

Нейрохимия, 2019, T. 36, № 1, стр. 41-48
Действие альфа-токоферола на активность ацетилхолинэстераз из разных источников
Е. М. Молочкина 1, Ю. А. Трещенкова 1
1 ФГБУН Институт химической физики им. Н.М. Эмануэля РАН
Москва, Россия
Поступила в редакцию 27.04.2018
После доработки 20.06.2018
Принята к публикации 07.06.2018
Аннотация
Выявлено значительное ингибирующее действие альфа-токоферола (ТФ) на растворимые коммерческие препараты ацетилхолинэстеразы (АХЭ) – человеческую рекомбинантную (hАХЭ) и АХЭ из Electric eel. Ингибирование осуществляется путем взаимодействия фитильной цепи с периферическим анионным сайтом АХЭ. Однако ТФ не ингибирует молекулярные формы фермента, важные для развития болезни Альцгеймера, в частности, АХЭ эритроцитов и “холинергическую” мембраносвязанную АХЭ мозга мышей, которая даже активируется введением ТФ in vivo. Выбирая модель для скрининга веществ с антихолинэстеразными свойствами при разработке новых средств терапии БА, следует иметь в виду, что не всегда ингибирование коммерческих препаратов ферментов свидетельствует об антихолинэстеразном действии в организме.
В настоящее время одним из наиболее социально значимых заболеваний является болезнь Альцгеймера (БА). К главным патогенетическим и промотирующим течение БА факторам относится окислительный стресс (ОС) [1, 2]. Веществом, тормозящим ОС в организме, служит витамин Е, основным компонентом которого является ТФ (рис. 1), в связи с чем последний нередко включают в схему лечения БА [3–7], хотя результаты применения ТФ неоднозначны, противоречивы и требуют дальнейших исследований.
Главное проявление БА – драматическая потеря памяти и когнитивных функций, связанная с падением уровня медиатора ацетилхолина (АХ) вследствие повреждения и гибели холинергических нейронов в соответствующих отделах мозга. Уровень АХ регулируется работой фермента ацетилхолинэстеразы (АХЭ). Основной молекулярной формой АХЭ головного мозга является АХЭ-Т, организованная в тетрамеры и локализованная в синаптической мембране посредством обогащенного пролином мембранного якоря (PRiMA) [8, 9]. Именно АХЭ-Т производит деградацию АХ (холинергическая АХЭ). Активность этой АХЭ в мозге при БА несколько понижена, хотя по общему количеству ферментного белка мозг больных практически не отличается от мозга здоровых [10]. При БА значительную часть АХЭ-Т составляет молекулярная форма АХЭ, неактивная в отношении гидролиза АХ в синаптической щели, но способная взаимодействовать с бета-амилоидом (главным патогенетическим фактором БА), усиливая его токсическое действие [11–13]. Как для поддержания уровня АХ, так и для предотвращения развития “амилоидного каскада” требуется ингибирование АХЭ. Именно ингибиторы АХЭ до настоящего времени остаются наиболее результативным средством терапии БА [14, 15].
Кроме АХЭ в организме человека (в том числе в мозге) имеется бутирилхолинэстераза (БХЭ), назначение которой до конца не выяснено. В норме содержание БХЭ в мозге человека крайне мало, однако, при БА оно возрастает параллельно с одновременным уменьшением АХЭ. Считается, что БХЭ берет на себя функцию гидролиза АХ, и, следовательно, ее ингибирование также полезно для терапии БА. Целесообразно ингибирование БХЭ и с точки зрения некоторых данных о взаимодействии БХЭ с бета-амилоидом, которое усиливает образование патологических фибрилл [16]. Тем не менее, существуют данные и представления о положительной роли БХЭ при БА, об отсутствии ее вредоносного взаимодействия с амилоидом. Сторонники такого взгляда считают более целесообразным применять препараты, избирательно ингибирующие АХЭ [17].
БА – многофакторное заболевание. Основными направлениями разработки новых лекарственных средств лечения БА являются в настоящее время синтез “гибридных” мультитаргетных препаратов, а также поиск для известных старых лекарств новых свойств и мишеней, связанных с развитием БА.
В соответствии с представлениями о патогенезе и развитии БА наиболее перспективные препараты для лечения этой патологии должны обладать антиоксидантными и антихолинэстеразными свойствами. В последнее время у многих растительных жирорастворимых фенольных соединений, у экстрактов эфирных масел и их компонентов, обладающих сильным АО действием, обнаружена способность существенно ингибировать холинэстеразы, в связи с чем эти вещества считаются перспективными для применения в терапии БА [18].
