

Нейрохимия, 2019, T. 36, № 2, стр. 101-118
Возможные механизмы комплексного влияния ацетилхолина на тета-активность, обучение и память
И. Г. Силькис 1
1 Федеральное государственное бюджетное учреждение науки
Институт высшей нервной деятельности и нейрофизиологии Российской академии наук,
Москва, Россия
Поступила в редакцию 02.08.2018
После доработки 19.09.2018
Принята к публикации 28.09.2018
Аннотация
Ухудшение обучения и памяти связывают с нарушением выраженности ритмической активности, а также синхронизации по фазе между тета-активностью в гиппокампе, энторинальной коре и других областях коры. Поскольку несколько структур, вовлеченных в генерацию тета-ритма, содержат холинергические клетки, концентрация ацетилхолина, должна влиять на параметры ритма и играть существенную роль в процессах обучения и памяти. В настоящей работе проведен анализ возможных механизмов влияния ацетилхолина на параметры тета-ритма в гиппокампе. Комплексный характер этих механизмов связан с влиянием на тета-активность как холинергического входа из медиальной перегородки в гиппокамп, так и холинергических клеток педункулопонтийного и орального понтийного ядер, а также ядра Мейнерта. Кроме того, на тета-активность влияет таламическое ядро реуниенс, проецируещееся в медиальную перегородку, поле СА1 гиппокампа и энторинальную кору. Влияние ядер гипоталамуса (супрамаммилярного, заднего и медиального маммиллярного) также осуществляется через их взаимодействие с гиппокампальной формацией и перегородкой. Часть структур, участвующих в генерации тета-активности, содержит длинноаксонные ГАМКергические клетки, действие которых на другие структуры является как ингибирующим, так и растормаживающим. Ацетилхолин, способствуя усилению возбуждения в сети, участвующей в генерации тета-ритма, может приводить к увеличению его выраженности (мощности), тогда как частота ритма определяется торможением и должна увеличиваться, если оно ослабляется.
Принятые сокращения:
БА – болезнь Альцгеймера;
БГ – базальные ганглии;
ЗГЯ – заднее ядро гипоталамуса;
ЛП – латеральная часть перегородки;
МП – медиальная часть перегородки;
МЯ – ядро Мейнерта;
ММЯ – медиальное маммиллярное ядро;
ПфК – префронтальная кора;
ОПЯ – оральное понтийное ретикулярное ядро;
ППЯ – педункулопонтийное ядро;
РЕ – таламическое ядро реуниенс;
СМЯ – супрамаммиллярное ядро;
ЭК – энторинальная кора.
Ритмическая активность в нейронных сетях гиппокампа и новой коры играет важную роль в памяти и обучении. Полагают, что гиппокампальный тета-ритм вовлечен в процессы внимания, навигации и кодирования информации, обучения и памяти [1]. Роль ритмической активности для обучения и памяти естественно объяснить тем обстоятельством, что синхронизация нейронных разрядов способствует модификации синаптической передачи (длительной потенциации и длительной депрессии), поскольку при совпадении во времени пре- и постсинаптической активности выполняется правило Хебба. Известно, что спонтанные или вызванные тета-осцилляции коррелируют с выделением ацетилхолина, причем срабатывание холинергических клеток в значительной степени синхронизировано с тета-ритмом [2]. О связи холинергической активации с тета-ритмом, длительной потенциацией (ДП) и обучением свидетельствуют данные о том, что внутривенное введение агонистов мускариновых холинорецепторов способствовало появлению тета-активности и увеличивало амплитуду ДП в поле СА1 гиппокампа, вызванную тетанизацией коллатералей Шаффера [3]. Оральное использование этих агонистов исправляло нарушения памяти при реакции избегания, вызванные антагонистом мускариновых рецепторов скополамином [3]. Важно отметить, что тета-активность зависит от концентрации ацетилхолина. Так, например, показано, что низкие дозы антагониста холинорецепторов атропина усиливали ритмическую активность, а высокие – подавляли [4].
В большинстве известных нам работ исследовалось влияние отдельных структур, содержащих холинергические нейроны, на тета-активность в гиппокампе. Основное внимание обычно уделяется участию в генерации тета-ритма холинергических нейронов медиальной перегородки/диагональной полоски Брока (МП/ДПБ) [1, 5]. Имеются и данные о том, что в генерации тета-ритма участвуют цепи, включающие также энторинальную кору (ЭК). Кроме того, имеются различные доказательства участия в ритмической активности гиппокампа восходящей системы ствола мозга [6]. Влияние исходит из ростральной части моста, холинергических клеток педункулопонтийного ядра (ППЯ), орального ретикулярного понтийного ядра (ОПЯ), супрамаммиллярного ядра (СМЯ) гипоталамуса, а также задних ядер гипоталамуса (ЗГЯ), которые проецируются в МП/ДПБ [6]. Холинергические нейроны понтийных ядер могут влиять на тета-ритм через прямые и непрямые проекции в МП [7]. Нейроны этих ядер являются частью восходящей системы, переносящей в септальный генератор тета-ритма возбуждение, вызванное поведенческой активностью [8, 9]. Через МП восходящая синхронизирующая активация передается в лимбические области [6]. Кроме того, ранее нами было указано на то, что гиппокампальный тета-ритм должен находиться под влиянием таламического ядра реуниенс (РЕ), поскольку клетки этого ядра проецируются в МП, поле СА1 гиппокампа и ЭК [10]. Усиление активности клеток ядра РЕ, вызванное, например, холинергическим входом из ППЯ, должно увеличить возбуждение нейронов гиппокампа и МП, что изменит функционирование клеток-мишеней МП в гиппокампе.
В структуры, участвующие в генерации тета-ритма, включены не только холинергические клетки, но и длинноаксонные ГАМКергические и глутаматергические нейроны. Хотя у этих структур имеются общие клетки-мишени, нам не известны, работы, в которых учитывалось бы взаимовлияние разных структур. Из вышеизложенного следует, что при анализе механизмов, лежащих в основе нарушений памяти и связанных с дефицитом ацетилхолина, необходимо учитывать процессы, протекающие в сложной нейронной сети, вовлеченной в генерацию ритмической активности, которая включает гиппокамп, МП/ДПБ, ППЯ, ОПЯ, ядро РЕ, СМЯ и другие ядра гипоталамуса. Для понимания этих механизмов следует принять во внимание результаты проведенного в работе [11] моделирования, которое показало, что в нейронной сети с взаимосвязанными тормозными клетками, в которую поступает достаточно сильное возбуждение, частота осцилляций зависит от силы торможения. Чем сильнее торможение, тем меньше частота осцилляций. От силы возбуждения зависит амплитуда (мощность) осцилляций. Как будет показано далее, нейронная сеть, включающая гиппокамп, перегородку, ЭК, ядро РЕ и маммиллярные ядра, удовлетворяет этим требованиям. Судя по результатам этого моделирования, изменения активности в каждой из структур должно влиять на параметры ритмической активности, в частности, тета-ритма.
Целью настоящей работы являлся анализ функциональной организации сети, участвующей в генерации тета-ритма и возможных механизмов, лежащих в основе влияния ацетилхолина на тета-активность в гиппокампальной формации.
ОСОБЕННОСТИ ФУНКЦИОНАЛЬНОЙ ОРГАНИЗАЦИИ НЕЙРОННОЙ СЕТИ, УЧАСТВУЮЩЕЙ В ГЕНЕРАЦИИ ТЕТА-АКТИВНОСТИ
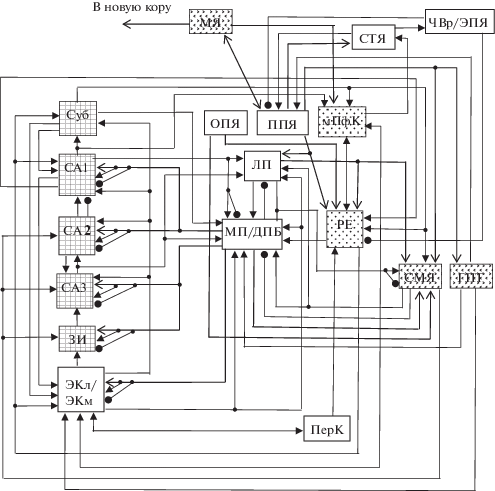
Участие нейронов медиальной перегородки в тета-активности. При анализе особенностей функциональной организации нейронной сети, участвующей в генерации тета-активности, использованы известные результаты экспериментальных исследований. Составленная с их учетом схема организации взаимодействий между структурами в нейронной сети, участвующей в генерации тета-ритмов, представлена на рис. 1. С целью упрощения, различные звенья сети представлены прямоугольниками, но в каждом из них имеются возбудительные и тормозные нейроны.
Рис. 1.
Схема взаимодействий между структурами в нейронной сети, участвующей в генерации тета-ритмов. ЗИ – зубчатая извилина, СА1, СА2, СА3 – поля гиппокампа; Суб. – субикулюм; ЭКл/ЭКм – латеральная и медиальная части энторинальной коры; ПерК – периринальная кора; ПфК – префронтальная кора; МП/ДПБ – медиальная часть перегородки/диагональная полоска Брока; ЛП – латеральная часть перегородки; ППЯ – педункулопонтийное ядро; ОПЯ – оральное понтийное ретикулярное ядро; РЕ – таламическое ядро реуниенс; СМЯ –супрамаммиллярное ядро; ГПТ – ядра гипоталамуса, включая маммиллярные; МЯ – ядро Мейнерта; ЧВр/ЭПЯ – выходные ядра базальных ганглиев: ретикулярная часть черного вещества и энтопедункулярное ядро; СТЯ – субталамическое ядро. Линии со стрелками – возбудительные входы; линии, оканчивающиеся черными кружками – тормозные входы; утолщенные линии с открытыми стрелками – холинергические входы. С целью упрощения различные звенья сети представлены прямоугольниками, но в каждом из них имеются возбудительные и тормозные нейроны.
Полагают, что холинергический вход из МП вероятнее участвует в холинергическом контроле кодирования информации, а не ее извлечении из памяти [12]. C пространственной памятью связывают не только холинергические, но и ГАМКергические нейроны МП. Принято считать, что нейроны обоих типов участвуют в генерации тета-ритма (4–12 Гц) [13, 14]. Показано, что повреждение ГАМКергического входа из МП в гиппокамп уменьшает выделение ацетилхолина и ухудшает пространственную память [15].
Хотя увеличение активности холинергических клеток МП предшествует усилению мощности тета ритма [16], однако отмечено, что эти клетки имеют низкую фоновую частоту (менее 4 Гц) даже во время тета-ритма [17]. Срабатывающие пачечными разрядами ГАМКергические нейроны МП играют важную роль в генерации тета-ритма за счет воздействия на тормозные интернейроны гиппокампа и возможного последующего растормаживания пирамидных клеток [18–23]. Активность ГАМКергических нейронов МП привязана по фазе или к провалу (178 градусов), или к пику (330 градусов) тета-ритма [18].
Наибольшая плотность ГАМКергических волокон из МП наблюдается в слоях радиальном и ориенс поля СА3 гиппокампа, а также в хилусе и гранулярном слое зубчатой извилины (ЗИ) [20]. Аксонные окончания ГАМКергических нейронов МП иннервируют разные типы интернейронов гиппокампа. Среди них содержащие вазоактивный полипептид интернейроны, которые располагаются в слоях лакунозум молекулярный и пирамидном поля СА1, содержащие холецистокинин, соматостатин и парвалбумин интернейроны в слое ориенс, а также содержащих калретинин и полипептид Y интернейроны, которые располагаются в радиальном слое поля СА1 и слое луцидум поля СА3 [13]. Обратно в МП проецируется небольшое количество тормозных нейронов поля СА1, содержащих калбиндин [24]. Обратно в перегородку проецируются и возбудительные нейроны полей СА3 и СА1 гиппокампа [13], что приводит к созданию замкнутых петель.
ГАМКергические нейроны МП базального переднего мозга, которые участвуют в координировании тета-осцилляций и проецируются на ГАМКергические интернейроны нескольких типов в полях СА1 и СА3 гиппокампа, характеризуются разными типами разрядов [25]. На наркотизированных крысах показано, что частота нейронов одной группы септальных нейронов увеличивалась во время генерации тета-ритма и они срабатывали в основном на нисходящей фазе тета-осцилляций в поле СА1, тогда как у нейронов другой группы частота была наибольшей во время восходящей фазы тета-ритма [25]. Эти данные позволили предположить, что разные группы ГАМКергических нейронов МП вносят вклад в координацию активности септо-гиппокампальной сети через параллельные пути в соответствии с расположением их мишеней в полях гиппокампа [25]. ГАМКергические нейроны МП, которые срабатывают в периоды негативной фазы тета-активности, описаны в работе [26]. Эти нейроны содержат парвалбумин и характеризуются наибольшей ритмической активностью по сравнению с другими нейронами МП, срабатывают короткими пачками (длительностью в среднем 38 мс) и специфически иннервируют область СА3, а не поля СА1, СА2 или ЗИ [26]. Эти клетки образуют синаптические контакты с аксо-аксонными и содержащими холицестокинин ГАМКергическими нейронами септо-темпорального сегмента поля СА3. Выдвинуто предположение, что эти нейроны координируют осцилляторную активность, связывая по фазе разряды специфических интернейронов поля СА3. Кроме того, они вносят вклад в селекцию ансамбля пирамидных клеток в негативной фазе тета-активности за счет растормаживания пирамидных клеток [26].
О существенном вкладе ГАМК-торможения в генерацию тета-ритма свидетельствует тот факт, что повреждение или фармакологические манипуляции с ГАМКергическими нейронами МП препятствовали появлению тета-активности в лимбической цепи [27], тогда как даже после избирательного повреждения всех холинергических клеток МП гиппокампальный тета-ритм полностью не подавлялся [28]. Однако его амплитуда существенно уменьшалась и в спектре мощности оставался лишь небольшой, хотя и значимый пик. При этом частота гиппокампального тета-ритма не изменялась [28]. В более поздних работах также было показано, что оптогенетическая стимуляция холинергических клеток усиливает мощность тета-ритма, но не влияет на его частоту [5]. Существует мнение, что для поддержания некоторой тета-активности в гиппокампе достаточно и ГАМКергических септо-гиппокампальных проекций, а холинергические нейроны служат для усиления выраженности гиппокампального тета-ритма за счет увеличения связывания по фазе септальных клеток [28–30]. Следует отметить, что один тормозный интернейрон может эффективно синхронизировать во времени спонтанную активность большого числа пирамидных клеток, в активности которых выделяется частота 4–7 Гц [31]. Это происходит за счет широкой дивергенции ветвлений одного интернейрона больше чем на тысяче пирамидных клеток.
Холинергические нейроны МП могут оказывать влияние на активность гиппокампа как непосредственно через прямые проекции, так и опосредованно, через влияние на другие нейроны МП. За счет прямых проекций ацетилхолин через модулирующее воздействие на возбудительные входы может усилить активность пирамидных клеток [32–34]. С другой стороны, за счет прямых проекций активация холинергических клеток МП может приводить к увеличению активности тормозных интернейронов и снижению активности основных клеток в разных полях гиппокампа [12]. Показано, что при оптогенетической стимуляции холинергического входа из МП за счет активации мускариновых рецепторов на корзинчатых клетках поля СА1 экспрессирующих вазоактивный интестинальный пептид, частота их разрядов увеличивается и возрастает амплитуда ТПСТ в пирамидных клетках поля СА1 [35]. При непрямом действии ацетилхолин может способствовать растормаживанию пирамидных клеток. Результирующий эффект воздействия ацетилхолина на нейроны гиппокампа, а также на обучение и память зависит от его концентрации [36]. При низких концентрациях ацетилхолина пирамидные клетки были наиболее активны [37, 38]. Этот эффект объясняют воздействием на никотиновые рецепторы, приводящем к увеличению активности тормозных интернейронов, которые проецируются на другие тормозные интернейроны, иннервирующие пирамидные клетки [20]. Кроме того, за счет воздействия на мускариновые рецепторы уменьшается активность тормозных интернейронов, непосредственно проецирующихся на пирамидные нейроны [20]. Судя по предложенным нами унифицированным правилам модуляции [39, 40], к снижению активности тормозных интернейронов может приводить воздействие на постсинаптические М2 или М4 рецепторы на этих интернейронах, способствующее индукции длительной депрессии (ДД) на их возбудительных входах. Показано также, что микроэлектрофоретическая аппликация агонистов М2 рецепторов приводила к существенному увеличению амплитуды популяционного спайка в ЗИ и в слое ориенс поля СА1 и к усилению синхронизации их активности с гиппокампальным тета-ритмом [41]. Поскольку показано, что этот эффект не был связан с увеличением возбудительной передачи, можно полагать, что в его основе лежало ослабление торможения. Авторы работы [41] полагают, что важную роль в наблюдавшихся эффектах играла холинергическая модуляция возвратного, а не афферентного торможения основных клеток гиппокампа интернейронами этой структуры. При высоких концентрациях ацетилхолина активируются мускариновые рецепторы, которые способствуют увеличению активности тормозных интернейронов, проецирующихся на пирамидные клетки, так что активность последних снижается [20]. Судя по предложенным нами ранее унифицированным правилам модуляции [39, 40], к такому эффекту должно было приводить воздействие на мускариновые рецепторы типов М1, М3 и М5 на интернейронах. Высокие концентрации ацетилхолина могут способствовать появлению тета-активности [20, 37, 38]. То, что активация мускариновых М1/М3 рецепторов увеличивала частоту срабатывания тормозных интернейронов и усиливала тета-ритм, показано в работе [42]. Появление тета-ритма при высокой концентрации ацетилхолина наблюдалось и в работе [2]. Индукции спонтанной тета-активности препятствовало внутригиппокампальное введение антагонистов М1 рецепторов, но не никотиновых рецепторов [43].
Ранее нами было указано на то, что изменение активности тормозных нейронов может приводить к длительному изменению эффективности тормозного входа к клетке-мишени, причем для модификации тормозной передачи требуется активация ГАМКб-рецепторов [44]. Однако показано, что несмотря на большое количество ГАМКб-рецепторов, ГАМК, выделяемая одним гиппокампальным интернейроном, обычно активирует только ГАМКа-рецепторы на клетке-мишени. Активация постсинаптических ГАМКб-рецепторов в гиппокампе облегчается лишь во время ритмической активности, поскольку при этом ГАМК выделяется одновременно несколькими интернейронами [45]. Результаты вышеуказанной работы позволяют предположить, что модификация тормозной передачи должно облегчаться при тета-ритме. Показано что вследствие вовлечения тормозных интернейронов МП синхронизация активности в тета-ритме увеличивалась [12]. По мнению авторов указанной работы, активация как прямого, так и опосредованного путей из МП в гиппокамп должна приводить к активности пирамидных клеток более точно привязанной к фазе тета-осцилляций. Часть ГАМКергических клеток, получающих иннервацию из МП, проецируется обратно в МП, образуя цепь обратной связи. Примечательно, что активность ГАМКергических клеток модулируется тета-активностью, а у холинергических клеток такой модуляции не наблюдалось [46].
Ранее полагали, что только холинергические и ГАМКергические проекции из МП участвуют в генерации гиппокампального тета-ритма [47]. Однако примерно четверть проекций из МП в ЗИ, поля СА3 и СА1 являются глутаматергическими [48]. Глутаматергические нейроны септума иннервируют пирамидные нейроны ЗИ, полей СА1 и СА3 [48] и при их стимуляции в нейронах гиппокампа наблюдались небольшие ВПСП [49]. В МП холинергические, ГАМКергические и глутаматергические нейроны образуют тесно взаимосвязанную нейронную сеть. Популяция глутаматергических нейронов МП по характеру разрядов является разнородной. Среди них имеются клетки, срабатывающие с частотой (примерно 20 Гц), что ниже, чем у ГАМКергических клеток (примерно 40 Гц) и холинергических нейронов (примерно 75 Гц), но в их спайковой активности имеются высокочастотные пачечные разряды [50]. Эти нейроны срабатывают коррелировано с низкочастотной ритмической активностью нейронов ЭК, пириформной коры, проецирующейся в ЭК, и префронтальной коры (ПфК), связанной с ядром РЕ [50]. В работе [51] выделены глутаматергические клетки МП с быстрыми, медленными, пачечными и кластерными разрядами. Значительное количество клеток с быстрыми разрядами имеют ритмическую спонтанную активность, сходную с той, которая характерна для ГАМКергических нейронов МП, и их активация приводила к быстрым ответам пирамидных клеток в поле СА3 [51]. Отмечено, что глутаматергические нейроны МП иннервируют в основном соседние ГАМКергические клетки и меньше холинергические нейроны [49, 52]. Таким образом, часть глутаматергические нейроны МП может влиять на тета активность за счет возбуждения септальных нейронов по локальным цепям. На мышах в свободном поведении показано, что активация глутаматергических нейронов МП сильно синхронизирует тета-активность в гиппокампе [49]. Введение глутамата в МП приводило к увеличению тета-активности как как в МП, так и в гиппокампе [53]. Синхронизация активности нейронов МП с тета-ритмом зависит от взаимодействий между тремя разными популяциями нейронов. Синхронизация была значительно больше у пар ГАМКергических нейронов. Однако когерентность их потенциалов действия с тета-ритмом была низкой [52]. Исходя из результатов приведенных выше работ, можно предположить, что увеличение возбуждения тормозных нейронов МП со стороны соседних глутаматергических клеток приводит к увеличению тормозного действия на тормозные интернейроны гиппокампа и последующему растормаживанию пирамидных клеток. Примечательно, что глутаматергические нейроны МП также подвержены токсическому воздействию бета-амилоидов [54], поэтому при БА может снижаться возбуждение пирамидных клеток гиппокампа со стороны МП, как за счет уменьшения прямого возбуждения, так и растормаживания.
Ацетилхолин влияет на активность нейронов МП. Воздействие на мускариновые М3 рецепторы способствовало возбуждению антидромно идентифицированных септо-гиппокампальных нейронов, среди которых были клетки с медленно-проводящими аксонами, предположительно холинергические, и клетки с быстро проводящими, предположительно, ГАМКергические [55]. Увеличение активности этих клеток за счет активации М3 рецепторов согласуется с правилами модуляции [39, 40]. Оптогенетическая стимуляция холинергических клеток МП приводила к изменению частоты 83% нейронов МП, не являющихся холинергическими, причем примерно 52% этих клеток были ГАМКергическими [56]. По-видимому, остальные 48% клеток являлись глутаматергическими. То, что ацетилхолин влияет на активность не только ГАМКергических, но и глутаматергических нейронов МП [57–59], приводит к изменению функционирования нейронной сети МП [56]. Показано, что избирательная и неизбирательная оптогенетическая активация МП приводит к сходным изменениям функционирования нейронов гиппокампа и осцилляторной активности [56]. Принято считать, что баланс между холинергической и ГАМКергической системами определяет, генерируется ли в гиппокампе синхронизированная тета-активность [6, 47]. Выявлены разные компоненты взаимодействия МП и гиппокампа. Медленно разряжающиеся холинергические нейроны МП привязаны к амплитуде тета-ритма за счет тонической деполяризации пирамидных клеток и корзинчатых интернейронов [60], тогда как ГАМКергические септальные нейроны с быстрыми спайками связаны с частотой тета-ритма, вследствие периодической гиперполяризации корзинчатых клеток и ритмического растормаживания пирамидных нейронов [21]. С таким механизмом действия ацетилхолина согласуются данные о том, что введение иммунотоксина сапорина в МП, разрушающего холинергические нейроны МП, уменьшает амплитуду, но не частоту тета-активности в поле СА1, вызванную введением карбахола в МП [61]. Разрушение холинергических нейронов МП приводило также к снижению мощности тета-активности, вызванной введением формалина в заднюю лапу [61]. При этом наблюдалось подавление поп-спайка в поле СА1 и усиление тета-ритма в активности тормозных интернейронов [61]. Авторы вышеуказанной работы предположили, что холинергические нейроны МП влияют на амплитуду, но не частоту тета-ритма, за счет цепей, вовлекающих тормозные интернейроны. Результаты работы [61] указывают на то, что мощность тета-активности, вызванной сенсорными стимулами, может быть усилена входом из МП. По-видимому, наиболее оптимальный протокол стимуляции МП должен имитировать паттерны разрядов медленных холинергических и быстрых ГАМКергических клеток и поэтому включать как низкие, так и высокие частоты. Возможность имитировать in vivo паттерны разрядов в тета-ритме в МП является критичным для исследования эффектов влияния МП на функционирование гиппокампа. Например, авторы работы [62] использовали стимуляцию с частотой 100 Гц внутри пачечного разряда с интервалами между пачками в тета-ритме (8 Гц).
Как уже указывалось, нейроны гиппокампа иннервируют клетки МП. Показано, что активация входов из гиппокампа в МП приводит к гиперполяризации холинергических нейронов и снижению частоты их разрядов [1]. Было предположено, что гиппокампальные волокна оканчиваются на тормозных интернейронах МП [1]. Не исключено, однако, что этот эффект связан с непосредственным тормозным действием длинноаксонных тормозных нейронов поля СА1 на холинергические клетки МП. Судя по результатам работы [1], вход из гиппокампа в МП, снижая активность холинергических нейронов может уменьшить выраженность гиппокампального тета-ритма. Возможно, такая отрицательная обратная связь способствует поддержанию мощности тета-ритма на определенном уровне и препятствует ее чрезмерному увеличению.
В гиппокампе выделяют два типа низкочастотной активности: тета-активность (4–6 Гц) и ритм (7–12 Гц), который некоторые авторы относят к альфа-активности. Ритм 7–12 Гц характерен для произвольной двигательной активности и исследовательского поведения, а ритм 4–6 Гц характерен для неподвижного состояния или парадоксального сна [13]. Использование неизбирательного антагониста мускариновых рецепторов атропина не влияло на ритм 7–12 Гц, тогда как генерация ритма 4–6 Гц прекращалась [22]. Эти данные согласуются с результатами экспериментов in vivo, в которых показано, что в МП имеется два типа нейронов с пачечными разрядами, но только у одного типа они устраняется скополамином или атропином, а у другого нет [63, 64]. С другой стороны, показано, что оба типа этой ритмической активности имеют чувствительные и нечувствительные к ацетилхолину компоненты [63].
Вклад педункулопонтийного, латеродорзального и орального ретикулярного понтийного ядер в тета-активность. На гиппокампальный тета-ритм должны влиять и холинергические клетки ядер моста. Введение карбахола в ППЯ или ОПЯ вызывало в гиппокампе тета активность [66–68]. Также можно индуцировать тета-ритм в гиппокампе за счет электрической стимуляции рострального тегментального понтийного ядра (РТЯ). Если инъекция карбахола в ОПЯ приводила к тета-ритму с ЛП 38.5 с, то инъекция в ППЯ – с ЛП 1.7 мин [67]. Судя по ЛП возникновения тета-ритма, его инициатором является ОПЯ, активность которого модулируется ацетилхолином. При этом инъекция карбахола в ППЯ вызывала более сильный и длительный эффект, чем инъекция в ОПЯ [69]. Авторы работы [70] считают ОПЯ первичным генератором тета-ритма. В цепи, запускаемой ОПЯ, имеются нейроны, на которых располагаются никотиновые α7 рецепторы. На это указывает тот факт, что воздействуя на α7 рецепторы, можно было менять параметры тета-ритма, вызванного стимуляцией ОПЯ [71]. Унилатеральная инактивация ППЯ введением в него прокаина приводила к ипсилатеральному подавлению тета-ритма в гиппокампе [70]. При этом на время около 30 мин после инъекции наблюдались уменьшение амплитуд пиков в преобразовании Фурье в области тета-частот и сдвиг в сторону более низких частот.
Как указывалось в нашей предшествующей работе [10], ППЯ может влиять на тета-ритм, поскольку нейроны этого ядра возбуждают клетки таламического ядра реуниенс (РЕ), которые проецируются в МП, поле СА1 гиппокампа и энторинальную кору (ЭК). Кроме того, активация ППЯ может влиять на МП через латеральную перегородку (ЛП) [72]. У 87.5% нейронов ППЯ активность менялась перед появлением тета-ритма. В ОПЯ процент клеток был меньше 14/22 (63.6%), но в этом ядре латентный период изменений был короче (0.57 с до появления тета-ритма), тогда как в ППЯ – 0.96 с [73]. Пропорция клеток, менявших частоту, была 56.3% в ППЯ и 22.7% в ОПЯ [73]. Авторы указанной работы предположили, что нейроны ОПЯ участвуют в появлении тета-ритма, а нейроны ППЯ – в его поддержании. Показано, что инактивация ППЯ с помощью прокаина приводит к снижению мощности тета-ритма, но не влияет на его частоту [70]. На частоту тета-ритма, вызванного стимуляцией ОПЯ, не влияло разрушение холинергических нейронов МП, но оно препятствовало увеличению амплитуды ритма [61]. Из этих данных следует, что холинергические нейроны МП усиливают выраженность тета-активности, вызванной стимуляцией ОПЯ. Не исключено, что в появлении тета-ритма участвует только часть нейронов ОПЯ, поскольку в нем выделено 2 типа клеток, по-разному реагирующих на ацетилхолин. При воздействии на постсинаптические мускариноввые рецепторы у 100% нейронов ОПЯ типа 1 и 73.7% нейронов типа 2 увеличивалась деполяризация, а у остальных 26.3% нейронов типа 2 увеличивалась гиперполяризация [74]. С учетом правил модуляции [39, 40] можно предположить, что в первом случае активировались М1 рецепторы, а во втором – М2 рецепторы.
Нейроны диссипативной части ППЯ реципрокно связаны с клетками субталамического ядра (СТЯ), которое получает возбуждение из мПфК. Нейроны СТЯ также могут разряжаться с тета-частотой [75]. Между активностью в СТЯ и мПфК имеется взаимодействие в области тета-частот (4–8 Гц), причем мощность тета-ритма в СТЯ коррелирует с временем реакции при обучении [76]. Вследствие наличия прямых связей активность в ППЯ и СТЯ является взаимозависимой. Так показано, что стимуляция каждого из ядер меняет активность другого [77, 78]. Через СТЯ мПфК может влиять на активность ППЯ.
Кроме того, на активность ППЯ могут влиять латеральный гипоталамус и выходные ядра БГ – ретикулярная часть черного вещества и энтопедункулярное ядро, поскольку в ППЯ имеется значительно число входов из этих структур [79]. Нейроны выходных ядер БГ являются ГАМКергическими, поэтому для нормальной активности ППЯ необходимо, чтобы торможение со стороны БГ было слабым. Для этого необходимо, чтобы концентрация дофамина во входном ядре БГ – стриатуме была достаточно велика [10]. В свою очередь, компактная часть ППЯ проецируется в заднюю латеральную область гипоталамуса, ядро Мейнерта (МЯ) в базальном переднем мозге [80]. При этом значительная часть холинергических нейронов ППЯ проецируется в гипоталамус и ЛП, тогда как число их проекций в МЯ невелико [81, 82]. Судя по организации взаимодействий МЯ с другими структурами (рис. 1), при его повреждении должна уменьшаться активность холинергических нейронов ППЯ, что приведет к снижению активности нейронов ядра РЕ, и последующему ослаблению активности нейронов МП (рис. 1). В свою очередь, это может явиться причиной снижения мощности тета-ритма в гиппокампе. Возможно, в результате таких же изменений повреждение МЯ приводило к подавлению ДП в перфорантном пути к ЗИ крыс и снижению респонсивности нейронов ЗИ [83], хотя, как отметили авторы указанной работы, МЯ не посылает прямых холинергических проекций в гиппокамп.
Вклад супрамаммиллярного, заднего и медиального маммиллярных ядер в генерацию тета-ритма. Структуры каудального промежуточного мозга, включая СМЯ, маммиллярные ядра (ММЯ) и ЗГЯ, занимают стратегическое положение во взаимодействии восходящего и нисходящего потоков между стволом мозга и лимбической системой, которая включает перегородку и гиппокамп [84]. Супрамаммиллярное ядро, реципрокно связанное с септо-гиппокампальной системой, является важным источником проекций в гиппокампальную формацию и играет важную роль в модуляции такой гиппокамп-зависимой активности, как тета-ритм и память [84]. Маммиллярное ядро также является частью системы генерации тета-ритма [85]. Хотя ранее полагали, что СМЯ и ММЯ играют роль в однонаправленной передаче активности в гиппокамп, современные экспериментальные данные свидетельствуют в пользу того, что цепь СМЯ – МП – гиппокамп – ММЯ находится под сложным двунаправленным контролем [86]. Эти структуры взаимозависимо влияют на частоту тета-ритма. Так, повреждение СМЯ [87] и ММЯ [88] уменьшало частоту тета-ритма в гиппокампе. Возможно, этот эффект был связан с увеличением активности ГАМКергических нейронов МП, поскольку показано, что инактивация СМЯ приводит к гиперктивности в МП/ДПБ [89]. Имеются данные о том, что стимуляция СМЯ приводит к тета-ритму в гиппокампе через воздействие на МП [90]. Инактивация МП, наоборот, увеличивала частоту тета-ритма в СМЯ/ММЯ, но при этом мощность тета-ритма в равной степени уменьшалась в гиппокампе и в СМЯ/ММЯ [86]. Ранее также было показано, что гиппокампальный вход смещает тета-активность в СМЯ в сторону низких частот [91, 92]. В области более высоких частот тета-активность в СМЯ не зависела от гиппокампа, тогда как в ММЯ частота тета-ритма ниже и зависит от гиппокампа [86]. Было высказано предположение, что увеличение частоты тета-ритма в СМЯ в отсутствие входа из гиппокампа может быть вызвано внутренним источником или неким неизвестным внешним источником [91]. В работе [86] также предположено, что вход из гиппокампа в СМЯ/ММЯ в тета-ритме взаимодействует с другим тета-ритмическим входом и модулирует его. Предполагается, что имеется, по крайней мере, еще один источник модуляции активности в СМЯ/ММЯ, который не зависит от системы МП-гиппокамп. Наблюдались случаи, когда тета-активность в СМЯ/ММЯ наблюдалась до или после инактивации МП [86]. Примечательно, что частоту тета-ритма в гиппокампе можно было увеличить в результате инактивация латеральной перегородки (ЛП) с помощью мусцимола [93]. Было высказано предположение, что этот эффект является следствием потери контроля СМЯ со стороны ЛП, что позволило тета-активности из СМЯ через МП влиять на гиппокамп с изначально присущей высокой частотой [86].
На нейроны ММЯ влияют нисходящие проекции из гиппокампа, причем клетки ММЯ срабатывают согласованно с тета-ритмом в гиппокампе [85]. В состоянии наркоза тета-активность в ММЯ коррелирует с активностью в поле СА1 [94]. Имеются данные, указывающие на то, что тета-активность в ММЯ является эфферентной копией тета-активности в гиппокампе и отличается от тета-активности в СМЯ, которая является суммой входов от тета-активности в гиппокампе и вне гиппокампа [86]. В дорзальной части ММЯ активность предшествует появлению тета-ритма в гиппокампе, а вентральная часть ММЯ больше зависит от наличия тета-ритма в гиппокампе [86]. Хотя мощность тета-активности в СМЯ/ММЯ ослабляется при инактивации МП [91], однако для функционирования ММЯ может быть более важен не гиппокампальный вход, а некий иной вход, тогда как гиппокамп играет модулирующую роль [85]. По-видимому, этот вход в ММЯ является существенным и для гиппокампа, так как недавно обнаружено, что у крыс в состоянии наркоза инактивация медиального ММЯ может приводить к прерыванию гиппокампального тета-ритма [85].
Однако, если тета-активность вызывали внешним раздражителем (щипанием хвоста), активность группы нейронов СМЯ следовала за тета-активностью в гиппокампе с постоянной задержкой. Предположили, что это могло происходить благодаря входу в СМЯ из МП [92]. Если при уретановом наркозе ритмическая активность генерируется в СМЯ и переносится в септум, а оттуда в гиппокамп, то в состоянии бодрствования действуют еще и другие (кроме СМЯ) восходящие пути и механизмы [95]. Возможно, вовлекается заднее ядро гипоталамуса (ЗГЯ), которое функционирует синергично с СМЯ и предположительно влияет на амплитудную модуляцию тета-ритма [95]. На активность нейронов в ЗГЯ, СМЯ и ММЯ влияла стимуляция ретикулярного ОПЯ, приводящая к появлению тета-активности в гиппокампе [96]. Во время тета-ритма нейроны ОПЯ активируют СМЯ, которое вовлекает в активность ГАМКергические и холинергические ритмические клетки МП. Ритмические септальные клетки модулируют активность пирамидных клеток и интернейронов гиппокампа, способствуя генерации тета-активности [95].
В СМЯ/ММЯ выделены фазические тета-on клетки, ритмические разряды которых были привязаны к фазе тета-активности [91]. В ЗГЯ также выделены тета-on клетки, у которых тоническая активность увеличивалась во время тета-активности в гиппокампе [91]. Нейроны СМЯ, разряды которых были привязаны к фазе гиппокампального тета-ритма [97], могли различаться по величине сдвига. Выделены 4 группы клеток СМЯ/ММЯ, которые имеют разные фазовые сдвиги по отношению к ритмической активности в гиппокампе. В первой и второй группах нейроны срабатывали со сдвигом по фазе 177 или 8 градусов по отношению к нейронам поля СА1 гиппокампа; у двух других групп сдвиг составлял 97 градусов по отношению к пику верхней или нижней фаз [84]. Не исключено, что эти нейроны являются частью разных цепей, в которые включены и тормозные клетки. После того, как тета-активность в гиппокампе устраняли с помощью введения прокаина в МП/ДПБ, стимуляция ОПЯ вызывала паттерны разрядов во всех тета-on клетках в ЗГЯ и в СМЯ, причем их частота увеличивалась [91]. По контрасту, введение прокаина в МП устраняло фазические разряды и уменьшало частоту разрядов тета-on клеток в ММЯ, которые были вызваны стимуляцией ОПЯ [91]. На основании этих данных было предположено, что ММЯ получает нисходящую информацию из септо-гиппокампальной системы, тогда как СМЯ и ЗГЯ являются восходящей частью этой системы, а нисходящий вход из этой системы ограничивает частоту разрядов нейронов СМЯ и ЗГЯ [91]. На крысах, наркотизированных уретаном, показано, что тета-активность в СМЯ и ЗГЯ может генерироваться независимо от одновременно регистрируемого гиппокампального тета-ритма [98]. Этот тета-ритм не зависел и от внешнего раздражителя (воздействия на хвост) [98]. Тета-ритм в срезах СМЯ и ЗГЯ можно было вызвать и карбахолом, причем эффект зависел от активации мускариновых рецепторов и блокировался их антагонистом атропином [98].
В СМЯ выделяют латеральную и медиальную части. Наибольшая плотность проекций в гиппокамп исходит из латерального СМЯ [99]. На крысах под уретановым наркозом показано, что микроинъекция карбахола или никотина в обе части СМЯ приводит к сходному паттерну тета-активности [99]. Инъекция карбахола в СМЯ, приводила к активации тета-ритма и подавление поп-спайка в поле СА1, тогда как микроинъекция в СМЯ антагонистов мускариновых и никотиновых рецепторов уменьшала эти эффекты [99]. Мы полагаем, что эти эффекты могли быть вызваны усилением активности глутаматергических клеток СМЯ под действием ацетилхолина. Предположение основывается на данных о том, что ингибирование глутаматергических нейронов СМЯ подавляет тета-активность [100].
Примечательно, что введение карбахола в СМЯ концентрационно-зависимо подавляет возбудимость пирамидных клеток поля СА1, которая отделена от тета-активности [101]. В этой работе обнаружено также, что наибольший эффект наблюдался при микроинъекции карбахола в латеральную часть СМЯ. Этому эффекту препятствовала инактивация МП с помощью прокаина [101]. Эти данные указывают на возможную роль холинергического влияния со стороны латерального СМЯ на модуляцию возбуждения пирамидных клеток поля СА1 через МП [101]. Введение антагониста только никотиновых рецепторов мекамиламина в латеральную часть СМЯ избирательно уменьшало ответы в поле СА1, вызванные стимуляцией ОПЯ или инъекцией формалина в заднюю лапу [99]. Эти данные также свидетельствуют в пользу того, что нейроны латерального СМЯ, на которых имеются никотиновые рецепторы, являются элементом влияния на активность в гиппокампе, вызванную стимуляцией ОПЯ или внешними стимулами [99].
Нейроны СМЯ образуют асимметричные синапсы на нейронах ЗИ, причем афферентация поступает не только на основные клетки, но и ГАМКергические интернейроны [102]. О важной роли такого афферентного торможения в гиппокампе свидетельствуют результаты моделирования сети гиппокампа, показавшие, в частности, что клетки в ЗИ способствуют поддержанию частоты срабатывания гранулярных клеток в пределах физиологических условий и зашумленного входа из ЭК [103]. Этот стабилизирующий эффект зависит от отношения между порогами основных клеток и интернейронов. По-видимому, возбудительный вход является сильным, так как показано, что стимуляция СМЯ усиливает перфорантный вход из ЭК в ЗИ, если вход из МП инактивируется [90]. Мишенями афферентов из СМЯ являются также пирамидные клетки полей СА2 и СА3а [90]. Показано, что инактивация СМЯ приводит к снижению активности в поле СА3 гиппокампа [89]. С учетом этого было выдвинуто предположение, что СМЯ контролирует поток информации в поле СА3 и может играть ключевую роль в пространственной памяти [89]. От содержащих калретинин нейронов СМЯ в перегородку поступают глутаматергические волокна, которые оканчиваются на холинергических клетках МП и калбиндин содержащих клетках ЛП [104]. Таким образом, формируется цепь, влияющая на параметры тета-ритма, в которую включена не только МП, но и ЛП.
В свою очередь, в СМЯ проецируются нейроны, находящиеся на границе МП и ЛП, причем 60% терминалей является ГАМКергическими, а 40% – глутаматергическими [105]. По данным работы [106] в СМЯ оканчиваются в основном (90%) аксонов холинергических нейронов МП, но есть небольшое количество глутаматергических и ГАМКергических окончаний. Проецирующиеся в СМЯ септальные нейроны получают возбуждение от глутаматергических нейронов ЭК [97]. В СМЯ аксоны септальных ГАМКергических нейронов оканчиваются на содержащих калретинин не ГАМКергических нейронах, которые проецируются в перегородку и гиппокамп [97]. Поэтому, возбуждение из ЭК может подавить связанные с тета-активностью нейроны в СМЯ за счет их ингибирования через перегородку [97].
В поле СА2 выделено два типа корзинчатых ГАМКергических интернейронов. Они имеют длинные аксоны и могут влиять на все поля СА, причем в полях СА1 и СА2 они обеспечивают афферентное торможение, а в поле СА3 – возвратное [107]. Таким образом тормозные нейроны поля СА2 могут координировать срабатывание пирамидных клеток во время тета-ритма. Поскольку нейроны поля СА2 получают возбуждение из СМЯ, гипоталамус может модулировать тета-активность в гиппокампе и через указанный путь [107].
Влияние таламических ядер реуниенс и ромбовидного на тета-активность. Полагают, что важную роль в когнитивной деятельности играет синхронизация ритмической активности нейронов гиппокампа и ПфК в диапазоне 4–8 Гц, а прерывание связей между этими структурами приводит к психическим заболеваниям. Связующим (координирующим) звеном между гиппокампом и новой корой является самое большое таламическое ядро вентральной части средней линии РЕ [9, 108]. С ядром РЕ реципрокно связанны нейроны ПфК, причем 8% нейронов связано и с гиппокампом, и с ПфК [109]. Полагают, что ядра средней линии РЕ и ромбовидное участвуют в обработке мультимодальной сенсорной информации [110].
Ядро РЕ получает возбуждение от нейронов ЛП, нейронов поля СА1/субикулюм, ППЯ, ЛДЯ и ряда областей коры [111]. В свою очередь, глутаматергические клетки дорзолатеральной части ядра РЕ проецируются в слой лакунозум-молекулярный поля СА1 (где располагаются апикальные дендриты пирамидный нейронов) и в субикулюм, а клетки вентромедиальной части ядра РЕ проецируются в МП [112, 113]. Входы из ядра РЕ в септо-гиппокампальную систему являются возбудительными [112]. На то, что входы из ядер РЕ и ромбовидного в гиппокамп являются сильными, указываю данные о возможности индукции ДП на входе из ядра РЕ в поле СА1 гиппокампа [109].
Клетки ядра РЕ реагируют на сенсорное раздражение. В зависимости от паттерна разрядов в ответ на сенсорное раздражение среди клеток ядра РЕ выделена группа, отвечающая изменением частоты; группа, срабатывающая пачечными разрядами, и группа, ответы которой связаны с когерентностью гиппокампального тета-ритма (5–12 Гц) [114]. Показано, что при тета-активности, вызванной раздражением хвоста, росла частота разрядов нейронов ядра РЕ, тогда как пачечная активность и когерентность почти не менялись [114]. При стимуляции хвоста в поле СА1 также наблюдается увеличение ритмической активности в диапазоне частот 3–7 Гц [4]. По-видимому, ядро РЕ играет определенную роль в этом эффекте. На это указывают результаты работы [114], в которой показано, что при наличии тета-ритма значительно уменьшались латентные периоды ВПСП в пирамидных нейронах поля СА1, вызванных стимуляцией ядра РЕ и ЭК, по сравнению с отсутствием тета-ритма. Появление тета-ритма в поле СА1 может быть связано с увеличением частоты разрядов нейронов ядра РЕ, которое должно приводить к усилению возбуждения их клеток-мишеней в поле СА1 и МП. Увеличение активности нейронов МП, в свою очередь, должно приводить к усилению тета-активности как в МП, так и в гиппокампе, как это продемонстрировано в работе [53] с помощью введения глутамата в МП.
Следует отметить, что ядро РЕ и СМЯ проецируются в разные части гиппокампа: СМЯ – в ЗИ, поля СА2 и СА3, а РЕ – в поле СА1 и субикулюм [9, 89]. На этом основании высказано предположение, что вход из СМЯ усиливает вход из ЭК в ЗИ, а вход из ядра РЕ усиливает входы из поля СА3 и ЭК в поле СА1 [9].
Имеются многочисленные свидетельства влияния ацетилхолина на активность таламуса, причем отмечено, что в ядре РЕ большая плотность холинергических волокон [115]. Как уже указывалось, в ядро РЕ проецируются нейроны ППЯ [111]. Кроме того, большинство таламических ядер средней линии (к которым относится и РЕ) получает иннервацию из ОПЯ [116], а нейроны ОПЯ выделяют ацетилхолин [117]. Поскольку нейроны ядра РЕ получают холинергическую иннервацию и затем возбуждают нейроны МП, можно ожидать, что при снижении холинергического воздействия и снижения вследствие этого активности клеток ядра РЕ, будут слабее возбуждаться и нейроны МП.
Если нейроны ядра РЕ преимущественно иннервирует холинергические, а не ГАМКергические клетки МП, можно ожидать, что при низкой активности клеток ядра РЕ будет уменьшаться мощность тета-ритма, но не его частота. Если нейроны ядра РЕ возбуждают также ГАМКергические клетки МП, можно ожидать, что при сниженном холинергическом воздействии на ядро РЕ эти ГАМКергические клетки также будут возбуждаться слабее, уменьшится эффективность торможения гиппокампа и увеличится частота тета-ритма. Судя по тому, что агонисты α7 рецепторов усиливали мощность тета-ритма, а при БА, характеризующейся дефицитом ацетилхолина и снижением плотности чувствительных к нему рецепторов, мощность тета-ритма снижена [118], более вероятно, что афференты из таламического ядра РЕ оканчиваются на холинергических клетках МП.
В центральном таламусе есть две группы нейронов, которые различным образом участвуют в ритмической активности дорзального гиппокампа. Нейроны одной группы, которая экспрессирует калретинин, в отличие от другой, экспрессирующей калбиндин, разряжаются с низкой частотой и их активность не увеличивается во время гиппокампального тета-ритма [119]. Клетки, экспрессирующие калретинин, с учетом их анатомических и физиологических свойств, отнесены к глутаматергическим нейронам, проецирующимся в ЭК [119]. Нейроны другой группы, экспрессирующей калбиндин, разряжались с более высокой частотой во время гиппокампального тета-ритма. Было высказано предположение, что таламические нейроны из разных групп избирательно вовлекаются в различные стадии процессов запоминания [119].
Кроме того, нейроны срединных ядер таламуса (к которым относится РЕ) получают холинергическую иннервацию из базального переднего мозга [120]. Холинергические нейроны базального переднего мозга реципрокно связаны и с холинергическими нейронами ППЯ. Показано, что расположенные в базальном переднем мозге нейроны МЯ и МП/ДПБ имеют различные проекции: МЯ связано с разными областями коры и миндалины, а МП – с гиппокампальным комплексом и орбитофронтальной корой [121].
Ранее нами было указано на то, что через ядро РЕ и ППЯ на параметры тета-ритма могут влиять базальные ганглии (БГ) [10]. Влияние осуществляется благодаря тому, что нейроны ядра РЕ и ППЯ находятся под тормозным контролем со стороны выходных ядер БГ (ретикулярной части черного вещества и энтопедункулярного ядра). Во входное ядро БГ (вентральный стриатум) проецируются нейроны поля СА1 и новой коры. Согласно предложенному нами механизму [10], тормозное влияние со стороны БГ должно ослабляться (усиливаться) под действием агонистов (антагонистов) дофаминовых рецепторов. В частности, при действии антагониста и усилении торможения нейронов ППЯ и ядра РЕ должно уменьшится возбуждение нейронов МП и поля СА1, а относительное усиление торможения должно привести к увеличению интервалов между разрядами, т.е. к снижению частоты ритма. Тот факт, что именно указанный эффект наблюдался в работе [122] в результате введения антагониста Д2 рецепторов в стриатум, согласуется со следствием предложенной нами модели. При этом наблюдался не только сдвиг частоты тета-ритма в ЭЭГ гиппокампа с 8–10 Гц, до 5–7 Гц, но и нарушалось поведение в пространственном лабиринте [122]. На связь между БГ и гиппокампом указывают и данные о том, что тета-активность, одновременно регистрируемая в стриатуме и в поле СА1 во время движения по выбранному рукаву у крыс, научившихся хорошо выполнять задачу в Т-лабиринте, была высоко когерентна, причем фазы активности были разнонаправленны [123]. Дофамин может влиять на тета-активность и через ОПЯ, нейроны которого получает и дофаминергическую иннервацию из вентрального поля покрышки [116]. В последние годы холинергическая теория деменции эволюционирует в направлении от фокусировки исключительно на памяти к расширенной когнитивной функции, модулируемой более сложными и взаимодействующими нейронными сетями [124]. Полагают, что на внимание, обучение и выполнение задачи влияет взаимодействие холинергической и дофаминергической систем и что нарушение этих двух нейромедиаторных систем увеличивает риск деменции при болезни Паркинсона [124].
Вовлечение энторинальной коры в ритмическую активность. Полагают, что важную роль в процессах памяти играют флуктуации в тета-ритме в цепи, связывающей ЭК с гиппокампом. Как указывалось выше, нейроны ЭК могут влиять на параметры тета-ритма, поскольку возбуждают нейроны перегородки и, в свою очередь, получают иннервацию из МП. Ацетилхолин влияет на активность нейронов ЭК, причем показано на срезах ЭК, что карбахол участвует в формировании тета-ритма в гиппокампальной цепи за счет воздействия на М1 рецепторы, но не на М2 или никотиновые рецепторы [125]. Кроме того, показано, что во время тета-активности ацетилхолин уменьшает возбуждение медиальной ЭК [126]. Поскольку в результате этого может уменьшиться и возбуждающее влияние ЭК на нейроны разных полей гиппокампа, это должно привести к изменению параметров тета-ритма. В свою очередь, тета-ритм влияет на эффективность латерального и медиального перфорантных входов из ЭК в ЗИ, причем по-разному. Так, тета-активность усиливала действие никотина на ответ нейронов ЗИ на стимуляцию медиального перфорантного пути, но препятствовало вызванному никотином увеличению ответа на стимуляцию латерального перфорантного пути [127].
В ЭК, как и в других частях лимбической системы. выделены тета-on и тета-off клетки [126]. Электрическая стимуляция ЗГЯ увеличивала частоту разрядов тета-on клеток в ЭК и значительно уменьшала частоту разрядов тета-off клеток [128]. При этом частота разрядов клеток ЭК, не связанных с тета-активностью, не менялась [128]. Микроинъекция прокаина в МП устраняла тета-активность в гиппокампе и тета-активность в ЭК, вызванную стимуляцией ЗГЯ [128]. Результаты указанной работы свидетельствуют в пользу сходного параллельного влияния восходящего синхронизирующего пути из ствола мозга на относящиеся к тета активности клетки в гиппокампе и ЭК [128].
Связанные с пространственным расположением клетки решетки в слоях 2 и 3 ЭКм преимущественно разряжаются во время провала и пика тета-активности соответственно [129]. При этом клетки решетки, срабатывающие во время провала в тета-активности, несут больше пространственной информации [129]. У этих клеток меньше частота разрядов, но они имеют тенденцию к срабатыванию пачечными разрядами [129]. Внутриклеточные исследования показали, что проецирующиеся в гиппокамп нейроны слоя 2 ЭК активировались частотами более 5 Гц, а нейроны слоя 3 хорошо активировались частотами менее 10 ГЦ и ингибировались при частотах более 10 Гц [130]. Поскольку нейроны слоя 2 ЭК проецируются в ЗИ и поле СА3, а нейроны нижней части слоя 3 – в поле СА1, из приведенных выше работ следует, что эти нейроны отличаются как по свойствам, так по вовлечению в разные участки цепей, участвующих в ритмической активности. Эта активность, однако, способствует модификации синаптической передачи. Так показано, что в пути от слоя 3 ЭК к пирамидным клеткам поля СА1 стимуляция в тета-ритме приводит к слабой ДП, которая усиливалась в присутствии антагониста ГАМКа-рецепторов бикукуллина [131]. Судя по результатам работы [131], афферентное торможение на перфорантном входе к пирамидным клеткам поля СА1 является сильным.
Примечательно, что активность на пике и провале тета-активности в пирамидном слое поля СА1 соотносят с кодированием информации и извлечением ее из памяти, соответственно, причем ацетилхолин регулирует баланс между кодированием и извлечением из памяти [132]. Антагонист мускариновых рецепторов скополамин не только ослаблял гамма-ритм на пике тета-активности, но и ухудшал кодирование [132]. Блокада мускариновых рецепторов уменьшала пространственную настройку тета-on и тета-off клеток ЭК, однако различие было больше для клеток решетки, срабатывающих на пике тета-ритма, т.е. в слое 3 ЭК. Результаты работы [132] указывают на важность активации мускариновых рецепторов для определения расположения в пространстве, а также на наличие разных популяций клеток решетки. По другим данным, в новом окружении, когда концентрация ацетилхолина увеличивается, расширяется паттерн активности клеток решетки и уменьшается частота тета-ритма [133].
РОЛЬ ТЕТА-АКТИВНОСТИ В ОБУЧЕНИИ И ПАМЯТИ И ВЛИЯНИЕ АЦЕТИЛХОЛИНА НА ЭТИ ПРОЦЕССЫ
Как указывалось во введении, холинергический вход может влиять на обучение и формирование следов памяти, поскольку способствует появлению тета-ритма, при котором активность пре- и постсинаптических клеток синхронизирована во времени, что приводит к улучшению условий для пластических перестроек межнейронных связей [134]. Поскольку в генерации ритмической активности и синхронизации участвуют разные структуры, каждая из них может вносить определенный вклад в выполнение этого условия. Принято считать, что эпизодическая память зависит от функционирования нейронной сети, включающей такие связанные структуры, как нижневисочная кора, гиппокамп, маммиллярные тела [135, 136]. Показано, что при БА по сравнению с контролем снижена не только концентрация ацетилхолина, но и синхронизация в тета-диапазоне между правой дорзолатеральной ПфК и правой задней нижней теменной долей [137]. Также ухудшена связь левой нижней теменной коры и левого гиппокампа, латеральной фронтальной области и передней цингулярной коры [137]. Разрыв связей между областями мозга является преклиническим признаком БА [138]. Показано, что повреждение маммиллярных тел приводит к нарушениям эпизодической памяти, причем этот эффект обостряется, если до повреждения был снижен уровень ацетилхолина в нижневисочной коре [135]. Поэтому, в условиях характерного для БА холинергического дефицита в нижневисочной коре, амнезия, вызванная повреждением гипоталамуса, выражена сильнее и ее трудно восстановить [135]. Судя по результатам работы [139], при БА сильно снижена тета-активность во фронтальной и теменной областях коры. В этих же областях уменьшена вызванная событием когерентность в тета-диапазоне [139]. Аналогичное снижение когерентности в тета-диапазоне обнаружено в работе [140]. Повреждение МП также приводит к снижению синхронизации по фазе в тета-активности фронтальной коры и дорзального гиппокампа [138]. О роли в рассматриваемых процессах ЭК, связывающей кору с гиппокампом, свидетельствуют данные о том, что наличие в ЭК характерных для БА тау образований приводит к ухудшению взаимодействий в нейронных цепях при ассоциативном обучении [141]. При БА амилоидные бляшки найдены почти во всех таламических ядрах, но они более многочисленны в ядрах, относящихся к лимбическим цепям, в частности, в ядре РЕ интраламинарного комплекса [142]. При БА значительное число нейрофибриллярных сплетений найдено и в СТЯ [143]. С учетом указанного выше характера взаимодействии между СТЯ, ППЯ и ядром РЕ, можно полагать, что если при БА активность СТЯ ослабляется из-за наличия бляшек, это должно снизить возбуждение холинергических клеток ППЯ. В результате возбуждение нейронов ядра РЕ дополнительно уменьшится. В свою очередь, это приведет к изменению активности МП и, следовательно, параметров тета-ритма.
Тета-активность появляется при исследовательском поведении [144]. Когда стимулы подавали на пике тета-активности, наблюдалась ДП, которая длилась 48 часов, а если стимулы подавали во время провала, ДП не индуцировалась [144]. Следует отметить, что с одной стороны тета-ритм способствует индукции ДП, а с другой стороны, увеличение активности нейронов вследствие ДП на их возбудительных входах, может увеличивать амплитуду тета-ритма. Так показано, что индукция ДП в синапсах, образованных коллатералями Шаффера на пирамидных клетках поля СА1, приводит к усилению мощности, но не частоты тета-ритма [145]. Во время исследовательского поведения ацетилхолин увеличивает активность клеток места в тета-ритме и усиливает ДП [146, 147]. Активируемый при обучении холинергический вход усиливает ДП в поле СА1 за счет воздействия на мускариновые М1 рецепторы [146, 148]. Полагают, что при лечении когнитивных дисфункций при БА, включая нарушения памяти, вещества, воздействующие на М1 рецепторы, являются одними из ключевых. В частности, показано, что агонист М1 рецепторов, увеличивает внутригиппокампальную когерентность в тета-диапазоне во время локомоций, связанных с поведением [149]. Кроме того показано, что оптогенетическая стимуляция холинергического септального входа регулирует пластичность ответов нейронов поля СА1 на стимуляцию коллатералей Шаффера через холинорецепторы разных типов в зависимости от времени между стимуляцией холинергического входа и афферентного входа [150]. Если стимуляция осуществлялась через 10 мс после стимуляции коллатералей Шаффера, наблюдалась ДП, зависящая от активации мускариновых рецепторов, причем эта пластичность пропадала под действием бета-амилоидов [150]. Если стимуляция холинергического входа осуществлялась в интервале 10–100 мс до стимуляции коллатералей Шаффера, наблюдалась ДП, зависящая от активации α7 рецепторов [150]. Ацетилхолин, выделившийся при появлении нового стимула, избирательно подавлял эффективность синаптической передачи в пути СА3-СА1, но сохранял в пути из ЭК в поле СА1, по которому передается сенсорная информация [151]. По мнению автора указанной работы кодирование осуществляется в основном в пирамидном слое на пике тета активности, совпадающим с поступлением сигналов из ЭК, а извлечение из памяти осуществляется во время провала в тета активности, совпадающего со входом из поля СА3. На крысах в свободном поведении показано, что ацетилхолин способствует такому кодированию, тогда как антагонист холинорецепторов скополамин ослабляет формирование различных отображений пространства в новом окружении и уменьшает способность изменения пространственных карт клеток-места [151]. Примечательно, что во время начальной фазы исследования нового пространственного окружения на пике тета волны усиливалась и эффективность входов из ЭК к пирамидным клеткам поля СА3, тогда как эффективность внутренних ассоциативных связей СА3-СА3 снижалась [152]. На провале тета-волны имело место обратное соотношение. Поскольку этот эффект блокировался антагонистом мускариновых рецепторов атропином [152], можно полагать, что снижение эффективности ассоциативных связей и увеличение эффективности коркового входа к нейронам поля СА3 на пике тета-волны вызвано воздействием на мускариновые рецепторы. В более позднем обзоре [153] также приведены доказательства того, что повреждение холинергического входа в гиппокамп или использование антагонистов холинорецепторов влияет на процессы, связанные с гиппокампом. С учетом этих данных было сделано заключение, что основная функция септо-гиппокампального холинергического входа состоит в уменьшении интерференции процессов кодирования информации и извлечения ее из памяти [153]. Таким образом, характер воздействия ацетилхолина на входы к пирамидным нейронам гиппокампа зависит от поведенческой активности и фазы тета-ритма.
Оказалось, что во время активного поведения стимуляции одного только холинергического входа в гиппокамп из МП недостаточно для того, чтобы вызвать существенные изменения в спайковой активности гиппокампальных нейронов [62]. Однако неизбирательная стимуляция МП вызывала в гиппокампе увеличение спайковой активности в тета-ритме и повышение синхронизации разрядов клеток места, а также сужение их пространственной настройки [62]. Оказалось, что деполяризация холинергических клеток МП вызывала активацию соседних не холинергических нейронов МП, а также интернейронов гиппокампа [62]. Эти данные указывают на важный вклад глутаматергического и ГАМКергического входов из МП в гиппокамп. При неизбирательной активации МП исследовательское поведение соотносилось с паттерном тета-активности разряжающихся пачками клеток, не являвшихся холинергическими. Выдвинуто предположение, что септальный вход не может изменить карту полей места, т.е. он не может изменить пространственное отображение, однако этот вход может быть важным при формировании полей места в новом окружении [62]. То, что спайковая активность холинергических нейронов МП меняется в зависимости от поведенческого состояния животного, показано рядом исследователей. Оказалось, что во время активного исследовательского поведения частота срабатывания холинергических клеток составляла 4.7 Гц, тогда как в не активном состоянии частота составляла 3.4 Гц [56]. Полагают, что в состоянии бодрствования сниженный эффект от воздействия ацетилхолина на активность гиппокампа может являться следствием уже существующей высокой концентрации ацетилхолина и сильного воздействия на мускариновые рецепторы. Поэтому дополнительная стимуляция септального входа слабо влияла на гиппокампальную систему [5].
У крыс с поврежденным ядром Мейнерта была увеличена мощность тета-ритма в коре и снижена в гиппокампе по сравнению с контролем [154]. Результаты этой работы указывают на разнонаправленный характер влияния холинергического входа из МЯ на функционирование гиппокампа и коры. Эти изменения коррелировали с ухудшением выполнения задачи в бассейне Морриса [154]. Использование избирательного ингибитора ацетилхолинестеразы, что должно привести к увеличению концентрации ацетилхолина, восстанавливало эти изменения ритмической активности, уменьшая мощность тета-ритма в коре и увеличивая в гиппокампе, а также уменьшало нарушения рабочей памяти и внимания [154].
Имеются свидетельства того, что СМЯ может контролировать тета-активность в гиппокампе только при некоторых типах поведения. [155]. Показано, например, что дисфункция СМЯ слабо влияет на тета-активность и обучение в водном лабиринте, но оказывает существенное влияние на выполнение других задач [156]. Отмечено, что СМЯ вовлекается в обучение не на ранней стадии, а позднее, причем распространение тета-активности возрастало от СМЯ к гиппокампу, а также увеличивалась когерентность тета-ритмов в СМЯ и гиппокампе [156]. В эгоцентрической пространственной координации участвуют ММЯ [157].
К настоящему времени накоплен ряд данных, свидетельствующих о вкладе таламического ядра РЕ в поведенческую активность [108, 158–160]. Повреждение ядер РЕ и ромбовидного или их оптогенетическая стимуляция влияли на выполнение задач, связанных с гиппокампом [159]. В незнакомом окружении после повреждения ядер РЕ и ромбовидного пространственные характеристики клеток места поля СА1 ухудшались и были нестабильными, однако в знакомом окружении пространственные характеристики не менялись [108]. На участие ядер РЕ и ромбовидного в пространственной памяти указывают и данные о том, что повреждение ядер РЕ и ромбовидного влияет на пространственную память в радиальном лабиринте, но не на зрительно-пространственное время реакции [161]. Поскольку, как отмечено выше, ядро РЕ влияет на параметры тета-ритма и синхронизацию активности гиппокампа и коры, не исключено, что в основе наблюдавшихся нарушений лежало именно отсутствие такой синхронизации.
Приведенные экспериментальные данные свидетельствуют в пользу того, что структуры, вовлеченные в генерацию тета-ритма, играют существенную роль в процессах обучения и памяти. Изменения параметров тета-ритмов в результате изменений функционирования содержащихся в этих структурах холинергических клеток могут лежать в основе нарушений памяти, характерных для болезни Альцгеймера.
ЗАКЛЮЧЕНИЕ
Ухудшение обучения и памяти связывают с нарушением выраженности ритмической активности, а также синхронизации по фазе между тета-активностью в гиппокампе, энторинальной коре и других областях коры. Поскольку несколько структур, вовлеченных в генерацию тета-ритма, содержат холинергические клетки, концентрация ацетилхолина, должна влиять на параметры ритма и играть существенную роль в процессах обучения и памяти. В настоящей работе проведен анализ возможных механизмов влияния ацетилхолина на параметры тета-ритма в гиппокампе. Комплексный характер этих механизмов связан с влиянием на тета-активность как холинергического входа из медиальной перегородки в гиппокамп, так и холинергических клеток педункулопонтийного и орального понтийного ядер, а также ядра Мейнерта. Кроме того, на тета-активность влияет таламическое ядро реуниенс, проецируещееся в медиальную перегородку, поле СА1 гиппокампа и энторинальную кору. Влияние ядер гипоталамуса (супрамаммилярного, заднего и медиального маммиллярного) также осуществляется через их взаимодействие с гиппокампальной формацией и перегородкой. Часть структур, участвующих в генерации тета-активности, содержит длинноаксонные ГАМКергические клетки, действие которых на другие структуры является как ингибирующим, так и растормаживающим. Ацетилхолин, способствуя усилению возбуждения в сети, участвующей в генерации тета-ритма, может приводить к увеличению его выраженности (мощности), тогда как частота ритма определяется торможением и должна увеличиваться, если оно ослабляется.
Список литературы
Melonakos E.D., White J.A., Fernandez F.R. // Hippocampus. 2016. V. 26. № 12. P. 1525–1541.
Zhang H., Lin S.C., Nicolelis M.A. // J. Neurosci. 2010. V. 30. P. 13431–13440.
Iga Y., Arisawa H., Ise M., Yasuda H., Takeshita Y. // Eur. J. Pharmacol. 1996. V. 308. № 1. P. 13–19.
Dawe G.S., Markevich V.A., Zarei M., Grigoryan G.A., Stephenson J.D. // Журн. высш. нерв. деят. им. И.П. Павлова 2006. Т. 56. № 2. С. 264–273.
Vandecasteele M., Varga V., Berenyi A., Papp E., Bartho P., Venance L., Freund T.F., Buzsaki G. // Proc. Natl. Acad. Sci. USA. 2014. V. 111. P. 13535–13540.
Bland B.H., Oddie S.D. // Neurosci. Biobehav Rev. 1998. V. 22. № 2. P. 259–273.
Pignatelli M., Beyeler A., Leinekugel X. // J. Physiol. Paris. 2012. V. 106. № 3–4. P. 81–92.
Kocsis B. // Brain Res. 2006. V. 1086. № 1. P. 92–97.
Vertes R.P. // Prog. Brain Res. 2015. V. 219. P. 121–144.
Силькис И.Г. // Нейрохимия. 2008. Т. 25. № 3. С. 184–190.
Whittington M.A., Traub R.D., Jefferys J.G.R. // Nature. 1995. V. 373. № 6515. P. 612–615.
Dannenberg H., Pabst M., Braganza O., Schoch S., Niediek J., Bayraktar M., Mormann F., Beck H. // J. Neurosci. 2015. V. 35. № 22. P. 8394–8410.
Teles-Grilo Ruivo L.M., Mellor J.R. // Front. Synaptic Neurosci. 2013. V. 5. Article 2.
Yoder R.M., Pang K.C. // Hippocampus. 2005. V. 15. № 3. P. 381–392.
Roland J.J., Stewart A.L., Janke K.L., Gielow M.R., Kostek J.A., Savage L.M., