обзоре представлены последние данные о функциях кофилина в клетке и сигнальных путях, вовлеченных в его регуляцию. Особое внимание уделено исследованиям связи кофилина с динамикой актина в таких процессах, как контроль синаптической пластичности, апоптоз нейронов, нейровоспаление. Показаны молекулярные механизмы активации/инактивации кофилина и особенности структуры актина в нервных клетках при нейродегенерации на моделях in vitro и in vivo. Рассмотрены новые направления в изучении кофилина и связанных с ним белков как прогностических маркеров и терапевтических мишеней при диагностике и лечении заболеваний нервной системы.
ВВЕДЕНИЕ
Актин-связывающие белки участвуют в регуляции динамики актинового цитоскелета в ответ на различные внутри- и внеклеточные сигналы [1]. К этой группе относится семейство актин-деполимеризующий фактор (ADF)/кофилины. В клетках млекопитающих экспрессируются три формы кофилинов: актин-деполимеризующий фактор, также известный как дестрин (destrin, Actin-Depolymerizing Factor, ADF), кофилин 1 или немышечный кофилин (cofilin 1, CFL1) и кофилин 2 (мышечный кофилин, cofilin 2, CFL2) [2]. Кофилин-2 может существовать, по крайней мере, в двух изоформах из-за альтернативного сплайсинга одного гена CFL2. Одна из этих изоформ (CFL2b) присутствует в скелетных мышцах и сердце, а другая (CFL2a) обнаружена в различных тканях. ADF/кофилины – это белки с молекулярной массой 15–19 кДа, которые имеют значительное структурное сходство. Каждый из них состоит из домена ADF-H с несколькими дополнительными аминокислотными остатками, включая метионин на N-конце полипептидной цепи и около десяти аминокислотных остатков на С-конце (согласно базе белков UniProt) [3]. Основная функция белков семейства ADF/кофилины – регуляция структуры актинового цитоскелета клетки, сборки и разборки актиновых филаментов [4, 5]. Последние исследование свидетельствуют о роли кофилина в патогенезе заболеваний нервной системы [6, 7]. Такие нейродегенеративные заболевания, как болезнь Альцгеймера, Паркинсона и Гентингтона, спинальная мышечная атрофия, амиотрофический латеральный склероз, прионные болезни и делеционно дупликационные синдромы, называемые многофакторными болезнями, перекрываются на уровне сигнального каскада ремоделирования актин–кофилин [8]. Данные заболевания получили название болезни актинового цитоскелета, или кофилинопатии [2, 9, 10].
В обзоре рассмотрены функции и молекулярные механизмы регуляции уровня и активности кофилина и его роль в функционировании нервной системы в норме и патологии.
ФУНКЦИИ КОФИЛИНА
Динамика актина. Кофилин с одной стороны способствует фрагментации актиновых филаментов, с другой – накоплению коротких фрагментов, которые могут использоваться в качестве “затравки” для сборки новых филаментов актина. Это зависит от концентрации кофилина относительно актина и других актин-связывающих белков [11, 12]. Исследования in vitro показали, что низкое отношение кофилин : актин (менее 1%) приводит к фрагментации актиновых филаментов, при более высоких соотношениях (от 1 : 10 до 1 : 2) кофилин стабилизирует F-актин [12] (табл. 1). Сборка комплекса кофилин-актин в нединамические палочки (cofilin-actin rods) является важным механизмом регуляции уровня ATP в клетке [13]. Следует отметить, что другие актин-связывающие белки (актин-связывающий белок 1, Aip1; тропомиозины; кортактин; родственный актину белок 2, Arp2; коронины) могут усиливать или ослаблять эффекты кофилина на динамику актинового цитоскелета [11, 14–16].
Таблица 1.
Концентрационная зависимость различных механизмов регуляторного влияния кофилина по отношению к актину
| Параметры | Внутриклеточная концентрация кофилина | |
|---|---|---|
| низкая (менее 1%) | высокая (10–50%) | |
| Форма актина, с которой происходит связывание | Фибриллярный (F) | Глобулярный (G) |
| Влияние на олигомеризацию актина | Диссоциация на мономеры | Полимеризация |
| Молекулярно-физиологический эффект | Разборка актиновых филаментов | Сборка актиновых филаментов |
Транспорт актина в ядро и процесс транскрипции. Актин играет важную роль в ремоделировании хроматина, образовании гетерогенных ядерных рибонуклеопротеидных комплексов и экспрессии генов [17]. Актин регулирует такие ядерные процессы, как транскрипция и активация генов [18, 19]. Интересно отметить, что актин не имеет специфических сигнальных последовательностей NLS (Nuclear Localization Signal; сигнал ядерной локализации), которые в цитоплазме узнаются NLS-рецепторным комплексом, импортином, и необходимы для транспорта в ядро. Кофилин в отличие от актина имеет последовательность NLS [20]. Недавно был идентифицирован импортин 9, который транспортирует из цитоплазмы в ядро актин в комплексе с кофилином [21]. Ингибирование кофилина приводит к значительному снижению активности транскрипции, при этом восстановление уровня ядерного актина независимо от кофилина не влияет на активность транскрипции, что указывает на важность кофилина как участника данного процесса [22]. Кроме того, снижение уровня кофилина приводит к неконтролируемой сборке сократительных актиновых филаментов, что способствует ядерной деформации и потере жизнеспособности клеток [19].
Участие в процессе апоптоза. Транслокация кофилина из цитоплазмы в митохондрии приводит к открытию митохондриальной поры, выходу цитохрома с и активации апоптоза [23]. При этом высвобождение цитохрома с не требует связывания актина, что указывает на непосредственную роль кофилина в апоптозе. Было показано, что в различных типах клеток (нейроны, нейтрофилы, клетки лимфомы, нейробластомы, рака предстательной железы) окисление кофилина и митохондриальная транслокация его дефосфорилированной формы индуцируют апоптоз клеток [23–26]. Кофилин может также участвовать в регуляции формирования апоптотических телец в процессе апоптоза [27].
Активация фосфолипазы D1. Последние исследования свидетельствуют о том, что фосфорилированный кофилин (неактивная форма) способен активировать фосфолипазу D1 (PLD1), что приводит к накоплению фосфатидной кислоты, которая через цепочку посредников также может влиять на сборку и разборку актинового цитоскелета [28]. Кроме того, активность PLD1 регулируется Rho-сигнальными путями, которые контролируют активность LIM киназы и последующее фосфорилирование кофилина [29]. Установлено, что фосфолипаза D1 участвует в хемотаксисе нейтрофилов и фагоцитов и миграции клеток в сторону факторов роста (опухолевые клетки, нейроны) [28, 30].
РЕГУЛЯЦИЯ АКТИВНОСТИ КОФИЛИНА
Фосфорилирование и дефосфорилирование кофилина. Кофилин может фосфорилироваться по серину 3 (S3) LIM-киназами 1 и 2 (LIMK1, LIMK2) и тестикулярной протеинкиназой (TESK) [4, 31–34]. При этом LIMK1 и TESK являются специфичными преимущественно для нервной ткани, тогда как LIMK2 распространена повсеместно [12]. Фосфорилированный кофилин представляет собой неактивную форму кофилина и не может связываться с F-актином и вызывать его фрагментацию. Следует отметить, что данный механизм регуляции активности кофилина играет важную роль в морфологии шипиков и регуляции синаптической пластичности [35].
Недавно был идентифицирован новый сайт фосфорилирования кофилина по тирозину 68 (Y68) при участии вирусной (v)-Src-киназы [36]. При этом фосфорилирование Y68 не влияет на активность кофилина, но увеличивает его убиквитилирование и протеасомальную деградацию, что приводит к снижению внутриклеточного уровня кофилина. Опосредованно приводят к фосфорилированию кофилина Rho GTP-азы, p38MAPK (p38 mitogen-activated protein kinase, p38 митоген-активируемая протеинкиназа) и протеинкиназа С [12].
Семейство слингшот протеинфосфатаз (slingshot, SSH-1, 2, 3) и протеинфосфатаза хронофин (chronophin) участвуют в дефосфорилировании фосфокофилина [2, 37, 38]. Способностью к дефосфорилированию кофилина обладают также серин/треониновые фосфатазы 1-го и 2-го типа, однако они обладают более широкой субстратной специфичностью [39]. Установлено, что связывание слингшот фосфатазы с F-актином приводит к увеличению ее активности более чем в 1200 раз. Cлингшот фосфатазы способны дефосфорилировать кофилин, что может приводить к фрагментации актиновых филаментов [40]. Кроме того, слингшот фосфатазы дефосфорилируют и таким образом ингибируют LIM-киназы. В свою очередь слингшот фосфатаза фосфорилируется и инактивируется под действием РАК4 (р21-активируемой протеинкиназы 4) и протеинкиназы D1 [40, 41].
Белок 14-3-3 связывает фосфорилированный кофилин и препятствует его дефосфорилированию под действием протеинфосфатаз [38, 42]. В то же время РАК4, а также МАРКАР-киназа 2 (MAP kinase-activated protein kinase 2, активированная митоген-активированным белком протеинкиназа 2) способны фосфорилировать и активировать LIM киназу [40, 43]. Интересно отметить, что TES киназа также подвергается фосфорилированию, вероятно, под действием МАР-киназ [44].
Убиквитилирование кофилина. Последние исследования указывают на роль убиквитин-протеасомальной системы в регуляции уровня и активности кофилина. Недавно была продемонстрирована модификация кофилина мультиубиквитиновыми цепями по лизину 63, которые вовлечены в сигнальные процессы, активацию киназ и белок-белковые взаимодействия [45]. Кроме того, установлено, что белок паркин, функционирующий как убиквитин (E3) лигаза и участвующий в деградации специфических белков в 26S протеасоме, может подавлять фосфорилирование кофилина за счет регуляции активности LIMK1 [46].
Связывание кофилина с фосфатидилинозитол-4,5-бифосфатом. Данные последних лет свидетельствуют о том, что кофилин способен связываться с фосфатидилинозитол-4,5-бифосфатом (ФИФ2) на внутренней поверхности плазматической мембраны клетки, что приводит к инактивации кофилина и нарушению его взаимодействия с актином [47, 48].
Окислительная модификация кофилина. Кофилин может подвергаться окислительной модификации. Окисление тиольных групп остатков цистеинов в молекуле кофилина приводит к образованию как внутри-, так и межмолекулярных дисульфидных связей. Окисленный кофилин слабо взаимодействует с LIM киназой, что приводит к увеличению концентрации дефосфорилированной формы кофилина. Кроме того, окисленный кофилин на клеточной мембране инактивируется присоединением мембранных липидов, тогда как восстановленная форма нечувствительна к ингибированию присоединением ФИФ2 [49]. Транспорт кофилина в митохондрии также зависит от его окислительной модификации.
Олигомеризация кофилина. В условиях in vitro было показано образование межмолекулярных дисульфидных связей, что приводит к димеризации и олигомеризации кофилина [50]. Кофилин может существовать в виде димера, тетрамера и более высокомолекулярных структур. Установлено, что in vivo димер кофилина является промежуточной формой с крайне небольшим сроком существования с последующим образованием устойчивых тетрамеров, которые являются функциональными единицами кофилина. На олигомеризацию кофилина влияют также процессы фосфорилирования/дефосфорилирования. Данные компьютерного моделирования показывают, что фосфорилирование кофилина по S3 вызывает конформационные изменения в зоне белок-белковых взаимодействий, препятствуя образованию олигомеров [51]. С помощью масс-спектрометрического анализа установлено, что кофилин олигомеризуется с образованием межмолекулярных дисульфидных связей in vitro, при этом при олигомеризации in vivo не показано образование S–S связей.
Влияние внутриклеточного рН на активность кофилина. Активность кофилина зависит от уровня внутриклеточного pH. Показано, что от уровня внутриклеточного рН зависит связывание молекул ФИФ2 с кофилином. Высокое значение pH приводит к уменьшению количества ФИФ2 вблизи мембраны и локальному увеличению уровня свободного кофилина и его активации. Кроме того, рН регулирует процессы связывания кофилина и актина и деполимеризацию актиновых филаментов. Следует отметить, что кофилин является более активным при щелочном значении рН (pH 8) [5]. Локальные изменения pH также влияют на связывание кофилина с кортактином. Увеличение pH приводит к отсоединению кофилина от кортактина, активации кофилина и клеточной инвазии [52].
Механочувствительная регуляция связывания кофилина с актином. Кофилин преимущественно связывается с менее эластичными актиновыми филаментами и опосредует их деградацию, при этом нити актина при растяжении защищены от кофилин-опосредованной фрагментации [53].
РОЛЬ КОФИЛИНА В ФУНКЦИОНИРОВАНИИ НЕРВНОЙ СИСТЕМЫ
Нейрональная пластичность. Высокую степень пластичности нервной системы обеспечивает реорганизация актинового цитоскелета [44, 54–56]. F-актин контролирует морфологические изменения шипиков, которые происходят в ответ на синаптическую активность: увеличение шипиков связано с повышенной синаптической активностью и зависит от сборки и стабилизации F-актина, в то время как сжатие шипиков требует разборки актиновых филаментов [57]. ADF/кофилин является ключевым регулятором динамики актина [58]. Исследования на ген-меченных мышах показали связь ADF/кофилина с нейрональной динамикой актина при развитии ЦНС [59]. При низких концентрациях ADF/кофилин способствует разборке F-актина путем увеличения диссоциации до мономерного G-актина [60]. Было установлено, что ADF/кофилин играет важную роль в морфологии шипиков [7], контроле синаптической пластичности [61] и высвобождении нейротрансмиттеров [62]. Инактивация кофилина 1 и ADF снижает обучение и вызывает тяжелые поведенческие аномалии [61–63]. Ряд исследований показал, что кофилин 1 выступает в качестве двунаправленного регулятора структурной пластичности шипиков [64, 65]. Высокая экспрессия неактивного кофилина 1 в культуре гиппокампа приводит к более зрелым шипикам и увеличению их плотности, что связано с долговременной потенциацией [7]. Избыточная экспрессия активного кофилина 1 индуцирует формирование незрелых шипиков при долговременной депрессии [66]. Последние исследования также показали увеличение шипиков и их плотности в срезах гиппокампа мышей, что является доказательством роли кофилина 1 в морфологии шипиков in vivo [61]. Кроме того, было установлено, что на уровне ADF/кофилина сходятся несколько сигнальных путей, что приводит к изменениям актинового цитоскелета синапсов и регуляции синаптической пластичности. Киназы PAK и LIMK1 способствуют инактивации ADF/кофилина при долговременной потенциации [67]. Следовательно, генетические нарушения PAK или LIMK1 увеличивают активность ADF/кофилина, изменяют морфологию и плотность шипиков, нарушая синаптическую пластичность [35, 61]. Кроме этого, LIMK1 фосфорилирует транскрипционный фактор CREB (cAMP response element-binding protein; белок, связывающийся с цAMФ-зависимым элементом), тем самым, стимулируя активацию CREB-зависимых сигнальных путей [68]. Делеция гена, кодирующего LIMK1 у мышей, приводит к аномалиям в структуре синапса и шипиков дендритов [69].
Функции ADF/кофилина в высвобождении нейромедиаторов и синаптической пластичности предполагают его решающую роль также и в поведении. Действительно, измененная морфология шипиков и дефекты в синаптической пластичности (снижение долговременной потенциации и отсутствие долговременной депрессии) отмечены при нарушении синтеза киназ LIMK1, PAK1 и 3 [35]. На мышиной модели было показано, что при дефиците кофилина 1 происходит нарушение долгосрочной пространственной памяти, в то время как краткосрочная пространственная память не затрагивается [61, 62]. Таким образом, нарушение регуляции активности ADF/кофилина может вызывать или способствовать возникновению невропатий, в частности синдрому Уильямса, сопровождаемого когнитивными нарушениями и моторными дисфункциями [61, 70]. В последних исследованиях показана потеря дендритных шипиков и нарушение их морфологии в нейронах коры головного мозга при церебральной малярии. При этом эти изменения коррелируют с уменьшенными уровня LIMK1, кофилина 1, фосфокофилина 1 и β-актина в головном мозге, а также с образованием кофилин-актиновых палочек [71].
Апоптоз нейронов. Последние данные свидетельствует о том, что ADF/кофилин играет также важную роль в регуляции апоптоза нейронов [2, 72]. Транслокация кофилина в митохондрии считается ранним этапом апоптотического каскада [24, 73]. Этот процесс требует дефосфорилирования и активации кофилина. Дефосфорилированный кофилин связывает G-актин и транслоцируется в митохондрии, что приводит к изменениям в динамике актинового цитоскелета, дисфункции митохондрий, высвобождению цитохрома с и апоптозу [74]. Дефосфорилирование кофилина может происходить при участии фосфатаз РР1 (protein phosphatase 1) и PP2A (protein phosphatase 2A), которые регулируются Rock1/PTEN/PI3K сигнальными путями [72]. При этом дефосфорилирование кофилина может способствовать формированию кофилин-актиновых палочек [75, 76]. Избыточная экспрессия LIMK1 защищает клетки от апоптоза путем инактивации кофилина и ингибирования каспазы-3 (рис. 1). Установлено, что нарушение динамики актина вызывает снижение потенциала митохондриальной мембраны и увеличение уровня активных форм кислорода (АФК) [77].
Рис. 1.
Молекулярные пути регуляции активности кофилина и его участие в функционировании нервных клеток.
