

Нейрохимия, 2019, T. 36, № 1, стр. 78-83
Влияние меседина на содержание биомаркеров оксидативного стресса в мозговой ткани при ишемии
А. Г. Тананян 1, М. Г. Баласанян 1, А. В. Байков 1, Л. М. Овсепян 2, Г. С. Казарян 2
1 Ереванский государственный медицинский университет им. М. Гераци, кафедра фармакологии
Ереван, Армения
2 Институт молекулярной биологии НАН РА
Ереван, Армения
Поступила в редакцию 06.11.2017
После доработки 12.07.2018
Принята к публикации 16.06.2018
Аннотация
Изучено влияние α2-адреноблокатора меседина (2-(2-метиламино-4-тиазолил)-1,4-бензодиоксана гидрохлорид) на содержание биомаркеров оксидативного стресса в мозговой ткани в условиях ишемических нарушений. Целью исследования явилось выявление возможных механизмов антигипоксантного действия меседина. В условиях нарушения мозгового кровообращения, вызванного перевязкой правой общей сонной артерии, исследованы сдвиги содержания конечного продукта перекисного окисления липидов (ПОЛ) – малонового диальдегида (МДА) и карбонильных производных белков – альдегид-динитрофенилгидразонов нейтрального характера (АДНФГuv), кетон-динитрофенилгидразонов нейтрального характера (КДНФГuv), альдегид-динитрофенилгидразонов основного характера (АДНФГvs) и кетон-динитрофенилгидразонов основного характера (КДНФГvs) в мозговой ткани крыс (n = 56). Результаты экспериментов позволили выявить один из возможных механизмов антигипоксантного действия меседина – его способность предотвращать накопление малонового диальдегида и окислительное карбонилирование белков в условиях ишемических нарушений мозга. Полученные данные указывают на то, что меседин может быть рассмотрен в качестве потенциального средства коррекции церебральной ишемии, поскольку наряду с выявленной ранее способностью улучшать мозговое кровообращение, предотвращать развитие морфологических сдвигов и нейроповеденческих расстройств, препарат смягчает агрессивное влияние оксидативного стресса, защищая мозговую ткань от последствий гипоксии.
ВВЕДЕНИЕ
В патогенезе цереброваскулярных заболеваний, являющихся одной из основных причин инвалидизации и смертности населения, решающая роль отводится оксидативному стрессу, включая свободнорадикальное и перекисное окисление, приводящее к повреждению клеточных мембран и гибели клеток [1]. Ишемия мозга, независимо от патогенеза, сопровождается каскадом патобиохимических реакций или ишемическим каскадом, который при недостаточной коррекции кровоснабжения мозга приводит к необратимым повреждениям нервной ткани [2]. Согласно литературным данным, ишемический каскад развивается с первых же минут ишемического инсульта и способствует развитию оксидативного стресса, характеризующегося, интенсивным генерированием активных форм кислорода (АФК), активацией свободнорадикальных процессов, повышением синтеза NO [3]. Этим обусловлена довольно высокая эффективность антигипоксантов и антиоксидантов, включая соединения, улучшающие локальную гемодинамику, синтетические переносчики кислорода, ГАМК-ергические соединения, агонисты дофаминовых рецепторов, стимуляторы энергетического обмена, регуляторы кислотно-щелочного баланса и др. в профилактике и лечении ишемических инсультов [4–6].
Ввиду накопления избыточного количества АФК развитие оксидативного стресса сопровождается повреждением липидов, нуклеиновых кислот, белков, углеводов, приводя к гибели нервных клеток. В молекулярных механизмах оксидативного стресса основная роль отводится перекисному окислению липидов (ПОЛ), продукты которого, наряду с другими индукторами окисления (АФК, свободное железо и др.), индуцируют окислительную модификацию белков (ОМБ) и нуклеиновых кислот [4, 6, 7].
Продукт ПОЛ – малоновый диальдегид (МДА), являющийся одним из наиболее ранних маркеров оксидативного стресса [8], повышает уровень других маркеров оксидативного стресса – продуктов ОМБ [7, 9].
ОМБ лежит в основе нарушений, а порой и полной потери специфических функций белка [10], в связи с чем окислительная модификация ферментов и структурных белков может играть важную роль в этиологии ряда патологий [11].