При отборе антихолинэстеразных веществ часто используют коммерческие препараты ферментов – растворимую АХЭ из Electric eel (Electrophorus Electricus) и человеческую рекомбинантную АХЭ (human recombinant AChE) (hАХЭ).
Целью нашей работы была оценка антихолинэстеразного действия ТФ на этих моделях и сравнение эффекта с его влиянием на активность АХЭ из других “источников”, а именно, на молекулярные формы АХЭ, которые могут быть вовлечены в развитие БА.
Задачами работы было изучение действия ТФ на активность растворимой АХЭ из Electric eel, растворимой рекомбинантной человеческой АХЭ, коммерческого препарата мембраносвязанной АХЭ эритроцитов человека, на активность АХЭ свежевыделенных эритроцитов крови мышей, а также на активность АХЭ цитоплазматической фракции и фракции синаптосом головного мозга мышей in vitro и in vivo. В задачи работы входило и изучение влияния ТФ на активность БХЭ.
МАТЕРИАЛЫ И МЕТОДЫ
В работе использовали альфа-токоферол фирмы Serva. Мы изучали действие ТФ в физиологических (терапевтических) концентрациях 10–5–10–4 М. Отнести данный диапазон концентраций к физиологическим позволил анализ литературных данных [19–22] по содержанию ТФ в органах и тканях экспериментальных животных, обеспечивающему потребности организма при стандартном рационе, а также оценка концентраций ТФ, которые вводили в организм животных в терапевтических целях для противодействия патологическим процессам, связанным в основном с усилением ПОЛ.
В работе использовали коммерческие препараты фермента АХЭ (3.1.1.7) – растворимую АХЭ из Electric eel (Sigma) V-S, рекомбинантную человеческую АХЭ (human recombinant AChE) (Sigma), АХЭ из эритроцитов человека мембраносвязанную (тип XIII) (Sigma), а также растворимую бутирилхолинэстеразу БХЭ (3.1.1.8) из сыворотки лошади (Sigma).
Для изучения действия ТФ in vivo мышам линии Balb (самцам массой 20–24 г) внутрибрюшинно вводили ТФ, растворенный в 0.5% этаноле, в физиологической дозе 10–4 моль/кг за сут до декапитации. Животным контрольной группы вводили этанол.
Выделение синаптосом и цитоплазматической фракции головного мозга мышей проводили методом дифференциального центрифугирования гомогената с очисткой на градиенте плотности сахарозы [23]. Для изучения действия ТФ на АХЭ in vitro cинаптосомы и цитоплазматическую фракцию выделяли из головного мозга мышей линии SHK.
Отметим, что использованные нами цитоплазматическая фракция и синаптосомы не гидролизовали бутирилтиохолин (БТХ), то есть не содержали измеримых количеств БХЭ.
Белок в субклеточных фракциях определяли методом Лоури [24].
Эритроциты выделяли из “цитратной” крови мышей центрифугированием (10 мин при 1500 оборотов/мин на центрифуге Elmi CM-6) с последующим трехкратным промыванием в физиологическом растворе NaCl.
Выделенные эритроциты подвергали гемолизу (осмотический шок плюс замораживание–оттаивание). В гемолизате, содержащем строму эритроцитов с локализованной в мембранах АХЭ, определяли активность фермента.
Измерение активности холинэстераз проводили спектрофотометрически по методу Элмана [25], используя ацетилтиохолин иодид (АТХ) (Serva) или бутирилтиохолин иодид (БТХ) (Alfa Aesar) в качестве субстрата, с непрерывной регистрацией кинетики образования продукта реакции на спектрофотометре SP–Ultra (Россия). Ферментативную реакцию проводили в кювете спектрофотометра при температуре 37°С, рН реакционной смеси = 8.2 (0.05 M Трис-HCl буфер). В зависимости от объекта исследования среда реакции содержала фермент 0.015–0.03 ед. в мл, цитозоль, суспензию синаптосом или гемолизат эритроцитов, а также 0.001 М ДТНБ (5,5'-дитиобис-(2-нитробензойная кислота), Sigma), в опытах in vitro – этанол (0.1%) (х. ч. Fluka) или добавленный в виде спиртового раствора ТФ (конечная концентрация 10–5–10–4 М). Реакцию начинали добавлением субстрата (50–1000 мкМ) через 15–30 мин преинкубации фермента с ТФ или этанолом при 37°С. Время реакции 5 мин. Запись кинетики проводили при длине волны 412 нм, в случае эритроцитов – 436 нм [26]. Кинетические параметры реакции гидролиза АТХ максимальную скорость Vmax, кажущуюся константу Михаэлиса Km, каж, (далее Кm), Vmax/Km рассчитывали как параметры уравнения Михаэлиса–Ментен из кривой зависимости начальной скорости увеличения оптической плотности от концентрации субстрата. При оценке скорости ферментативной реакции учитывали соответствующие blank-контроли (изменение оптической плотности в отсутствие фермента или субстрата).