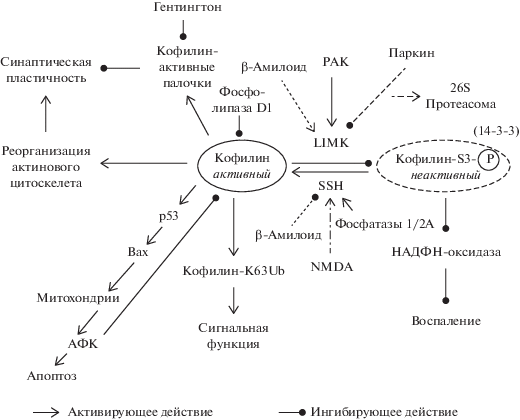
При глутаматной эксайтотоксичности, приводящей к гибели нейронов, было установлено, что кофилин может взаимодействовать с Вах, действуя как его переносчик в митохондрии, что способствует деполяризации митохондриальной мембраны, высвобождению апоптотических факторов и гибели нейронов. Исследования показывают, что только семейство slingshot фосфатаз, а именно фермент slingshot 1L фосфатаза участвует в процессе активации кофилина во время эксайтотоксичности. Действительно, кофилин-опосредованная Вах транслокация является ключевым событием в эксайтотоксичной гибели нейронов. Ингибирование кофилина или slingshot 1L фосфатазы обладает выраженным нейропротективным эффектом при N-метил-D-аспартат (NMDA)-опосредованной гибели нейронов [78]. Кроме того, было показано образование межмолекулярных дисульфидных связей, что приводит к димеризации и олигомеризации кофилина при воздействии на нейроны глутамата [79].
Окислительный стресс может быть как механизмом активации кофилина, так и механизмом его окисления и инактивации. АФК способны повреждать остатки цистеина в молекуле кофилина, в результате чего происходит образование внутримолекулярных дисульфидных связей. Такая структурная модификация нарушает кофилин-актиновое взаимодействие и высвобождает кофилин для транслокации в митохондрии. Таким образом, подавляя активность кофилина либо с помощью фосфорилирования, либо с помощью фармакологических ингибиторов, можно предотвратить гибель нейронов [76].
Показано, что дефосфорилированный кофилин образует комплекс с р53 и способствует его митохондриальной и ядерной локализации, что активирует транскрипцию генов и запуск процесса апоптоза. Установлено, что снижение уровня кофилина ингибирует митохондриальную и ядерную транслокацию р53 у трансгенных мышей APP/PS1 (модель болезни Альцгеймера). Кроме того, проапоптотический путь кофилин-р53 подвергается отрицательной регуляции с помощью PLD1 за счет полной инактивации кофилина и ингибирования образования комплекса кофилин/р53. Установлено, что активный кофилин не способен индуцировать апоптоз в клетках, генетически не кодирующих р53 [80].
Нейровоспаление. Первыми клетками ЦНС, которые активируются при различных поражениях головного мозга являются микроглия и астроциты [81, 82]. Астроциты играют важную роль в поддержании и повышении выживаемости нейронов за счет повышения уровня глутатиона и супероксиддисмутазы [83]. Активация глиальных клеток характеризуется морфологическими и функциональными изменениями, которые приводят к увеличению клеточной пролиферации, миграции и продукции противовоспалительных цитокинов [84].
Подавление воспалительной реакции при острой ишемии может уменьшить повреждающее воздействие и возникновение неврологических осложнений [85]. Тем не менее, воспалительная реакция при хронической ишемии необходима для репарации, регенерации и функционального восстановления [86]. В исследованиях на клеточной линии микроглии RA2 инактивация кофилина приводила к ингибированию активности NADPH-оксидазы и формирования АФК [87]. Кроме того, было установлено, что фагоцитарная активность клеток микроглии сильно зависит от активации кофилина [88]. В целом, в настоящее время достаточно много доказательств, подчеркивающих регуляторную роль кофилина в активации глиальных клеток. При этом необходимо отметить, что для восстановления миелиновой оболочки необходим кофилин [76, 89].
Последние исследования показали существенную роль экзосом в процессе нейровоспаления, одним из основных компонентов которых является кофилин. Интересно отметить, что кофилин способствует деполимеризации актина, который может индуцировать образование экзосом при участии цитоплазматической мембраны [90, 91].
Следовательно, временное ингибирование кофилина при острых воздействиях может предотвратить развитие воспаления. Было установлено, что внутримозговое кровоизлияние является наиболее тяжелой формой инсульта, что во многом обусловлено воспалительной реакцией при активации микроглии и токсическими эффектами гемина, эндогенного продукта разрушения гемоглобина. Было показано, что гемин вызывает увеличение экспрессии кофилина и продукции NO в клетках микроглии. Отмечено увеличение уровня индуцибельной NO-синтазы (iNOS), фактора некроза опухоли-альфа (TNF-α) при действии гемина, частично опосредованное кофилином. Эти исследования показывают, что кофилин имеет важное значение при индуцированном гемином воспалении, окислительном стрессе и миграции микроглии. Таким образом, ингибирование кофилина может быть потенциальной терапией при травмах головного мозга, вызванной токсичностью гемина при геморрагическом инсульте [92].
Нейродегенерация. В мозге пациентов с болезнью Альцгеймера установлено значительное увеличение соотношения фосфокофилин 1/кофилин 1, что указывает на инактивацию кофилина 1. Кроме того, на мышиных моделях было показано, что инактивация кофилина 1 может быть связана с возрастными изменениями. Остается не до конца изученным вопрос – инактивация кофилина 1 является причиной или следствием болезни Альцгеймера. Согласно данным Barone с сотр., болезнь Альцгеймера может активировать процессы старения через фосфорилирование кофилина 1 [54]. Механизмы, приводящие к активации кофилина 1 или фосфорилированию/инактивации, могут быть различны и зависеть частично от уровня β-амилоида (Aβ) и его агрегатного состояния [26, 93]. При этом растворимый и фибриллярный Aβ могут по-разному влиять на кофилин 1, способствуя либо его активации [94], либо его инактивации [95]. До сих пор остается неясным, происходят ли эти события одновременно или последовательно. С молекулярной точки зрения, Aβ пептиды могут также по-разному воздействовать на киназу LIMK1, активность которой нарушается при болезни Альцгеймера и других нейродегенеративных заболеваниях [11, 96]. Было показано, что инактивация фосфатазы SSH1 в мозге больных болезнью Альцгеймера подтверждает гипотезу о том, что равновесие между активностями LIMK1 и SSH1 изменяется при данном заболевании. Найдены значительные корреляции между фосфокофилином 1 и отношением фосфокофилин 1/ кофилин 1 и инактивацией фосфатазы SSH1 или активацией киназы LIMK1 на мышиных моделях при старении. Таким образом, нарушение регуляции SSH1/LIMK1 /кофилин 1 происходит в мозге как при старении, так и при болезни Альцгеймера [54].