Таким образом, активация свободнорадикального и перекисного окисления липидов и ОМБ при патологических состояниях позволяет рассматривать указанные процессы в качестве фармакологической мишени антигипоксантов и антиоксидантов [12].
Все вышеизложенное явилось основой для исследования влияния синтезированного в Институте тонкой органической химии им. А.Л. Мнджояна НАН РА α2-адреноблокатора меседина (2-(2-метиламино-4-тиазолил)-1,4-бензодиоксана гидрохлорид) [13], обладающего антигипоксантной активностью, на уровень маркеров гипоксии [11, 14].
Ранее проведенными нами исследованиями установлено, что меседин в условиях экспериментального нарушения кровоснабжения мозга улучшает локальный корковый мозговой кровоток [15], предотвращает развитие морфологических сдвигов, и устраняет нейроповеденческие последствия локальной ишемии [16, 17].
МЕТОДЫ ИССЛЕДОВАНИЯ
Эксперименты проводились на белых беспородных крысах-самцах (n = 56) весом 170–220 г. Ишемия мозга моделировалась односторонней перевязкой правой общей сонной артерии (ПОСА) в условиях хлоралгидратной анестезии (400 мг/кг, в/б) [18–20]. Оксидативный стресс у животных оценивался интенсивностью процесса ПОЛ определением уровня МДА в аскорбатзависимой системе и оценкой ОМБ по содержанию карбонильных производных белков.
В основе метода определения интенсивности процесса ПОЛ в аскорбатзависимой системе окисления лежит реакция взаимодействия МДА с тиобарбитуровой кислотой, которая протекает при высокой температуре в кислой среде и приводит к образованию цветного триметинового комплекса [21].
Для количественного определения МДА в мозговой ткани животных отдельно отбиралось каждое полушарие, для гомогенизации добавлялся Трис HCl буфер в пересчете 10 мл на 1 г ткани. 0.1 мл гомогената добавляли в инкубационную смесь, содержащую 0.3 мл 40 мМ Трис HCl буфера (pH 7.4), 0.3 мл соли Мора (Fe(NH4)2(SO4)2 · 6H2O 10–6 M) и 0.3 мл 0.8 мМ аскорбиновой кислоты. Инкубация, при которой протекал процесс ПОЛ, осуществлялась в течение 30 мин на водяной бане при 37°C в условиях постоянного встряхивания пробирок [22]. После этого реакция прекращалась добавлением 1 мл 30% трихлоруксусной кислоты, а осадок отделялся центрифугированием в течение 10 мин при 3000 об./мин. К 1 мл надосадочной жидкости добавляли 0.2 мл 0.6 М HCl и 0.8 мл 0.12 M тиобарбитуровой кислоты, после чего смесь нагревали при 100°C в течение 10 мин. Интенсивность окрашивания оценивалась спектрофотометрически при длине волны 535 нм. Расчет количества МДА производился по общепринятой формуле с использованием коэффициента молярнoй экстинкции ε = 1.56 × 105 моль–1 см–1.
Для осуществления вышеупомянутого метода животные (n = 28) были разделены на 3 группы:
I – животные контрольной (ложнооперированной) группы (n = 6), не подвергшиеся ПОСА до декапитации.
II – группа ишемизированных животных (n = 11), получавших физиологический раствор непосредственно после ПОСА и декапитированных через 60 мин.
III – группа животных (n = 11), получавших меседин в/б сразу после ПОСА в дозе 10 мг/кг и подвергнутых декапитации через 60 мин.
Для оценки окислительной модификации белков опрелялся уровень карбонильных производных белков по методу R.L. Levine в модификации Е.Е. Дубининой [23]. Метод оценки ОМБ основан на реакции взаимодействия карбонильных производных окисленных аминокислотных остатков белков с 2,4-динитрофенилгидразином (2,4-ДНФГ) с образованием 2,4-динитрофенилгидразонов [10, 24].
