

Нейрохимия, 2019, T. 36, № 2, стр. 140-148
Проапоптотический эффект мелатонина на функционирование неспецифической митохондриальной поры (мPTP) в митохондриях мозга крыс
Ю. Л. Бабурина 1, И. В. Одинокова 1, О. В. Крестинина 1
1 ФГБУН Институт теоретической и экспериментальной биофизики РАН
Пущино, Россия
Поступила в редакцию 07.06.2018
После доработки 20.07.2018
Принята к публикации 08.08.2018
Аннотация
Мелатонин (МЕЛ) является гормоном, вырабатываемом шишковидной железой. Легко проникая через клеточную мембрану, он способен накапливаться в митохондриях в высоких концентрациях и улучшать их функциональное состояние. Однако существуют также данные, показывающие, что действие МЕЛ на митохондрии может сильно варьировать, т.е. имеет высокую специфичность в зависимости от видов тканей, условий введения, состава и структурных особенности молекул-мишеней. В настоящей работе мы исследовали влияние МЕЛ на функциональное состояние митохондрий мозга крыс при его непосредственном добавлении к митохондриям в условиях открытия неспецифической поры митохондрий (мРТР), а также на изменение уровней регуляторов мРТР (VDAC и CNPазы) и основных субъединиц комплексов электронной транспортной цепи (ЭТЦ). Было обнаружено, что непосредственное добавление МЕЛ в концентрации 10 нМ и 100 нМ к митохондриям приводит к изменению уровня субъединиц I и IV комплексов ЭТЦ, инициации открытия мРТР, а также к уменьшению уровней белков регуляторов функционирования мРТР. Полученные данные позволяют показать возможный потенциал МЕЛ как проапоптотического агента, что может иметь важное биологическое и фармакологическое значение.
Мелатонин (N-ацетил-5-метокситриптамин или, согласно аббревиатуре IUPAC, N-[2-(5-метокси-1Н-индол-3-ил)этил]ацетамид, МЕЛ) – это нейроэндокринный гормон, вырабатываемый шишковидной железой (эпифизом). МЕЛ широко распространен в природе, его присутствие показано в клетках и тканях позвоночных, беспозвоночных, растений, одноклеточных эукариот, водорослей и даже бактерий [1–5]. Благодаря своей амфифильной природе, молекулы МЕЛ способны легко проникать через мембраны, что позволяет ему свободно распространяться по всему организму и достигать всех субклеточных структур [6]. Обладая всеми свойствами, характерными для гормонов, МЕЛ также имеет особенности, отличающие его от классических гормонов. Синтезируемый шишковидной железой, он переносится в свободной или альбумин-связанной форме [7–9] и может действовать через специфические G-белок-связанные мембранные рецепторы (MT1 или MTRN1a, MT2 или MTRN1b и MT3) [10, 11], а также, предположительно, через ядерные ретиноидные рецепторы RZR/ROR [12, 13]. Следует отметить, что механизмы действия МЕЛ по удалению свободных радикалов считают рецептор-независимыми [14].
Свободное распространение и функциональное разнообразие МЕЛ обуславливает широкий диапазон его функций. На данный момент, помимо функции регуляции циркадных ритмов, показано его противовоспалительное, антиоксидантное, онкостатическое действие [15]. Существует также множество данных о влиянии МЕЛ на функции митохондрий. Как известно, митохондрии играют ключевую роль в большом количестве физиологических процессов в клетке. Нарушение функций митохондрий рассматривается как основной фактор процессов старения, ишемии/реперфузии, септического шока и нейродегенеративных заболеваний [16, 17]. МЕЛ способен, проникая через клеточные мембраны, накапливаться в митохондриях в высоких концентрациях, в сотни раз превышающих его концентрацию в цитозоле [18, 19]. Многочисленные исследования показывают, что МЕЛ способен улучшать функциональное состояние митохондрий. Хроническое введение МЕЛ в фармакологической дозе предотвращает нарушение функций митохондрий в моделях экспериментального диабета и интоксикации, демонстрируя митохондрий-специфическую активность МЕЛ [20–22]. Являясь хорошо известным антиоксидантом, МЕЛ способен эффективно связывать свободные радикалы всех типов [23], однако, кроме непосредственного снижения токсического действия активных форм кислорода (АФК), действие МЕЛ направлено на восстановление активности ферментов-антиоксидантов и предотвращение увеличения проницаемости внутренней мембраны митохондрий. Эксперименты in vitro показали, что МЕЛ увеличивает активность митохондриальной ЭTЦ, защищая митохондрии от окислительного стресса, вызванного т-бутилгидропероксидом [24]. Было показано, что действие МЕЛ в митохондриях может быть направлено на регуляцию Ca2+-индуцированной, циклоспорин А-чувствительной поры (Mitochondrial Permeability Transition Pore, мPTP), которая формируется вследствие накопления сверхпороговых концентраций Ca2+ или в ответ на окислительный стресс и которая считается начальной стадией апоптоза [25]. В этих условиях в митохондриях наблюдается разобщение дыхания и фосфорилирования, ингибирование АТР-синтетазной активности, активация АТР-азной активности, и как следствие, увеличение гидролиза АТР [25]. Обнаружено, что введение МЕЛ способно не только предотвращать индукцию мPTP, но также и поддерживать митохондриальный потенциал (ΔΨm) и сохранять ΔΨm-зависимое образование АТФ [26]. Недавно мы показали защитное действие МЕЛ при его хроническом введении на индукцию мPTP при старении [27, 28].