Амилоидные включения, сформированные компонентами белковых сетей, поражение которых наблюдается при нейродегенеративных заболеваниях, окружены комплексом кофилин-актин. Нейриты нейронов при остром или хроническом стрессе формируют состоящие из кофилина и актина в соотношении 1 : 1 пучки филаментов (кофилин-актиновые палочки), нарушающие аксоплазматический транспорт и синаптические функции. Эти палочки содержат связанные дисульфидными мостиками молекулы кофилина, и их индукция происходит при любых воздействиях, ведущих к окислительному стрессу. Палочки формируются быстро (5–30 мин) более чем в 80% нейронов гиппокампа или коры в культуре при эксайтотоксичном содержании глутамата или после действия ингибиторов гипоксии/ишемии или митохондриальных функций [9, 10]. Актин в палочках является менее динамичным, чем фибриллярный актин в других структурах цитоскелета. Так как мембранный потенциал митохондрий зависит от уровня клеточного ATP, гидролиз которого связан с обменом актина, образование палочек временно защищает нейриты от снижения уровня ATP [97]. Но длительное присутствие этих палочек в клетках может приводить к нейродегенеративным заболеваниям, хотя как агрегация кофилина и актина оказывает патологические эффекты на ЦНС, остается неясным [97, 98]. Медленное формирование 50% от максимального ответа в течение 6 часов происходит в 20% нейронов гиппокампа при действии растворимого Aβd/t димера или тримера β-амилоида человека. Точно так же действуют и провоспалительные цитокины (TNFα, IL1β, IL6). Нейроны мышей – модели прионных заболеваний (PrP(C)null) – формируют палочки в ответ не на провоспалительные цитокины, а на действие глутамата, что подтверждает как разные гипотезы генеза болезни Альцгеймера, так и пересечение в одном и том же звене ремоделирования актина независимо индуцируемых нейродегенеративных заболеваний [9, 99]. Было показано, что кофилиновые палочки нарушают целостность дендритных микротрубочек в клеточной культуре гиппокампа крыс, способны блокировать внутриклеточный транспорт митохондрий и эндосом. Важно отметить, что формирование кофилиновых палочек индуцирует значительные потери дендритных шипиков. Все это может способствовать нейродегенерации и старению мозга [98]. На модели культуры нейронов и мышиной модели болезни Альцгеймера было показано, что нарушение структуры фибриллярного F-актина в синапсах наблюдается в самом начале развития болезни до проявления клинических признаков. Установлено, что разрушение структуры синаптического F-актина напрямую влияет на дефицит памяти [100]. Кроме того, при болезни Альцгеймера растворимые олигомеры β-амилоида приводят к нарушению синаптической пластичности. Было показано, что в мозге мышей β-амилоид взаимодействует с рецептором PirB (парный иммуноглобулин-подобный рецептор B), его аналог в мозге человека – LilrB2 (лейкоцит иммуноглобулин-подобный рецептор В2). В результате взаимодействия β-амилоида с рецептором происходит увеличение активности кофилина [101]. Ингибирование рецептора PirB или применение его растворимой формы приводит к формированию новых функциональных синапсов у мышей. Таким образом, растворимая форма LilrB2 в настоящее время рассматривается в качестве терапевтического агента при лечении больных с болезнью Альцгеймера [101, 102].
Нарушение динамики актинового цитоскелета отмечено и при болезни Гентингтона во всех типах нервных клеток, но особенно в нейронах [103]. Интересно отметить, что гентингтон (хантингтон) способствует разрушению кофилин-актиновых палочек в ядре. В клетках, в которых синтезировалась мутантная форма гентингтона, было отмечено увеличение уровня актин-кофилиновых палочек, что указывает на их роль в патогенезе болезни Гентингтона [104].
Кроме того, было установлено, что белок паркин, синтез которого нарушен при болезни Паркинсона, может регулировать активность кофилина. На клетках нейробластомы человека была показана способность белка паркин подавлять фосфорилирование кофилина за счет убиквитилирования киназы LIMK1. При этом в клетках линии HEK293 гиперэкспрессия LIMK1 приводила к снижению автоубиквитилирования белка паркин. Такая функциональная зависимость между паркин и LIMK1 указывает на роль данных ферментов в патогенезе болезни Паркинсона [46]. Показано, что уровень кофилина варьирует в лимфоцитах пациентов с болезнью Паркинсона, что может быть использовано при диагностике данного заболевания [105].
Многие нейрональные расстройства, такие как лиссэнцефалия, эпилепсия и шизофрения вызваны нарушением миграции нейронов в развивающемся мозге. Установлено, что нарушение радиальной миграции при участии ADF/кофилина приводит к отсутствию промежуточных слоев коры [106]. Кроме того, гиперактивный кофилин может быть повреждающим фактором, который участвует в разборке F-актина, дестабилизации кадгериновых белков и нарушении гематоэнцефалического барьера в условиях гипоксии [107]. Тем не менее, в настоящее время мало in vivo исследований, которые показывают корреляцию между активацией кофилина и нарушением ГЭБ при ишемическом и геморрагическом инсульте [76].
ЗАКЛЮЧЕНИЕ
Таким образом, кофилин можно рассматривать как многофункциональный белок, регулирующий различные сигнальные пути. При этом активность кофилина регулируется несколькими молекулярными механизмами, основными из которых считаются фосфорилирование и дефосфорилирование при действии LIM киназ и SSH фосфатаз. Участие кофилина и актина в таких процессах, как контроль синаптической пластичности, апоптоз нейронов, нейровоспаление и нейродегенерация, указывает на важность изучения сигнальных путей, вовлеченных в регуляцию уровня и активности кофилина в нервной ткани. При участии кофилина в нейронах происходит изменение морфологии шипиков, выход проапоптотических белков, формирование кофилин-актиновых палочек, активация микроглии. Остается открытым вопрос – изменение активности кофилина является причиной или следствием заболеваний нервной системы. Понимание молекулярных механизмов активации/инактивации кофилина в нервных клетках необходимо для поиска новых маркерных белков и разработки соответствующей терапии данных заболеваний.
Список литературы
Ridley A.J. // Cell. 2011. V. 145. № 7. P. 1012–1022.