Для количественного определения 2,4-динитрофенилгидразонов в мозговой ткани животных отдельно отбиралось каждое полушарие и для гомогенизации добавлялся 0.25 М раствор сахарозы в пересчете 5 мл на 0.5 г ткани. Во избежание искажения показателей уровня карбонильных производных белков из полученных гомогенатов удалялись нуклеиновые кислоты путем осаждения добавлением к 0.5 мл гомогената 4.5 мл 10% раствора стрептомицина сульфата, оставляя при комнатной температуре в течение 15 мин. Затем смесь центрифугировалась в течение 10 мин при 800 g. К 0.1 мл надосадочной жидкости добавляли 0.9 мл 20% трихлоруксусной кислоты (для осаждения белков), 1 мл 0.1 М раствора 2.4-ДНФГ и 1 мл 2 М HCl. Далее смесь инкубировалась при комнатной температуре в течение 1 часа после чего центрифугировалась 15 мин при 3000 об./мин. Полученный осадок трижды промывался 0.5 мл смесью этанола (96%) и этилацетата в соотношении 1 : 1 для экстракции липидов и 2.4-ДНФГ, не прореагировавших с карбонильными производными окисленных белков, а затем центрифугировался 15 мин при 3000 об./мин. Промытый осадок высушивался для удаления остатков этанол-этилацетата, затем растворялся в 2.5 мл 8 М раствора мочевины с добавлением 1 капли HCl. Раствор помещался в водяную баню на 10 мин, после чего охлаждался до комнатной температуры. Карбонильные производные окисленных белков регистрировались спектрофотометрически: альдегид-динитрофенилгидразоны нейтрального характера (АДНФГuv) и кетон-динитрофенилгидразоны нейтрального характера (КДНФГuv) в ультрафиолетовой области спектра при длинах волн 356 и 370 нм сответственно, а альдегид-динитрофенилгидразоны основного характера (АДНФГvs) и кетон-динитрофенилгидразоны основного характера (КДНФГvs) в области видимого света при 430 и 530 нм, соответственно. Количество полученных в результате карбонилирования продуктов выражалось в условных единицах оптической плотности (е. о. п) в расчете на 1 г ткани [25].
Для осуществления вышеупомянутого метода животные (n = 28) были разделены на 3 группы:
I – животные контрольной (ложнооперированной) группы (n = 8), не подвергшиеся ПОСА до декапитации.
II – группа ишемизированных животных (n = 10), получавших физиологический раствор непосредственно после ПОСА и декапитированных через 60 мин.
III – группа животных (n = 10), получавших меседин в/б сразу после ПОСА в дозе 10 мг/кг и подвергнутых декапитации через 60 мин.
Статистическая обработка данных производилась с использованием программы Microsoft Excel 2010, с помощью однокомпонентного аналитического метода ANOVA, достоверность полученных отклонений оценивалась по t-критерию Стъюдента. Данные представлены M ± σx.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Изучение процесса ПОЛ в условиях экспериментальной ишемии показало, что нарушения кровообращения мозга сопровождаются повышением уровня МДА в гомогенатax мозгoвой ткани. Количество МДА у контрольных животных при аскорбатзависимом ПОЛ составило 1.86 ± 0.21 мкмоль/г ткани в правом и 1.86 ± 0.25 мкмоль/г ткани в левом полушариях. По сравнению с контрольными животными у ишемизированных особей через 1 час после перевязки правой общей сонной артерии был зарегистрирован более высокий уровень МДА, равный 3.95 ± 0.33 мкмоль/г ткани в правом и 3.52 ± 0.28 мкмоль/г ткани в левом полушарии (p < 0.0005).
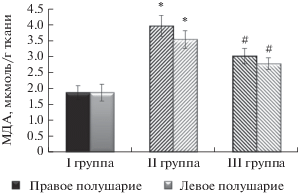
Результаты исследования показали, что внутрибрюшинное введение меседина в дозе 10 мг/кг ишемизированным животным непосредственно после ПОСА, в течение часа достоверно (p < < 0.0005) снижает количество МДА в обоих полушариях мозга: на 45.45% – в правом и на 45.18% – в левом полушариях по сравнению с ишемизированными животными, не получавшими меседин (рис. 1).
Рис. 1.