Не содержащий в своей молекуле боковой цепи аналог токоферола тролокс (Aldrich) использовали в виде водного и этанольного растворов.
Для обработки данных и расчетов кинетических параметров использовали программу SigmaPlot для Windows, v. 11.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
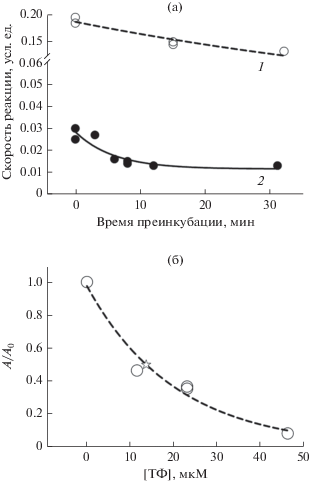
Действие ТФ на активность hАХЭ. На рис. 2 представлены данные по влиянию физиологической концентрации ТФ на активность растворимой рекомбинантной человеческой АХЭ, которая нередко используется для скрининга антихолинэстеразных веществ. Можно видеть (рис. 2а), что имеет место значительное ингибирование, которое развивается во времени – на стационарный уровень скорость реакции выходит не мгновенно, а после относительно продолжительного контакта фермента с ТФ. Это позволяет рассматривать ТФ как медленно связывающийся ингибитор АХЭ. На рис. 2б представлена зависимость стационарной скорости при близкой к насыщающей концентрации субстрата от концентрации ТФ. Концентрация, при которой наблюдается 50%-ное ингибирование [I]50, составляет 11 мкМ.
Рис. 2.
Влияние ТФ на активность человеческой рекомбинантной АХЭ. (37°С, рН 8.2, концентрация АХЭ 0.015 ед./мл, концентрация АТХ 0.74 мМ). (а) 1 – скорость реакции в отсутствие ТФ, (2) – в присутствии 46.1 мкМ ТФ в зависимости от времени преинкубации перед добавлением субстрата. (б) Зависимость скорости реакции (в стационарных условиях – после 15 мин преинкубации) от концентрации ТФ. А0 – скорость реакции в отсутствие ТФ, А – скорость реакции в присутствии ТФ).
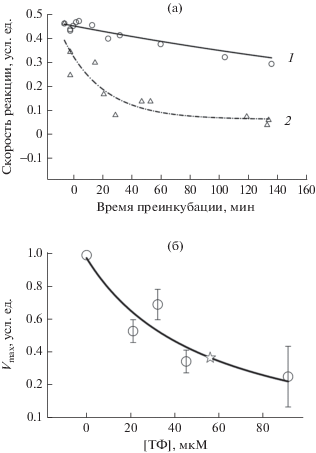
Действие ТФ на активность АХЭ из Electric eel. На рис. 3 приведен пример ингибирования альфа‑токоферолом в физиологической концентрации АХЭ из Electric eel, которая тоже часто используется для скрининга антихолинэстеразных веществ. Видно (рис. 3а) что в этом случае ингибирование также развивается во времени. На стационарный уровень скорость реакции выходит после довольно длительной (более длительной, чем для hАХЭ) преинкубации фермента с ТФ. Как и в случае hАХЭ, ТФ ведет себя как медленный обратимый ингибитор.
Рис. 3.
Влияние ТФ на активность АХЭ из Electric eel. (37°С, рН 8.2, концентрация АХЭ 0.03 ед./мл, концентрация АТХ 0.35 мМ). (а) 1 – скорость реакции в отсутствие ТФ, 2 – в присутствии 41.5 мкМ ТФ в зависимости от времени инкубации перед добавлением субстрата. (б) Зависимость максимальной скорости реакции (в стационарных условиях – после 30 мин преинкубации) от концентрации ТФ.