Однако следует отметить, что действие МЕЛ на митохондрии может варьироваться в зависимости от экспериментальных условий, а также имеет высокую специфичность в отношении разных органов и тканей [29]. Так, наряду с антиапоптотическим действием МЕЛ, было показано, что МЕЛ индуцирует митохондриально-опосредованный апоптоз в клетках HL-60 [30]. Кроме того, он может проявлять себя в качестве восстановительного или окислительного агента, в зависимости от структурных особенности молекул-мишеней и их среды, времени инкубации и используемых концентраций [31]. В данной работе было исследовано влияние непосредственного добавления различных концентраций МЕЛ (10 и 100 нМ) к суспензии митохондрий мозга крыс на функциональное состояние митохондрий, изменения уровней субъединиц комплексов ЭТЦ, а также на изменение содержания митохондриальных белков таких как VDAC и CNPаза, принимающих участие в функционировании мPTP.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение митохондрий мозга. Митохондрии мозга крысы выделяли по методу Симса [32], модифицированному в нашей лаборатории. Изолированный мозг измельчали, очищали от кровеносных сосудов и разрушали в десятикратном объеме среды, содержавшей 320 мМ сахарозу, 10 мМ Трис-HCl (pH 7.4), 0.5 мМ К+-EDTA, 0.5 мМ EGTA, 0.2% бычий сывороточный альбумин (БСА) (все реактивы фирмы Sigma, США), с помощью стеклянного гомогенизатора. Гомогенат центрифугировали при 2000 g в течение 3 мин, осадок удаляли, а супернатант центрифугировали повторно для более полного удаления ядер и поврежденных клеток. Осадок митохондрий, полученный центрифугированием при 12 500 g в течение 10 мин, промывали при 11500 g в течение 10 мин средой выделения, не содержащей EGTA, EDTA и БСА, и суспендировали в той же среде. Все процедуры выполняли при температуре 4°С. Концентрацию белка определяли по методу Бредфорд. Концентрация белка в изолированных митохондриях мозга составляла 25–30 мг/мл.
Определение митохондриальных функций. Митохондриальные параметры измеряли в термостатируемой ячейке с вмонтированными микроэлектродами, селективными к ионам Са2+ (“Нико Аналит”, Россия). С помощью Са2+-селективного электрода оценивали транспорт Са2+ митохондриями. Селективные электроды измеряют концентрацию соответствующих ионов во внешней среде, т.е. уменьшение концентрации ионов Са2+ в среде инкубации митохондрий означает их вход в митохондрии, а увеличение – выход. Митохондрии инкубировали в среде, содержащей 125 мМ KCl, 10 мМ Tris-HCl (рН 7.4), 0.4 мМ KH2PO4, 5 мМ сукцинат калия. В качестве субстрата дыхания использовали глутамат (5 мМ) и малат (5 мМ). Дыхательный контроль (respiratory control index, RCI) измеряли в закрытой ячейке после добавления 200 мкМ АДФ. Концентрация митохондриального белка в ячейке составляла 1 мг/мл. Открытие мРТР в митохондриях индуцировали последовательным увеличением концентрации внешнего Са2+ на 50 мкМ до достижения пороговой концентрации, при которой происходил выход Са2+ из митохондрий. МЕЛ добавляли в выбранной концентрации однократно после инкубации с митохондриями. Для численной оценки параметров митохондрий использовали значения пороговой концентрации Са2+, необходимого для инициации мРТР.