Bernstein B.W., Bamburg J.R. // Trends Cell Biol. 2010. V. 20. № 4. P. 187–195.
Shishkin S., Eremina L., Pashintseva N., Kovalev L., Kovaleva M. // Int. J. Mol. Sci. 2016. V. 18. № 12. P. 10.
Bamburg J.R. // Annu. Rev. Cell Dev. Biol. 1999. V. 15. P. 185–230.
Yeoh S., Pope B., Mannherz H.G., Weeds A. // J. Mol. Biol. 2002. V. 315. № 4. P. 911–925.
Rust M.B., Gurniak C.B., Renner M., Vara H., Morando L., Görlich A., Sassoè-Pognetto M., Banchaabouchi M.A., Giustetto M., Triller A., Choquet D., Witke W. // EMBO. 2010. V. 29. P. 1889–1902.
Gu J., Lee C.W., Fan Y., Komlos D., Tang X., Sun C., Yu K., Hartzell H.C., Chen G., Bamburg J.R., Zheng J.Q. // Nat. Neurosci. 2010. V. 13. № 10. P. 1208–1215.
Бapaнoв B.C. // Acta Naturae. 2009. T. 1. № 3. C. 77–89.
Саватеева-Попова Е.В., Никитина Е.А., Медведева А.В. // Генетика. 2015. Т. 51. № 5. С. 613–624.
Bamburg J.R., Bernstein B.W. // Cytoskelet. Hoboken NJ. 2016. V. 73. № 9. P. 477–497.
Vantroys M., Huyck L., Leyman S., Dhaese S., Vandekerkhove J., Ampe C. // Eur. J. Cell Biol. 2008. V. 87. № 8–9. P. 649–667.
Andrianantoandro E., Pollard T.D. // Mol. Cell. 2006. V. 24. № 1. P. 13–23.
Minamide L.S., Striegl A.M., Boyle J.A., Meberg P.J., Bamburg J.R. // Nat. Cell Biol. 2000. V. 2. № 9. P. 628–636.
Chan C., Beltzner C.C., Pollard T.D. // Curr. Biol. CB. 2009. V. 19. № 7. P. 537–545.
Kuhn T.B., Bamburg J.R. // Adv. Exp. Med. Biol. 2008. V. 644. P. 232–249.
Gandhi M., Achard V., Blanchoin L., Goode B.L. // Mol. Cell. 2009. V. 34. № 3. P. 364–374.
Pederson T. // J. Cell Biol. 2008. V. 180. № 6. P. 1061–1064.
Miyamoto K., Gurdon J.B. // Cell. Mol. Life Sci. CMLS. 2013. V. 70. № 18. P. 3289–3302.
Kanellos G., Frame M.C. // J. Cell Sci. 2016. V. 129. № 17. P. 3211–3218.
Munsie L.N., Desmond C.R., Truant R. // J. Cell Sci. 2012. V. 125. Pt 17. P. 3977–3988.
Dopie J., Skarp K.-P., Rajakylä E.K., Tanhuanpää K., Vartiainen M.K. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. № 9. P. E544–E552.
Obrdlik A., Percipalle P. // Nucl. Austin Tex. 2011. V. 2. № 1. P. 72–79.
Klamt F., Zdanov S., Levine R.L., Pariser A., Zhang Y., Zhang B., Yu L.-R., Veenstra T.D., Shacter E. // Nat. Cell Biol. 2009. V. 11. № 10. P. 1241–1246.
Chua B.T., Volbracht C., Tan K.O., Li R., Yu V.C., Li P. // Nat. Cell Biol. 2003. V. 5. № 12. P. 1083–1089.
Zhu B., Fukada K., Zhu H., Kyprianou N. // Cancer Res. 2006. V. 66. № 17. P. 8640–8647.
Woo J.A., Jung A.R., Lakshmana M.K., Bedrossian A., Lim Y., Bu J.H., Park S.A., Koo E.H., Mook-Jung I., Kang D.E. // Cell Death Differ. 2012. V. 19. № 9. P. 1413–1423.
Mannherz H. // Eur. J. Cell Biol. 2005. V. 84. № 4. P. 503–515.
Han L., Stope M.B., de Jesús M.L., Oude Weernink P.A., Urban M., Wieland T., Rosskopf D., Mizuno K., Jakobs K.H., Schmidt M. // EMBO J. 2007. V. 26. № 19. P. 4189–4202.
Schmidt M., Voss M., Weernink P.A., Wetzel J., Amano M., Kaibuchi K., Jakobs K.H. // J. Biol. Chem. 1999. V. 274. № 21. P. 14648–14654.
Lehman N., Di Fulvio M., McCray N., Campos I., Tabatabaian F., Gomez-Cambronero J. // Blood. 2006. V. 108. № 10. P. 3564–3572.
Huang T.Y., DerMardirossian C., Bokoch G.M. // Curr. Opin. Cell Biol. 2006. V. 18. № 1. P. 26–31.
Scott R.W., Olson M.F. // J. Mol. Med. 2007. V. 85. № 6. P. 555–568.
Toshima J., Toshima J.Y., Amano T., Yang N., Narumiya S., Mizuno K. // Mol. Biol. Cell. 2001. V. 12. № 4. P. 1131–1145.
Toshima J., Toshima J.Y., Takeuchi K., Mori R., Mizuno K. // J. Biol. Chem. 2001. V. 276. № 33. P. 31449–31458.
Huang W., Zhou Z., Asrar S., Henkelman M., Xie W., Jia Z. // Mol. Cell. Biol. 2011. V. 31. № 3. P. 388–403.
Yoo Y., Ho H.J., Wang C., Guan J.-L. // Oncogene. 2010. V. 29. № 2. P. 263–272.
Niwa R., Nagata-Ohashi K., Takeichi M., Mizuno K., Uemura T. // Cell. 2002. V. 108. № 2. P. 233–246.
Gohla A., Bokoch G.M. // Curr. Biol. CB. 2002. V. 12. № 19. P. 1704–1710.
Ambach A., Saunus J., Konstandin M., Wesselborg S., Meuer S.C., Samstag Y. // Eur. J. Immunol. 2000. V. 30. № 12. P. 3422–3431.
Soosairajah J., Maiti S., Wiggan O., Sarmiere P., Moussi N., Sarcevic B., Sampath R., Bamburg J.R., Bernard O. // EMBO J. 2005. V. 24. № 3. P. 473–486.
Eiseler T., Döppler H., Yan I.K., Kitatani K., Mizuno K., Storz P. // Nat. Cell Biol. 2009. V. 11. № 5. P. 545–556.
Birkenfeld J., Betz H., Roth D. // Biochem. J. 2003. V. 369. Pt 1. P. 45–54.
Kobayashi M., Nishita M., Mishima T., Ohashi K., Mizuno K. // EMBO J. 2006. V. 25. № 4. P. 713–726.
Sarmiere P.D., Bamburg J.R. // J. Neurobiol. 2004. V. 58. № 1. P. 103–117.
Gainullin M.R., Zhukov I.Y., Zhou X., Mo Y., Astakhova L., Ernberg I., Matskova L. // Sci. Rep. 2017. V. 7. № 1. http://www.nature.com/articles/s41598-017-09540-3. (Accessed February 13, 2018)
Lim M.K., Kawamura T., Ohsawa Y., Ohtsubo M., Asakawa S., Takayanagi A., Shimizu N. // Exp. Cell Res. 2007. V. 313. № 13. P. 2858–2874.
Gorbatyuk V.Y., Nosworthy N.J., Robson S.A., Bains N.P.S., Maciejewski M.W., Dos Remedios C.G., King G.F. // Mol. Cell. 2006. V. 24. № 4. P. 511–522.
Zhao H., Hakala M., Lappalainen P. // Biophys. J. 2010. V. 98. № 10. P. 2327–2336.
Klemke M., Wabnitz G.H., Funke F., Funk B., Kirchgessner H., Samstag Y. // Immunity. 2008. V. 29. № 3. P. 404–413.
Pfannstiel J., Cyrklaff M., Habermann A., Stoeva S., Griffiths G., Shoeman R., Faulstich H. // J. Biol. Chem. 2001. V. 276. № 52. P. 49476–49484.
Goyal P., Pandey D., Brünnert D., Hammer E., Zygmunt M., Siess W. // PLoS One. 2013. V. 8. № 8. P. e71769.
Magalhaes M.A.O., Larson D.R., Mader C.C., Bravo-Cordero J.J., Gil-Henn H., Oser M., Chen X., Koleske A.J., Condeelis J. // J. Cell Biol. 2011. V. 195. № 5. P. 903–920.
Tojkander S., Gateva G., Husain A., Krishnan R., Lappalainen P. // eLife. 2015. V. 4. https://elifesciences.org/articles/06126. (Accessed February 2, 2018)
Barone E., Mosser S., Fraering P.C. // Biochim. Biophys. Acta BBA – Mol. Basis Dis. 2014. V. 1842. № 12. P. 2500–2509.
Park S., Jung Y. // Anat. Cell Biol. 2010. V. 43. № 3. P. 201.
Landry C.D., Kandel E.R., Rajasethupathy P. // Trends Neurosci. 2013. V. 36. № 9. P. 535–542.
Matsuzaki M., Honkura N., Ellis-Davies G.C.R., Kasai H. // Nature. 2004. V. 429. № 6993. P. 761–766.
Hild G., Kalmár L., Kardos R., Nyitrai M., Bugyi B. // Eur. J. Cell Biol. 2014. V. 93. № 5–6. P. 238–251.
Flynn K.C., Hellal F., Neukirchen D., Jacob S., Tahirovic S., Dupraz S., Stern S., Garvalov B.K., Gurniak C., Shaw A.E., Meyn L., Wedlich-Söldner R., Bamburg J.R., Small J.V., Witke W., Bradke F. // Neuron. 2012. V. 76. № 6. P. 1091–1107.
Blanchoin L., Pollard T.D. // J. Biol. Chem. 1999. V. 274. № 22. P. 15538–15546.
Rust M.B. // Cell. Mol. Life Sci. 2015. V. 72. № 18. P. 3521–3529.
Wolf M., Zimmermann A.-M., Görlich A., Gurniak C.B., Sassoè-Pognetto M., Friauf E., Witke W., Rust M.B. // Cereb. Cortex. 2015. V. 25. № 9. P. 2863–2875.
Goodson M., Rust M.B., Witke W., Bannerman D., Mott R., Ponting C.P., Flint J. // PLoS Genet. 2012. V. 8. № 10. P. e1002970.
Zhou Q., Homma K.J., Poo M. // Neuron. 2004. V. 44. № 5. P. 749–757.
Morishita W., Marie H., Malenka R.C. // Nat. Neurosci. 2005. V. 8. № 8. P. 1043–1050.
Pontrello C.G., Sun M.-Y., Lin A., Fiacco T.A., DeFea K.A., Ethell I.M. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. № 7. P. E442–E451.
Chen L.Y., Rex C.S., Casale M.S., Gall C.M., Lynch G. // J. Neurosci. Off. J. Soc. Neurosci. 2007. V. 27. № 20. P. 5363–5372.
Mori T., Okano I., Mizuno K., Tohyama M., Wanaka A. // Brain Res. Mol. Brain Res. 1997. V. 45. № 2. P. 247–254.
Meng Y., Zhang Y., Tregoubov V., Janus C., Cruz L., Jackson M., Lu W.Y., MacDonald J.F., Wang J.Y., Falls D.L., Jia Z. // Neuron. 2002. V. 35. № 1. P. 121–133.
Hoogenraad C.C., Akhmanova A., Galjart N., De Zeeuw C.I. // BioEssays. 2004. V. 26. № 2. P. 141–150.
Simhadri P.K., Malwade R., Vanka R., Nakka V.P., Kuppusamy G., Babu P.P. // Ann. Neurol. 2017. V. 82. № 3. P. 429–443.
Li G., Cheng Q., Liu L., Zhou T., Shan C., Hu X., Zhou J., Liu E., Li P., Gao N. // Cell Commun. Signal. 2013. V. 11. № 1. P. 50.
Rehklau K., Gurniak C.B., Conrad M., Friauf E., Ott M., Rust M.B. // Cell Death Differ. 2012. V. 19. № 6. P. 958–967.
Yang E., Kim H., Lee J., Shin J.S., Yoon H., Kim S.J., Choi I.H. // Cell. Mol. Neurobiol. 2004. V. 24. № 2. P. 181–192.
Chen B., Jiang M., Zhou M., Chen L., Liu X., Wang X., Wang Y. // Brain Res. 2012. V. 1486. P. 1–13.
Alhadidi Q., Bin Sayeed M.S., Shah Z.A. // Transl. Stroke Res. 2016. V. 7. № 1. P. 33–41.
Gourlay C.W., Ayscough K.R. // J. Cell Sci. 2005. V. 118. Pt 10. P. 2119–2132.
Posadas I., Pérez-Martínez F.C., Guerra J., Sánchez-Verdú P., Ceña V. // J. Neurochem. 2012. V. 120. № 4. P. 515–527.
Samstag Y., John I., Wabnitz G.H. // Immunol. Rev. 2013. V. 256. № 1. P. 30–47.
Liu T., Wang F., LePochat P., Woo J.-A.A., Bukhari M.Z., Hong K.W., Trotter C., Kang D.E. // Sci. Rep. 2017. V. 7. № 1. http://www.nature.com/articles/s41598-017-09996-3. (Accessed February 13, 2018)
Barreto G.E., Sun X., Xu L., Giffard R.G. // PLoS One. 2011. V. 6. № 11. P. e27881.
Taylor R.A., Sansing L.H. // Clin. Dev. Immunol. 2013. V. 2013. P. 1–10.
Chen Y., Vartiainen N.E., Ying W., Chan P.H., Koistinaho J., Swanson R.A. // J. Neurochem. 2001. V. 77. № 6. P. 1601–1610.
Wang J. // Prog. Neurobiol. 2010. V. 92. № 4. P. 463–477.
Lee Y., Lee S.-R., Choi S.S., Yeo H.-G., Chang K.-T., Lee H.J. // BioMed Res. Int. 2014. V. 2014. P. 1–9.
Jin R., Liu L., Zhang S., Nanda A., Li G. // J Cardiovasc. Transl. Res. 2013. V. 6. № 5. P. 834–851.
Rasmussen I., Pedersen L.H., Byg L., Suzuki K., Sumimoto H., Vilhardt F. // BMC Immunol. 2010. V. 11. № 1. P. 44.
Gitik M., Kleinhaus R., Hadas S., Reichert F., Rotshenker S. // Front. Cell. Neurosci. 2014. V. 8. P. 104.
Nawaz S., Sánchez P., Schmitt S., Snaidero N., Mitkovski M., Velte C., Brückner B.R., Alexopoulos I., Czopka T., Jung S.Y., Rhee J.S., Janshoff A., Witke W., Schaap I.A., Lyons D.A., Simons M. // Dev. Cell. 2015. V. 34. № 2. P. 139–151.
Gupta A., Pulliam L. // J. Neuroinflammation. 2014. V. 11. P. 68.
Brites D., Fernandes A. // Front. Cell. Neurosci. 2015. V. 9. P. 476.
Sayeed M.S.B., Alhadidi Q., Shah Z.A. // J. Neuroimmunol. 2017. V. 313. P. 46–55.
Tu S., Okamoto S., Lipton S.A., Xu H. // Mol. Neurodegener. 2014. V. 9. № 1. P. 48.
Maloney M.T. // J. Neurosci. 2005. V. 25. № 49. P. 11313–11321.
Heredia L., Helguera P., de Olmos S., Kedikian G., Sola Vigo F., LaFerla F., Staufenbiel M., de Olmos J., Busciglio J., Caceres A., Lorenzo A. // J. Neurosci. 2006. V. 26. № 24. P. 6533–6542.
Bellani S., Mescola A., Ronzitti G., Tsushima H., Tilve S., Canale C., Valtorta F., Chieregatti E. // Cell Death Differ. 2014. V. 21. № 12. P. 1971–1983.
Bernstein B.W., Chen H., Boyle J.A., Bamburg J.R. // Am. J. Physiol. Cell Physiol. 2006. V. 291. № 5. P. C828–839.
Cichon J., Sun C., Chen B., Jiang M., Chen X.A., Sun Y., Wang Y., Chen G. // J. Biol. Chem. 2012. V. 287. № 6. P. 3919–3929.
Walsh A.J., Cook R.S., Manning H.C., Hicks D.J., Lafontant A., Arteaga C.L., Skala M.C. // Cancer Res. 2013. V. 73. № 20. P. 6164.
Kommaddi R.P., Das D., Karunakaran S., Nanguneri S., Bapat D., Ray A., Shaw E., Bennett D.A., Nair D., Ravindranath V. // J. Neurosci. 2018. V. 38. № 5. P. 1085–1099.
Bochner D.N., Sapp R.W., Adelson J.D., Zhang S., Lee H., Djurisic M., Syken J., Dan Y., Shatz C.J. // Sci. Transl. Med. 2014. V. 6. № 258. P. 258ra140.
Kim T., Vidal G.S., Djurisic M., William C.M., Birnbaum M.E., Garcia K.C., Hyman B.T., Shatz C.J. // Science. 2013. V. 341. № 6152. P. 1399–1404.
Hotulainen P., Hoogenraad C.C. // J. Cell Biol. 2010. V. 189. № 4. P. 619–629.
Bamburg J.R., Bernstein B.W. // F1000 Biol. Rep. 2010. http://www.f1000biology.com/reports/10.3410/ B2-62. (Accessed January 26, 2018)
Mila S., Giuliano Albo A., Corpillo D., Giraudo S., Zibetti M., Bucci E.M., Lopiano L., Fasano M. // Biomark. Med. 2009. V. 3. № 2. P. 117–128.
Bellenchi G.C., Gurniak C.B., Perlas E., Middei S., Ammassari-Teule M., Witke W. // Genes Dev. 2007. V. 21. № 18. P. 2347–2357.
Liu L., Xue Y., Liu Y., Wang Y. // J. Neurosci. Res. 2008. V. 86. № 5. P. 1153–1168.
Дополнительные материалы отсутствуют.