Фермент АХЭ, как известно [27], дестабилизируется и частично теряет активность в присутствии тиолсодержащих соединений, в том числе ДТНБ. На рис. 2а и 3а видно, что АХЭ действительно несколько теряет активность при инкубации в среде, содержащей ДТНБ, а ТФ, обладая выраженным ингибирующим эффектом, по-видимому, оказывает в то же время и стабилизирующее действие на фермент.
Тип ингибирующего действия ТФ оценивали по влиянию на максимальную скорость реакции Vmax и кажущуюся константу Михаэлиса Km АХЭ из Electric eel, которые определяли как параметры уравнения Михаэлиса–Ментен, характеризующего экспериментально полученную зависимость скорости реакции от концентрации субстрата АТХ в стационарных условиях (после 30 мин преинкубации фермента в присутствии ТФ). Оказалось, что ТФ изменяет Vmax, не влияя на Km, которая в присутствии ТФ составляет 90 ± 6 мкМ по сравнению с 96 ± 2 мкМ в контроле, то есть ведет себя как неконкурентный ингибитор.
На рис. 3б представлена зависимость Vmax от концентрации ТФ. Концентрация ТФ, при которой происходит 50%-ное ингибирование [I]50, а значит, поскольку ингибирование неконкурентное, Ki, составляет 56 мкМ. Сравнивая [I]50, можно отметить, что ТФ ингибирует hАХЭ несколько более эффективно, чем АХЭ из Electric eel.
Неконкурентный тип ингибирования говорит о взаимодействии ТФ с периферическим анионным сайтом (ПАС) АХЭ. Для терапии БА такой тип ингибирования помимо поддержания уровня АХ может обеспечивать противодействие токсическому взаимодействию АХЭ с бета-амилоидом, в котором задействован именно этот сайт молекулы фермента [11–13].
Действие ТФ на БХЭ. На активность БХЭ из лошадиной сыворотки ТФ не оказывал влияния. При близкой к насыщающей концентрации субстрата скорость реакции в опыте практически не отличалась от контроля (44 ± 3 усл. ед. по сравнению с 42 ± 3 при концентрации ТФ 10–4 М).
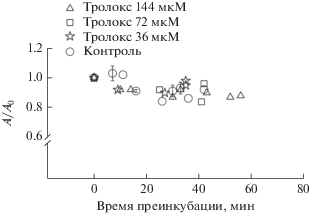
Действие тролокса на АХЭ из Electric eel. Представляло интерес изучить влияние на АХЭ тролокса, молекула которого является по существу фрагментом молекулы ТФ без фитильной цепи, содержащим –ОН группу, ответственную за антиоксидантную способность ТФ. На рис. 4 даны результаты соответствующего эксперимента. Очевидно, что тролокс не оказывает ингибирующего действия на АХЭ. Можно считать, что описанное выше ингибирование АХЭ под действием ТФ обусловлено взаимодействием боковой фитильной цепи его молекулы с ПАС фермента. Подтверждением этой роли фитильной цепи могут служить полученные ранее [28] данные об эффективном ингибировании АХЭ из electric eel ТФ-сукцинатом, в котором этерифицирована –ОН группа хроманового кольца. Различиями в свойствах ПАС АХЭ и БХЭ [29] может объясняться отсутствие ингибирующего эффекта ТФ на БХЭ.
Рис. 4.
Отсутствие влияния тролокса на АХЭ из Electric eel (37°С, рН 8.2, концентрация АХЭ 0.03 ед./мл, концентрация АТХ 0.72 мМ). А0 – скорость реакции в отсутствие тролокса без преинкубации. А – скорость реакции в зависимости от времени преинкубации в присутствии 0, 36, 72 и 144 мкМ тролокса.
Таким образом, выявлено выраженное ингибирующее действие физиологических концентраций ТФ на активность “модельных” ацетилхолинэстераз – человеческой рекомбинантной АХЭ и АХЭ из Electrophorus electricus.
Казалось бы, способность ТФ ингибировать АХЭ дает в совокупности с известным АО действием дополнительное обоснование для его использования в терапии БА. Однако следует иметь в виду, что АХЭ – фермент, имеющий целый ряд изоформ, и в плане изучения антихолинэстеразных свойств перспективных лекарственных препаратов интерес представляют прежде всего изоформы, которые могут быть вовлечены в развитие БА. В первую очередь к ним относится АХЭ головного мозга, в основном ее мембраносвязанная форма АХЭ-Т, ассоциированная с мембраной нервных окончаний.