Набухание митохондрий мозга крысы определяли по изменению рассеивания в митохондриальной суспензии при длине волны 540 нм (спектрофотометр Tecan I Control Infinite 200). Стандартная среда инкубации содержала 125 мM KCl, 10 мM Tris, 2 мM KH2PO4, 5 мM сукцинат калия, 2.5 мкM ротенон. Концентрация белка в кювете составляла 0.35 мг/мл, измерения проводили при 25°С.
Электрофорез и Вестерн блот анализ. Для получения образцов при определении изменений уровней митохондриальных комплексов, аликвоты (2 мг/мл) нативных митохондрий мозга крысы помещали в пробирку и растворяли в буфере Лэммли. Образцы нагревали до 37°С в течение 3 мин. Для определения уровней митохондриальных белков (VDAC и CNPазы) из ячейки отбирали аликвоту объемом 50 мкл, и добавляли буфер Лэммли для солюбилизации митохондриальных белков. Образцы нагревали до 95°С в течение 5 мин. На каждую дорожку геля наносили по 20 мкг митохондриального лизата. В качестве маркеров использовали наборы фирмы Bio-Rad (США), содержащие маркерные белки от 10 до 250 кДа. Перенос белков из геля на нитроцеллюлозную мембрану (Bio-Rad, США, 0.2 мкм) осуществляли на аппарате для полусухого переноса (Bio-Rad), используя буфер для переноса (Bio-Rad) методом Вестерн блот.
Изменения в уровнях субъединиц ЭTC были обнаружены с помощью коктейля антител Total Oxphos Rodent WB Antibody Cocktail (ab 110413, моноклональные антитела). Коктейль состоит из альфа-субъединицы комплекса V (CV-ATP5A-55 кДа), core protein 2 комплекса III (CIII-UQCRC2-48 кДа), субъединицы I комплекса IV (CIV-MTCO1-40 кДа), субъединицы 30 комплекса II (CII-SDHB-30 кДа), субъединицы NDUF88-20 кДа комплекса I (CI-NDUFB8). Моноклональные антитела к COX IV (Calbiochem) и к Tom20 при разбавлении 1 : 1000 (Cell Signaling, USA) использовали для нормирования белковой нагрузки.
Моноклональные анти-CNP-антитела (анти-CNP Ab) получали, как описано [33], и использовали при разведении 1 : 10 000, антитела к VDAC (Calbiochem) – 1 : 1000. Иммунореактивность детектировали с использованием соответствующих вторичных антител, конъюгированных с пероксидазой хрена (Jackson Immuno Research, West Grove, PA, USA). Пероксидазная активность определялась с помощью ECL (Bio-Rad, Hercules, CA, USA) с использованием ChemiDoc Touch Imaging System (Bio-Rad). Количественный анализ проводили с помощью денситометрии (программа Image Lab, Bio-Rad, Hercules, CA, USA).
Статистический анализ. Для статистического анализа брали среднее значение параметров из 5–6 экспериментов ± SD. Статистическую достоверность считали с использованием t-критерия Стьюдента.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
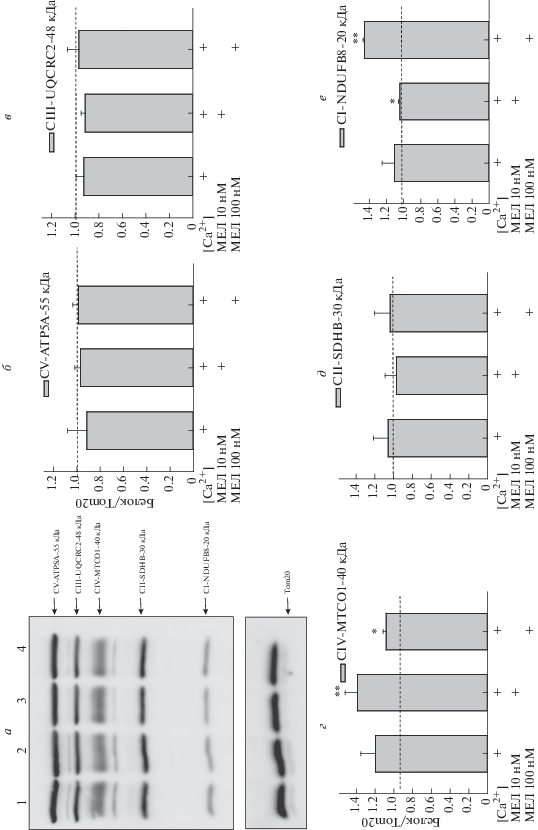
Способность МЕЛ действовать как скавенджер (поглотитель) свободных радикалов обусловлена его электронно-донорными свойствами [34]. Так как ЭТЦ связана с окисительным фосфорилированием, увеличение синтеза АТФ является одним из последствий действия МЕЛ в митохондриях. Поскольку, в литературе имеются сведения о способности МЕЛ увеличивать активность комплексов ЭТЦ, снижая формирование АФК в комплексах I и IV [35], мы провели исследование влияния МЕЛ при непосредственном его добавлении к митохондриям на изменение уровней главных субъединиц комплексов ЭТЦ как в контрольных условиях, так и в условиях открытия мPTP. Для этого к митохондриям однократно добавляли МЕЛ в выбранных концентрациях, инкубировали в среде (состав среды и условия инкубации описаны в разделе “Материалы и методы”), после чего готовили образцы для последующего анализа. Согласно литературным данным, добавление МЕЛ в концентрации 10 нМ к митохондриям мозга крыс способно увеличивать активность комплексов ЭТЦ, а в концентрации 100 нМ – значительно усиливать синтез АТФ [36], поэтому для исследования мы выбрали эти концентрации. На рис. 1, панель а представлен иммуноблот, отражающий результаты исследования, на панелях б–е – количественный анализ изменений уровня комплексов митохондриальной ЭТЦ. Как видно из рисунка, добавление выбранных концентраций МЕЛ вызывает увеличение уровня IV и I комплексов, что согласуется с литературными данными. Мы не заметили каких-либо значимых изменений в других комплексах в наших экспериментальных условиях. Следует отметить, что недавно мы показали что в митохондриях сердца крыс при острой сердечной недостаточности, МЕЛ был способен влиять на комплексы ЭТЦ [37]. Таким образом, в митохондриях в условиях мPTP возможно специфическое взаимодействие МЕЛ с комплексами I и IV, что позволяет защищать их от повреждающего действия АФК и окислительного стресса [36].
Рис. 1.
Изменения в митохондриальных комплексах ЭТЦ в присутствии/отсутствие МЕЛ при нагрузке митохондрий мозга пороговыми [Ca2+]. Образцы белка экстрагировали и подвергали Вестерн блот анализу. Изменения в митохондриальных комплексах исследовали с использованием коктейля антител Total OXPHOS Rodent WB Antibody Cocktail. a – Мембрана иммуноблота, окрашенная соответствующими антителами. Дорожка 1 – контроль, 2 – в присутствии пороговой [Ca2+], 3, 4 – в присутствии пороговой [Ca2+], а также 10 и 100 нМ МЕЛ соответственно, б – диаграмма, отражающая количественные изменения в альфа-субъединице комплекса V (CV-ATP5A-55 кДа), в – core protein 2 (CIII-UQCRC2-48 кДа) комплекса III, г – субъединицы I комплекса IV (CIV-MTCO1-40 кДа), д – субъединицы 30 комплекса II (CII-SDHB-30 кДа), e – субъединицы NDUF88-20 кДа комплекса I (CI-NDUFB8). В качестве контроля белковой нагрузки использовали антитела к Tom20. Количественные изменения отражены в единицах относительно контроля. * p < 0.05, ** p < 0.01.
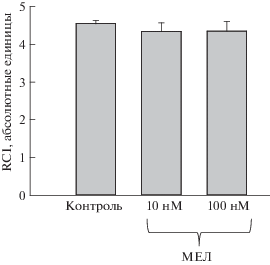
Далее мы измерили дыхательный контроль (respiratory control index, RCI) митохондрий мозга в наших экспериментальных условиях. RCI показывает эффективность митохондрий способствовать окислительному фосфорилированию и, таким образом, указывает на сопряжение между потреблением кислорода и продукцией АТФ. На рисунке показаны значения RCI для митохондрий в контрольных условиях и при непосредственном добавлении в среду инкубации различных концентраций МЕЛ. Представленные данные говорят об интактности изолированных митохондрий и сопряженности системы синтеза/гидролиза ATP. Как видно из рис. 2, никаких существенных изменений в RCI при добавлении МЕЛ к митохондриям мозга крысы не наблюдалось, что указывает на отсутствие изменений в митохондриальной мембране в данных условиях. Можно предположить, что МЕЛ при непосредственной инкубации с митохондриями не способен оказывать воздействие на систему сопряжения окислительного фосфорилирования и производства АТФ в митохондриях.
Рис. 2.
Влияние МЕЛ на дыхательный контроль (RCI) митохондрий мозга крыс. Митохондрии мозга крысы инкубировались в стандартной среде, как описано в разделе “Материалы и методы”). К суспензии митохондрий добавляли 100 мкМ АДФ, измерение проводили в закрытой ячейке.
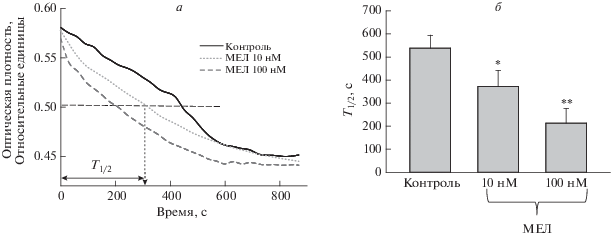
Ранее в наших исследованиях мы показали, что хроническое потребление животными МЕЛ способствует увеличению пороговой [Са2+] для открытия мPTP, снижает скорость набухания митохондрий, а также препятствует освобождению проапоптотических факторов, таких как цитохром с и митохондриальной CNPазы [28]. В рамках данного исследования мы проверили влияние МЕЛ на параметры функционирования мPTP при непосредственном его добавлении к митохондриям. Одним из главных параметров, характеризующих пору, является способность митохондрий удерживать Ca2+ в матриксе, количественной характеристикой данного параметра является пороговая концентрация Са2+ т.е. та концентрация, при которой происходит активация мРТР. На рис. 3а можно увидеть кривые изменения потоков Ca2+ в митохондриях мозга крыс в контрольных условиях и при добавлении МЕЛ (10 нМ и 100 нМ). На панели б представлены значения пороговых концентраций Ca2+ в данных условиях. Как видно из рисунка, пороговая концентрация Ca2+ достоверно снижается в присутствии 10 нМ МЕЛ на 14%, а в присутствии 100 нМ МЕЛ – на 21%. Это указывает на то, что МЕЛ облегчает открытие мPTP в митохондриях мозга крыс. Чтобы подтвердить данный эффект, мы исследовали другой параметр, характеризующий открытия мРТР – кальций-индуцированное набухание митохондрий. Добавление Ca2+ в пороговой концентрации к суспензии митохондрий, инкубированных в стандартной среде, вызывает уменьшение светорассеяния, что свидетельствует о набухании, и митохондриальные мембраны становятся проницаемыми для веществ с низкой молекулярной массой. Мы сравнили набухание митохондрий мозга крыс в контрольных условиях, и в присутствии МЕЛ. Количественно процесс набухания характеризовался временем достижения полумаксимального сигнала светорассеяния (T1/2). На рис. 4 показано, что МЕЛ увеличивает скорость набухания митохондрий, причем 100 нМ МЕЛ оказывает более сильное воздействие, чем 10 нМ. Так, в контроле T1/2 составляет в среднем 500 секунд, что на 40% больше, чем при добавлении 10 нМ МЕЛ, и более чем в 2 раза больше, чем при добавлении 100 нМ МЕЛ. Эти данные подтверждают описанные выше результаты исследования пороговой концентрации Ca2+, и позволяют сделать вывод, что непосредственное добавление МЕЛ к митохондриям приводит к инициации открытия мРТР. В то же время, как отмечалось ранее, в исследованиях нашей и других, хроническое введение МЕЛ приводит к противоположному эффекту, предотвращая открытие мPTP в митохондриях [26–28]. В связи с этим необходимо отметить, что ранее уже появлялись работы о противоречивости действий МЕЛ в зависимости от разных экспериментальных условий, состояния органелл, а также от специфичности органов и тканей [38, 39]. Так, Мартинис и соавторы показали, что в изолированных митохондриях печени крысы добавление МЕЛ в присутствии пороговых концентраций Ca2+ вызывало индукцию мPTP, увеличение окислительного стресса и освобождение проапоптотических факторов. Авторы связывают это явление с окислительным стрессом, вызванным пороговыми концентрациями Ca2+ и МЕЛ, что приводит к образованию перекиси водорода, и, как следствие к окислению тиоловых групп, глутатиона и пиридиновых нуклеотидов [29]. Следует отметить, что при накоплении в матриксе митохондрий критических концентраций АФК, когда антиоксидантные системы перестают справляться с защитой, возможен кратковременный переход мPTP в состояние низкой проводимости (superoxide flashes), при этом происходит выход АФК из митохондрий [40, 41].
Рис. 3.
Сравнительный анализ пороговой [Ca2+] в присутствии/отсутствие МЕЛ в митохондриях мозга крыс. Стандартная среда инкубации описана в разделе “Материалы и методы”. Перед добавлением пороговой [Ca2+] митохондрии инкубировались с МЕЛ в течение 5 минут. а – Транспорт Са2+ в митохондриях, б – количественный анализ пороговой [Ca2+]. *p < 0.05 сравнение относительно соответствующего контроля.
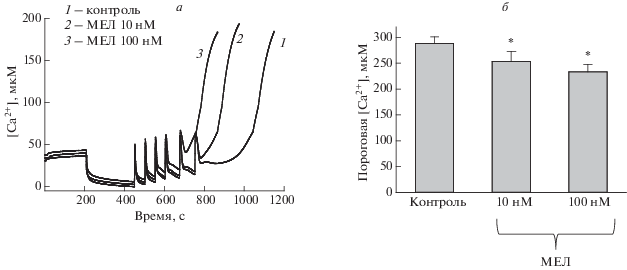
Рис. 4.
Влияние МЕЛ на набухание митохондрий мозга крыс. Набухание митохондрий крыс инициировали добавлением 300 нМ Са2+ на 1 мг белка и измеряли как изменение адсорбции митохондриальной суспензии при 540 нм при комнатной температуре. Панель а – кривые набухания митохондрий мозга в отсутствие/присутствии мелатонина, б – полупериод набухания (Т1/2) митохондрий. Концентрация белка в кювете составляла 0.35 мг/мл. *p < 0.05, **p < 0.01 сравнение относительно соответствующего контроля.
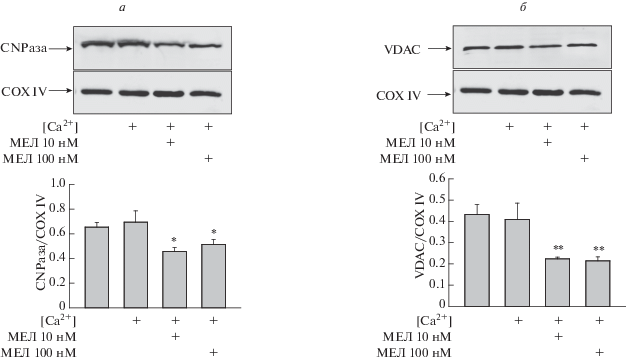
Выход же проапоптотических белков из митохондрий при функционировании мPTP рассматривают как ключевое событие митохондриального пути апоптоза. Недавно мы показали, что при открытии мРТР наряду с выходом проапоптотических белков происходит и выход CNPазы [42], белка, функции которого в митохондриях до недавнего времени оставались малоизученными. Кроме того, мы показали, что CNPаза является регулятором мPTP, а ее субстраты, 2',3'-цАМФ и 2',3'-цНАДФ, способны индуцировать открытие мРТР [43]. В рамках настоящей работы мы проверили, как изменяется уровень CNPазы в условиях открытия мРТР в присутствии различных концентраций МЕЛ. Результаты, представленные на рис. 5а, показывают, что в присутствии только пороговых концентраций Ca2+, уровень CNPазы не меняется по сравнению с контрольными условиями, что согласуется с нашими данными, полученными ранее [43]. Однако, в присутствии МЕЛ, уровень CNPазы значительно снижается (~30%), причем этот эффект не зависит от концентрации добавленного МЕЛ, достоверных различий между изменением уровня CNPазы при 10 и 100 нМ МЕЛ мы не наблюдали. Ранее мы показали, что уменьшение экспрессии CNPазы в митохондриях приводило к инициации открытия мPTP [43, 44], а также, что хроническое потребление МЕЛ способствовало удержанию CNPазы внутри митохондрий, возможно, для защиты клеток от повреждений [28]. Следовательно, можно предположить, что CNPаза может являться одной из мишеней для МЕЛ в митохондриях, а механизм воздействия МЕЛ на CNPазу, по-видимому, различается в случае хронического накопления и при непосредственном добавлении МЕЛ к митохондриям. Ранее мы показали, что в митохондриях мозга крысы VDAC колокализован с CNPазой [45], кроме того, известно, что VDAC принимает участие в регуляции мPTP [46, 47], поэтому мы проверили изменение уровня VDAC в аналогичных условиях. Как показано на рис. 5б, влияние МЕЛ на изменение содержания VDAC оказался схожим с действием МЕЛ на уровень CNPазы. Уровень VDAC не менялся при открытии мРТР, однако, снижался в два раза при добавлении как 10, так и 100 нМ МЕЛ. Этот эффект можно объяснить, по-видимому, существованием комплекса белков в митохондриальных мембранах, включающий в себя CNPазу и VDAC, который способен принимать участие в регуляции мPTP, и может являться мишенью непосредственного действия МЕЛ в митохондриях.
Рис. 5.
Изменение содержания CNPазы (а) и VDAC (б) в митохондриях мозга крыс в отсутствие/присутствии пороговой [Ca2+] и мелатонина. В качестве контроля белковой нагрузки использовали антитела к COX IV. Верхняя часть рисунка – иммуноблоты, окрашенные соответствующими антителами, нижняя часть – диаграммы, количественно отражающие изменения в содержании CNPазы в абсолютных единицах, нормированных на COX IV. * p < 0.05, ** p < 0.01 сравнение относительно соответствующего контроля.
ЗАКЛЮЧЕНИЕ
Суммируя вышеизложенное, можно заключить, что непосредственное добавление МЕЛ в концентрации 10 и 100 нМ к изолированным митохондриям мозга приводит к изменению уровней субъединиц комплексов I и IV ЭТЦ, инициации открытия мPTP, а также к уменьшению уровней белков регуляторов функционирования мРТР. Следует отметить, что данные результаты, полученные на изолированных митохондриях, предполагают продолжение исследований на более сложных биологических системах, однако, вместе с литературными данными, позволяют показать возможный потенциал МЕЛ как проапоптотического агента, что может иметь важное биологическое и фармакологическое значение.
Список литературы
Roopin M., Levy O. // J. Pineal Res. 2012. V. 53. P. 259–269.
Migliori M.L., Romanowski A., Simonetta S.H., Valdez D., Guido M., Golombek D.A. // J. Pineal Res. 2012. V. 53. P. 38–46.
Byeon Y., Park S., Kim Y.S., Park D.H., Lee S., Back K. // J. Pineal Res. 2012. V. 53. P. 107–111.
Gomez F.J., Raba J., Cerutti S., Silva M.F. // J. Pineal Res. 2012. V. 52. P. 349–355.
Stehle J.H., Saade A., Rawashdeh O., Ackermann K., Jilg A., Sebesteny T., Maronde E. // J. Pineal Res. 2011. V. 51. P. 17–43.
Slominski A., Tobin D.J., Zmijewski M.A., Wortsman J., Paus R. // Trends Endocrinol. Metab. 2008. V. 19. P. 17–24.
Morin D., Simon N., Depres-Brummer P., Levi F., Tillement J.P., Urien S. // Pharmacology. 1997. V. 54. P. 271–275.
Pardridge W.M., Mietus L.J. // J. Neurochem. 1980. V. 34. P. 1761–1763.
Cardinali D.P., Lynch H.J., Wurtman R.J. // Endocrinology. 1972. V. 91. P. 1213–1218.
Dubocovich M.L., Delagrange P., Krause D.N., Sugden D., Cardinali D.P., Olcese J. // Pharmacol. Rev. 2010. V. 62. P. 343–380.
Dubocovich M.L., Markowska M. // Endocrine. 2005. V. 27. P. 101–110.
Smirnov A.N. // Biochemistry (Mosc.). 2001. V. 66. P. 19–26.
Becker-Andre M., Wiesenberg I., Schaeren-Wiemers N., Andre E., Missbach M., Saurat J.H., Carlberg C. // J. Biol. Chem. 1994. V. 269. P. 28531–28534.
Kilic U., Yilmaz B., Ugur M., Yuksel A., Reiter R.J., Hermann D.M., Kilic E. // J. Pineal Res. 2012. V. 52. P. 228–235.
Reiter R.J., Mayo J.C., Tan D.X., Sainz R.M., Alatorre-Jimenez M., Qin L. // J. Pineal Res. 2016. V. 61. P. 253–278.
Miquel J., Economos A.C., Fleming J., Johnson J.E., Jr. // Exp. Gerontol. 1980. V. 15. P. 575–591.
Papa S., Skulachev V.P. // Mol. Cell. Biochem. 1997. V. 174. P. 305–319.
Acuna-Castroviejo D., Escames G., Leon J., Carazo A., Khaldy H. // Adv. Exp. Med. Biol. 2003. V. 527. P. 549–557.
Lopez A., Garcia J.A., Escames G., Venegas C., Ortiz F., Lopez L.C., Acuna-Castroviejo D. // J. Pineal Research. 2009. V. 46. P. 188–198.
Cheshchevik V.T., Dremza I.K., Lapshina E.A., Zabrodskaya S.V., Kujawa J., Zavodnik I.B. // Cell Biochem. Funct. 2011. V. 29. P. 481–488.
Zavodnik I.B., Lapshina E.A., Cheshchevik V.T., Dremza I.K., Kujawa J., Zabrodskaya S.V., Reiter R.J. // J. Physiol. Pharmacol. 2011. V. 62. P. 421–427.
Cheshchevik V.T., Lapshina E.A., Dremza I.K., Zabrodskaya S.V., Reiter R.J., Prokopchik N.I., Zavodnik I.B. // Toxicol. Appl. Pharmacol. 2012. V. 261. P. 271–279.
Petrosillo G., Casanova G., Matera M., Ruggiero F.M., Paradies G. // FEBS Lett. 2006. V. 580. P. 6311–6316.
Martin M., Macias M., Escames G., Leon J., Acuna-Castroviejo D. // FASEB J. 2000. V. 14. P. 1677–1679.
Halestrap A. // Nature. 2005. V. 434. P. 578–579.
Jou M.J. // J. Pineal Res. 2011. V. 50. P. 427–435.
Krestinina O.V., Baburina Y.L., Azarashvili T.S. // Biol. Membrany. 2014. V. 31. P. 95–103.
Baburina Y., Odinokova I., Azarashvili T., Akatov V., Lemasters J.J., Krestinina O. // Bba-Biomembranes. 2017. V. 1859. P. 94–103.
Martinis P., Zago L., Maritati M., Battaglia V., Grancara S., Rizzoli V., Agostinelli E., Bragadin M., Toninello A. // Amino Acids. 2012. V. 42. P. 1827–1837.
Bejarano I., Redondo P.C., Espino J., Rosado J.A., Paredes S.D., Barriga C., Reiter R.J., Pariente J.A., Rodriguez A.B. // J. Pineal Res. 2009. V. 46. P. 392–400.
Osseni R.A., Rat P., Bogdan A., Warnet J.M., Touitou Y. // Life Sci. 2000. V. 68. P. 387–399.
Sims N.R. // J. Neurochem. 1990. V. 55. P. 698–707.
Stricker R., Lottspeich F., Reiser G. // Biol. Chem. Hoppe Seyler. 1994. V. 375. P. 205–209.
Tan D.X., Manchester L.C., Reiter R.J., Plummer B.F., Limson J., Weintraub S.T., Qi W. // Free Radic. Biol. Med. 2000. V. 29. P. 1177–1185.
Acuna-Castroviejo D., Martin M., Macias M., Escames G., Leon J., Khaldy H., Reiter R.J. // J. Pineal Res. 2001. V. 30. P. 65–74.
Martin M., Macias M., Leon J., Escames G., Khaldy H., Acuna-Castroviejo D. // Int. J. Biochem. Cell Biol. 2002. V. 34. P. 348–357.
Odinokova I., Baburina Yu., Kruglov A., Fadeeva I., Zvyagina A., Sotnikova L., Akatov V. and Krestinina O. // Int. J. Mol. Sci. 2018. V. 19. P. 1–16.
Buyukavci M., Ozdemir O., Buck S., Stout M., Ravindranath Y., Savasan S. // Fundam. Clin. Pharmacol. 2006. V. 20. P. 73–79.
Srinivasan V., Spence D.W., Pandi-Perumal S.R., Trakht I., Cardinali D.P. // Integr. Cancer Ther. 2008. V. 7. P. 189–203.
Hou T., Zhang X., Xu J., Jian C., Huang Z., Ye T., Hu K., Zheng M., Gao F., Wang X., Cheng H. // J. Biol. Chem. 2013. V. 288. P. 4602–4612.
Ma Q., Fang H., Shang W., Liu L., Xu Z., Ye T., Wang X., Zheng M., Chen Q., Cheng H. // J Biol. Chem. 2011. V. 286. P. 27573–27581.
Baburina Y., Azarashvili T., Grachev D., Krestinina O., Galvita A., Stricker R., Reiser G. // Neurochemistry International. 2015. V. 90. P. 46–55.
Azarashvili T., Krestinina O., Galvita A., Grachev D., Baburina Y., Stricker R., Evtodienko Y., Reiser G. // Am. J. Physiol. Cell Physiol. 2009. V. 296. P. C1428–С1439.
Krestinina O., Azarashvili T., Baburina Y., Galvita A., Grachev D., Stricker R., Reiser G. // Neurochemistry Int. 2015. V. 80. P. 41–50.
Baburina Y., Azarashvili T., Grachev D., Krestinina O., Galvita A., Stricker R., Reiser G. // Neurochem Int. 2015. V. 90. P. 46–55.
Azarashvili T., Stricker R., Reiser G. // Biol. Chem. 2010. V. 391. P. 619–629.
Baines C.P., Kaiser R.A., Sheiko T., Craigen W.J., Molkentin J.D. // Nat. Cell Biol. 2007. V. 9. P. 550–555.
Дополнительные материалы отсутствуют.