Влияние меседина на содержание МДА в мозговой ткани. * p < 0.0005 по сравнению с контрольной группой, # p < 0.0005 по сравнению с группой ишемизированных животных (M ± σx). I – животные контрольной (ложнооперированной) группы (n = 6), не подвергшиеся ПОСА до декапитации, II – группа ишемизированных животных (n = 11), получавших физиологический раствор непосредственно после ПОСА и декапитированных через 60 мин, III – группа животных (n = 11), получавших меседин в/б сразу после ПОСА в дозе 10 мг/кг и подвергнутых декапитации через 60 мин.
Исследование процесса ОМБ в условиях экспериментальной ишемии показало, что цереброваскулярные нарушения, вызванные ПОСА, сопровождаются статистически достоверным (p < 0.005) повышением основных показателей, характеризующих окисление белков, в гомогенатах мозговой ткани по сравнению с контрольной группой.
Как видно из табл. 1, у контрольных животных в обоих полушариях уровни исследуемых показателей (АДНФГuv, АДНФГvs, КДНФГuv и КДНФГvs) не отличались.
Таблица 1.
Влияние меседина на содержание карбонильных производных белков (е. о. п./г ткани)
| Карбонильныe производныe белков в е.о.п./г ткани | I группа (n = 8) |
II группа (n = 10) |
III группа (n = 10) |
|||
|---|---|---|---|---|---|---|
| правое полушарие |
левое полушарие |
правое полушарие |
левое полушарие |
правое полушарие |
левое полушарие |
|
| АДНФГuv | 7.62 ± 0.87 | 7.59 ± 0.81 | 32.52 ±5.20* | 32.71 ± 5.99* | 10.40 ± 0.97# | 11.09 ± 1.15# |
| АДНФГvs | 6.48 ± 0.38 | 6.53 ± 0.39 | 27.69 ± 4.35* | 25.38 ± 3.89* | 13.40 ± 0.81# | 13.52 ± 0.66# |
| КДНФГuv | 6.37 ± 1.04 | 6.42 ± 1.08 | 20.33 ± 2.90* | 19.80 ± 2.61* | 9.57 ± 1.59# | 9.86 ± 1.79# |
| КДНФГvs | 6.07 ± 0.81 | 6.13 ± 0.74 | 17.54 ± 2.36* | 17.30 ± 3.36* | 10.72 ± 1.52# | 10.63 ± 1.03# |
В группе ишемизированных животных через 60 мин после ПОСА в правом и левом полушариях отмечалось более чем 4-х кратное превышение содержания АДНФГuv по сравнению с контрольной группой. Регистрировалось также и повышение уровня АДНФГvs почти в 4 раза (табл. 1).
Аналогичные изменения, но менее выраженные, отмечались и в содержании КДНФГuv и КДНФГvs. Так, в обоих полушариях уровень КДНФГuv возрос более, чем в 3 раза, а уровень КДНФГvs – в 2.8 раза (табл. 1).
Как показали эксперименты, внутрибрюшинное введение меседина в дозе 10 мг/кг непосредственно после ПОСА, в течение часа предотвращает повышение интенсивности окисления белков, о чем свидетельствует понижение уровня АДНФГuv в 3 раза, АДНФГvs – в 2, КДНФГuv – в 2 и КДНФГvs – в 1.6 раза в обоих полушариях по сравнению с группой ишемизированных животных (табл. 1).
Итак, применение меседина в условиях экспериментальной ишемии мозга приводит к значительному снижению уровня основных показателей, характеризующих ОМБ: АДНФГuv (на 88.84 и 86.07%), АДНФГvs (на 67.37 и 62.92%) КДНФГuv (на 77.08 и 74.29%) и КДНФГvs (на 59.46 и 59.71%) в правом и левом полушариях соответственно, по сравнению с ишемизированными животными, не получавшими препарат.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Выявленная эффективность лекарств с антигипоксантным и антиоксидантным действием диктует необходимость дальнейшего поиска средств коррекции церебральных ишемических нарушений среди соединений с подобной активностью [2, 6]. В указанном плане исследование меседина было обосновано полученными ранее данными относительно его антигипоксантной активности [13], превосходящей аналогичные эффекты не только α2-адреноблокаторов идазоксана и бетидина [26], но и хорошо известных антигипоксантов, какими являются винпоцетин и пирацетам [27, 28].