Репортерами одного из ключевых патогенетических факторов БА – окислительного стресса в организме считаются эритроциты [30–32]. Установлен вклад состояния эритроцитов крови при БА в нарушение снабжения мозга кислородом, усугубляющее течение болезни. Состояние мембран [33] эритроцитов и способность амилоида к связыванию с эритроцитами [34] рассматриваются как маркеры нейродегенеративных нарушений. Активность локализованной в эритроцитах АХЭ является одним из показателей их структурно-функционального состояния.
Исходя из сказанного, для изучения влияния физиологических концентраций ТФ мы выбрали АХЭ головного мозга и эритроцитарную АХЭ. Результаты представлены в табл. 1
Таблица 1.
Влияние физиологических концентраций альфа-токоферола (ТФ) на активность АХЭ эритроцитов крови и субклеточных фракций головного мозга
| № | Источник АХЭ | Параметр | Контроль | Опыт | Эффект |
|---|---|---|---|---|---|
| 1 | Синаптосомы (фракция нервных
окончаний головного мозга мышей). ТФ in vivo за 24 ч 100 мкмоль/кг |
Vmax, усл. ед. | 417 ± 40 | 478 ± 1 | Активация |
| Km, мкМ | 82 ± 3.2 | 26.0 ± 0.1 | |||
| Vmax/Km | 5.1 ± 0.7 | 18.2 ± 1.0 | |||
| n = 5 | n = 5 | ||||
| 2 | Цитоплазма головного мозга мышей. ТФ in vivo за 24 ч 100 мкмоль/кг |
Vmax, усл. ед. | 314 ± 11 | 303 ± 10 | Не влияет |
| Km, мкМ | 42 ± 6 | 45 ± 6 | |||
| Vmax/Km | 7.5 ± 1.3 | 6.7 ± 1.1 | |||
| n = 5 | n = 5 | ||||
| 3 | Синаптосомы головного мозга мышей. ТФ in vitro 45 мкМ |
Скорость при насыщающей концентрации субстрата, усл. ед. | 410 ± 7 | 404 ± 3 | Не влияет |
| n = 4 | n = 4 | ||||
| 4 | Цитоплазма головного мозга мышей. ТФ in vitro 45 мкМ |
Скорость при насыщающей концентрации субстрата, усл. ед. | 770 ± 20 | 750 ± 20 | Не влияет |
| n = 4 | n = 4 | ||||
| 5 | Мембраносвязанная АХЭ свежевыделенных эритроцитов мышей. ТФ in vitro 55 мк М | Vmax, усл. ед. | 180 ± 14 | 156 ± 12 | Слабое ингибирование, статистически недостоверно |
| Km, мкМ | 132 ± 38 | 123 ± 37 | |||
| n = 5 | n = 5 | ||||
| 6 | АХЭ эритроцитов человека
мембраносвязанная, Sigma. ТФ in vitro 46 мкМ |
Скорость при насыщающей концентрации субстрата, усл. ед. | 164 ± 8 | 144 ± 8 | Слабое ингибирование, статистически недостоверно |
| n = 4 | n = 4 |
В таблице даны средние значения ± стандартная ошибка SE. n – в графах 1, 2, 5 – число точек (концентраций субстрата), по которым определяли параметры уравнения Михаэлиса–Ментен. Каждая точка – среднее из 3–4-х параллельных измерений скорости реакции. В графах 3, 4, 6 n – число параллельных определений активности. Усл. ед. (условные единицы скорости реакции) – прирост оптической плотности при 412 или 436 нм (для эритроцитов) в мин, умноженный на 103 (графы 1–5) или на 102 (графа 6) и отнесенный к концентрации белка в реакционной смеси (мг/мл) (графы 1–4), к концентрации гемоглобина, выраженной в единицах оптической плотности при 540 нм (графа 5), или к содержанию твердого препарата фермента (мг/мл) (графа 6).
Влияние ТФ на активность АХЭ субклеточных фракций головного мозга и эритроцитов. В графе 1 табл. 1 приведены данные по влиянию ТФ на активность АХЭ нервных окончаний (синаптосом) головного мозга. Видно, что через сутки после внутрибрюшинного введения физиологической дозы ТФ мышам имеет место активация фермента, обусловленная в основном уменьшением константы Михаэлиса, которая отражает сродство фермента к субстрату.