Все вышеизложенное явилось основой для исследования влияния меседина на уровень маркеров гипоксии.
Результаты экспериментального исследования свидетельствуют о том, что в условиях церебральных ишемических нарушений, вызванных перевязкой правой общей сонной артерии, в мозговой ткани отмечается повышение количества МДА, причем направленность отмеченных изменений особо не отличается в правом (с перевязкой) и контралатеральном полушариях. Одновременно с накоплением конечного продукта перекисного окисления липидов в исследуемой модели ишемии мозга индуцируется также и процесс карбонилирования белков, о чем свидетельствует повышение количества динитрофенилгидразонов, причем преимущественно за счет первичных маркеров оксидативного стресса – альдегидных производных, что указывает на персистенцию фрагментов белков, нарушенных в результате активации процессов окисления в мозговой ткани. Хотя отмечается также и повышение количества вторичных маркеров – кетоновых производных, степень выраженности карбонилирования с образованием кетоновых агрегантов модифицированных белков уступает их изменению по типу концевого окисления, что согласуется с данными, ранее полученными другими авторами [29].
Таким образом, проведенные исследования свидетельствуют о том, что выявленная ранее антигипоксантная активность меседина проявляется также в условиях цереброваскулярных нарушений, вызванных перевязкой общей сонной артерии. Доказательством этого является способность меседина значительно предотвращать повышение уровня МДА, а также АДНФГuv, АДНФГvs, КДНФГuv и КДНФГvs, что объясняет возможные механизмы антигипоксантного действия – предотвращение перекисного окисления липидов и окислительной модификации белков.
Все это свидетельствует о том, что применение меседина в качестве потенциального средства, корригирующего цереброваскулярные ишемические нарушения обосновано, так как наряду с выявленной способностью улучшать мозговое кровообращение, предотвращать развитие ишемических структурных изменений и нейроповеденческих расстройств [30], исследованное соединение смягчает агрессивное влияние оксидативного стресса, защищая мозговую ткань в условиях гипоксии.
ВЫВОДЫ
1. Односторонняя перевязка правой общей сонной артерии сопровождается повышением уровня малонового диальдегида почти в 2 раза в правом и на 89.25% в левом полушарии.
2. Внутрибрюшинное введение меседина в дозе 10 мг/кг способствует снижению уровня малонового диальдегида в условиях ишемических нарушений мозга на 45.45% в правом и 45.18% в левом полушарии.
3. В условиях ишемизации мозга отмечается усиление процесса окислительной модификации белков: повышение содержания АДНФГuv более чем в 4 раза, АДНФГvs почти в 4 раза, КДНФГuv более чем в 3 раза и КДНФГvs – в 2.8 раза в правом и левом полушариях.
4. Внутрибрюшинное введение меседина в дозе 10 мг/кг снижает уровень основных показателей, характеризующих окислительную модификацию белков: АДНФГuv (на 88.84 и 86.07%), АДНФГvs (на 67.37 и 62.92%) КДНФГuv (на 77.08 и 74.29%) и КДНФГvs (на 59.46 и 59.71%) в правом и левом полушариях, соответственно.
5. Вышеперечисленные эффекты меседина указывают на выраженную антигипоксантную активность соединения.
Список литературы
Birben E., Sahiner U.M., Sackesen C., Erzurum S., Kalayci O. // World Allergy Organ J. 2012. V. 5. № 1. P. 9–19.
Ганцгорн Е.В., Хлопонин Д.П., Макляков Ю.С. // Медицинский вестник Юга России. 2013. № 2. С. 4–12.
Маслюкова А.В., Томилова И.К., Баклушина Е.А. // Вестник ИвГМА. 2015. Т. 20. № 1. С. 37–44.
Муравлева Л.Е., Молотов-Лучанский В.Б., Клюев Д.А., Бакенова Р.А., Култанов Б.Ж., Танкибаева Н.А., Койков В.В., Омарова Г.А. // Современные проблемы науки и образования. 2010. № 1. С. 74–78.
Chen H., Yoshioka H., Kim G.S., Jung J.E., Okami N., Sakata H., Maier C.M., Narasimhan P., Goeders C.E., Chan P.H. // Antioxid. Redox Signal. 2011. V. 14. № 8. P. 1505–1517.
Shirley R., Ord E.N.J., Work L.M. // Antioxidants. 2014. V. 3. № 3. P. 472–501.
Мартусевич А.К., Карузин К.А. // Биорадикалы и антиоксиданты. 2015. Т. 2. № 2. С. 5–18.
Некрасов Э.В. // Бюл. физ. и пат. дых. 2012. № 46. С. 98–108.
Арапова А.И., Фомина М.А. // Вестник ПГУ. Биология. 2016. № 1. С. 75–80.
Wong C.M., Marcocci L., Liu L., Suzuki Y.J. // Antioxid. Redox Signal. 2010. V. 12. № 3. P. 393–404.
Dalle-Donne I., Rossi R., Giustarini D., Milzani A., Colombo R. // Clin. Chim. Acta. 2003. V. 329. № 1–2. P. 23–38.
Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., Milzani A. // J. Cell. Mol. Med. 2006. V. 10. № 2. P. 389–406.
Ширинян Э.А., Арутюнян С.А., Гукасяан Т.Г. // Вестник МАНЭБ. 2003. Т. 8. № 4. С. 132–134.
Suzuki Y.J., Carini M., Butterfield D.A. // Antioxid. Redox Signal. 2010. V. 12. № 3. P. 323–325.
Тананян А.Г., Баласанян М.Г., Ширинян Э.А. // Ежегодная отчетная научная конференция ЕГМУ. Сборник научных статей. Ереван: ЕГМУ, 2013. Т. 1. С. 209–213.
Tananyan A.G., Balasanyan M.G. // Eur. Neuropsychopharmacol. 2014. V. 24. № 2. P. 610.
Тананян А. Г., Баласанян М. Г., Хостикян Н. Г., Марданян Л. С., Ерицян Э. Л. // Медицинская наука Армении. 2017. Т. LVII. № 2. С. 33–42.
Мясищева О.В., Покровский М.В., Гуреев В.В., Анциферов О.В., Мартынов М.А. // Научные ведомости БелГУ. Серия: Медицина. Фармация. 2014. Т. 26-1. № 11. С. 123–126.
Bacigaluppi M., Comi G., Hermann D.M. // Open Neurol. J. 2010. V. 4. P. 34–38.
Bajko Z., Balasa R., Motataianu A., Maier S., Chebut O.C., Szatmari S. // ISRN Neurol. 2013. V. 2013. P. 1–8.
Khoubnasabjafari M., Ansarin K., Jouyban A. // Bioimpacts. 2015. V. 5. № 3. P. 123–127.
Владимиров Ю.А., Арчаков А.И., Франк Г.М. // Перекисное окисление липидов в биологических мембранах. М.: Наука, 1972. 252 с.
Дубинина Е.Е. // Продукты метаболизма кислорода в функциональной активности клеток (жизнь и смерть, созидание и разрушение). Физиологические и клинико-биохимические аспекты. СПб.: Мед. пресса, 2006. 397 с.
Боев К.В., Василенко Д.В., Маслов А. И. // Universum: химия и биология. 2014. № 1. С. 4.
Абаленихина Ю.В., Фомина М.А., Чурилов Г.И., Иванычева Ю.Н. // Фундаментальные исследования. 2012. № 11. С. 1315–1319.
Медведев О.С., Мирзоян С.А., Ширинян Э.А., Петросян А.А., Гукасян Т.Г., Арутюнян С.А. // Эксп. клин. фармакол. 2000. № 4. С. 20–23.
Solanki P., Prasad D., Muthuraju S., Sharma A.K., Singh S.B., Ilavzhagan G. // Food Chem. Toxicol. 2011. V. 49. № 4. P. 917–922.
Patyar S., Prakash A., Modi M., Medhi B. // Pharmacol Rep. 2011. V. 63. № 3. P. 618–628.
Ильичева А.С., Фомина М.А. // Рос. мед.-биол. вестн. им. акад. И.П. Павлова. 2015. № 1. С. 45–51.
Тананян А.Г. // Медицинская наука Армении. 2017. Т. LVII. № 1. С. 89–98.
Дополнительные материалы отсутствуют.