В то же время (графа 2 табл. 1) растворимая АХЭ цитоплазматической фракции, практически идентичная мембранной по аминокислотному составу субъединиц, но не заякоренная в мембране, не подвергается влиянию введенного in vivo ТФ. Выраженный при этом активирующий эффект введенного in vivo ТФ на мембранную АХЭ нервных окончаний головного мозга мышей наводит на мысль о роли состояния мембран в реализации его действия.
Согласно данным по фармакокинетике [35], период полувыведения ТФ составляет 48 часов. В случае нашего опыта по введению ТФ in vivo декапитация мышей и выделение субклеточных фракций производились через 24 ч после инъекции препарата. Можно предполагать, что в это время введенный ТФ присутствовал в ткани мозга в концентрации, соизмеримой с введенной дозой, и, несомненно, должно было иметь место его влияние на структуру мембраны, обусловленное как изменением интенсивности ПОЛ, так и непосредственным взаимодействием ТФ с мембраной. Известно, что уже после 10 минут инкубации ТФ в суспензии мембран при комнатной температуре наступает изменение текучести липидной фазы [36]. В нашем случае в опыте in vitro время инкубации составляло 30 мин при 37°С. Как видно из таблицы, ТФ не влиял на активность АХЭ синаптосом при воздействии in vitro в этих условиях. По-видимому, состояние мембраны не играет роли в изменении активности АХЭ синаптосом при действии ТФ. Чем же можно объяснить наблюдаемую при введении ТФ in vivo активацию фермента? Имея в виду сведения об участии ТФ в регуляции экспрессии многих генов [37–39], можно предположить, что под действием ТФ происходит модификация экспрессии гена, кодирующего PRiMA, посредством которого АХЭ закрепляется в мембране, результатом чего может быть изменение количества или свойств мембраносвязанного ферментного белка.
В эритроцитах АХЭ находится в виде димеров, закрепленных в мембране посредством липидного “якоря” гликозилфосфатидилинозита (GPI) [40]. В одной из работ [22] приводятся результаты по активации АХЭ эритроцитов в результате длительного введения крысам ТФ in vivo. Не исключено, что в этом случае ТФ изменяет свойства GPI якоря, влияя, например, через систему регуляции ПОЛ на состав жирных кислот фосфолипида.
Из таблицы видно, что так же, как на мембраносвязанную АХЭ синаптосом, ТФ в физиологической концентрации не влияет достоверно на фермент эритроцитов in vitro – как на коммерческий препарат, содержащий строму эритроцитов, так и на АХЭ свежевыделенных клеток, хотя и проявляет слабую тенденцию к ингибированию. Скорее всего, как и в случае синаптосом, практическое отсутствие эффекта связано с локализацией ТФ и АХЭ в мембране, которая, по-видимому, не позволяет им взаимодействовать нужным для ингибирования образом.
Выше было показано, что в ингибировании растворимого кристаллического фермента принимает участие фитильный “хвост” молекулы ТФ. Известно, что ТФ фиксируется в мембране путем образования водородной связи между гидроксильной группой ТФ и кислородом либо карбонильной, либо фосфатной группы молекулы фосфолипида мембраны, при этом фитильный участок погружается в бислой параллельно цепочкам фосфолипидов [41]. Таким образом, “ингибирующий” участок ТФ и ПАС фермента разобщены. В последнее время большое внимание уделяется роли и строению мембранных липидных кластеров (рафтов) – доменов, обогащенных сфинголипидами и холестерином. Есть сведения о том, что мембранная АХЭ в основном локализуется и функционирует в рафтах [42]. В то же время существуют данные о том, что ТФ локализуется преимущественно в областях мембраны, обогащенных фосфолипидами, содержащими ненасыщенные жирные кислоты, где он выполняет свою основную антиоксидантную функцию [43]. Это может вносить свой вклад в пространственное разобщение, которое не позволяет ТФ оказывать ингибирующее действие на АХЭ. Таким образом, можно представить себе, почему ТФ не ингибирует мембраносвязанную АХЭ in vitro.
Возникает вопрос, почему ТФ ингибирует растворимую кристаллическую hАХЭ, АХЭ из Electric eel, но не влияет на активность растворимой АХЭ цитоплазматической фракции мозга. Имея в виду, что цитоплазматическая фракция содержит липопротеины и липидпереносящие белки, с которыми ТФ может связываться с высоким сродством [44, 45], можно предположить, что их связывание с ТФ препятствует его ингибирующему действию на АХЭ. Однако, разумеется, остается вероятность того, что причина различия кроется в разнице аминокислотных последовательностей и структуры этих холинэстераз, хотя известно, что они имеют высокую степень идентичности.
ЗАКЛЮЧЕНИЕ
Обнаружено, что в физиологических концентрациях (10–5–10–4 М) ТФ ведет себя как эффективный, медленно связывающийся ингибитор растворимых hАХЭ и АХЭ из Electric eel. Ингибирование протекает по неконкурентному типу за счет связывания фитильного “хвоста” ТФ с периферическим анионным сайтом АХЭ.
ТФ в физиологических концентрациях не ингибирует АХЭ из “источников”, которые могут реально участвовать в развитии БА. In vitro ТФ практически не влияет на активность мембраносвязанной АХЭ эритроцитов человека и мышей, не действует на активность мембранной АХЭ синаптосом и растворимой АХЭ цитоплазматической фракции головного мозга мышей. При введении ТФ in vivo имеет место выраженная активация АХЭ фракции синаптосом. Такой эффект может ослаблять обусловленное антиоксидантными свойствами положительное действие ТФ при терапии БА.
При разработке новых лекарственных средств лечения БА требуется проявлять осторожность, выбирая ту или иную модель для скрининга веществ с антихолинэстеразными свойствами, имея в виду, что не всегда ингибирование коммерческих препаратов ферментов свидетельствует об антихолинэстеразном действии в организме.
Список литературы
Kim G.N., Kim J.E., Rhie S.J., Yoon S. // Exp. Neurobiol. 2015. V. 24. № 4. P. 325–340.
Aliev G., Obrenovich M.E., Reddy V.P., Shenk J.C., Moreira P.I., Nunomura A., Zhu X., Smith M.A., Perry G. // Mini. Rev. Med. Chem. 2008. V. 8. № 13. P. 1395–1406.
Bittner D.M. // J. of Clinical Psychopharmacology. 2009. V. 29. № 5. P. 511–512.
Grimm M.O.W., Mett J., Hartmann T. // Int. J. Mol. Sci. 2016. V. 17. № 11. pii: E1785.
Farina L., Llewellyn D., Isaac. M.G.E.K.N.,Taber N. // Cochrane Database of Systematic Reviews. 2017. Issue 4. Art. №: CD002854.
La Fata G., Weber P., Mohajer M.H. // Nutrients. 2014. V. 6. P. 5453–5472.
Lloret A., Badia M.C., Mora N.J., Pallardo F.V., Alonso M.D., Vina J. // J. Alzheimer Dis. 2009. V. 17. № 1. P. 143–149.
Massolie J. // Neurosignals. 2002. V. 11. P. 130–143.
Perrier A.L., Massoulie J., Krejci E. // Neuron. 2002. V. 33. P. 275–285.
Campanary M.-L., Navarette F., Ginsberg D., Manzanares J., Saez-Valero J., Garcia-Ayllon M.-S. // J. of Alzheimer’s Dis. 2016. V. 53. P. 831–841.
Ferrari G.V., Canales M.A., Shin I., Weiner L.M., Silman I., Inestrosa N.C. // Biochemistry. 2001. V. 40. P. 10447–10457.
Inestrosa N.C., Sagal J.P., Colombres M. // Subcell. Biochem. 2005. V. 38. P. 299–317.
Inestrosa N.C., Dinamarca M.C., Alvarez A. // FEBS J. 2008. V. 275. № 4. P. 625–632.
Renn B.N., Asghar-Ali A.A., Thielke S., Catic A., Martini S.R., Mitchell B.G., Kunik M.E. // Am. J. Geriatr. Psychiatry. 2018. V. 26. № 2. P. 134–147.
Wang Yu., Wang H., Chen H.-Zh. // Current Neuropharmacology. 2016. V. 14. P. 346–375.
Darvesh S., Reid G.A. // Chem. Biol. Ineract. 2016. V. 259 (Pt B). P. 307–312.
Diamant S., Podoly E., Friedler A., Ligumsky H., Livnah O., Soreq H. // PNAS. 2006. V. 3. № 23. P. 8628–8633.
Stasiuk M., Janiszewska A., Kozubek A. // Nutrients. 2014. V. 6. P. 1823–1831.
Takahashi K., Takisawa S., Shimokado K., Kono N., Arai H., Ishigami A. // Eur. J. Nutr. 2017. V. 56. P. 1317–1327.
Vatassery G.T. // Lipids. 1978. V. 13. № 11. P. 828–831.
Vatassery G.T., Angerhofer C.K., Knox C.A., Deshmukh D.S. // Biochim. Biophys. Acta. 1984. V. 792. № 2. P. 118–122.
Shadnia S., Dasgar M., Taghikhani S., Mohammadirad A., Khorasani R., Abdollahi M. // Toxicol. Mech. Methods. 2007. V. 17. № 2. P. 109–115.
Hajos F. // Brain Res. 1975. V. 93. № 3. P. 485–489.
Lowry O. H., Rosebrough N. J., Fare A. L., Randall R. J // J. Biol. Chem. 1951. V. 193. № 1. P. 265–275.
Ellman G.L., Courtney K.D., Andres V. Jr., Featherstone R.M. // Biochem. Pharmacol. 1961. № 7. P. 88–96.
Worek F., Mast U., Kiderlen D., Diepold C., Eyer P. // Clin. Chim. Acta. 1999. V. 88. № 1–2. P. 73–90.
Steinberg N., Roth E., Silman I. // Biochemistry International. 1990. V. 21. № 6. P. 1043–1050.
Chelliah J., Smith J.D., Fariss M.W. // BBA. 1994. V. 1206. P. 17–26.
Johnson G., Moore S.W. // Current Pharmaceutical Design. 2006. V. 12. P. 217–225.
Kosenko E.A., Aliev G., Tikhonova L.A., Li Y., Poghosyan A.C., Kaminsky Y.G. // CNS Neurol. Disord. Drug Targets. 2012. V. 7. P. 926–932.
Carelli-Alinovi C., Misiti F. // Frontiers in Aging Neuroscience. 2017. V. 9. Article 276. doi 10.3389/fnagi2017.00276
Gilca M., Lixandru D., Gaman L., Vîrgolici B., Atanasiu V., Stoian I. // Alzheimer. Dis. Assoc. Disord. 2014. V. 28. P. 358–363.
Фаткуллина Л.Д., Молочкина Е.М., Зорина О.М., Подчуфарова Д.Е., Гаврилова С.И., Федорова Я.Б., Клюшник Т.П., Бурлакова Е.Б. // Журн. неврологии и психиатрии им. С.С. Корсакова. 2013. Т. 113. № 6. С. 62–67.
Lan J., Liu J., Zhao P., Xue R., Zhang N., Zhang P., Zhao P., Zheng F., Sun X. // Age and Ageing. 2015. V. 44. № 3. P. 458–464.
Joshi. Y.B., Pratico D. // Biofactors. 2012. V. 38. № 2. P. 90–97.
Belov V.V., Maltseva E.L., Palmina N.P. // Chemical Reactions in Gas, Solid and Liquid Phases. Synthesis, Properties and Application. / Ed. Zaikov G.E. N.Y.: Nova Publisher Inc., 2011. P. 29–43.
Azzi A. // Free Radic. Biol. Med. 2007. V. 43. P. 16–21.
Brigelius-Flohe R. // Free Rad. Biol. Med. 2009. V. 46. P. 545–554.
Azzi A., Gysin L, Kempna P., Munteanu A., Villacorta L., Visarius T., Zingg J.H. // Biol. Chem. 2004. V. 385. № 7. P. 585–591.
Hicks D., John D., Makova N.Z., Henderson Z., Nalivaeva N.N., Turner A.J. // J. Neurochem. 2011. V. 116. № 5. P. 742–746.
Atkinson J., Epand R.-F., Epand R.-M. // Free Radic. Biol. Med. 2008. V. 44. P. 739–764.
Xie H.Q., Leung K.W., Chen V.P., Chan G.K., Xu S.L., Guo A.J., Zhu K.Y., Zheng K.Y., Bi C.W., Zhan J.Y., Chan W.K., Choi R.C., Tsim K.W. // Chem. Biol. Interact. 2010. V. 187. № 1–3. P. 78–83.
Atkinson J., Harroun Th., Wassall R., Stillwell W., Katsaras J. // Mol. Nutr. Food Res. 2010. V. 54. P. 641–651.
Stocker A., Azzi A. // Antioxidants and Redox Signaling. 2000. V. 2. № 3. P. 397–404.
Nava Ph., Ceccini M., Chirico S., Gordon H., Morley S., Manor D., Atkinson J. // Bioorganic and Medicinal Chemistry. 2006. V. 14. P. 3721–3736.
Дополнительные материалы отсутствуют.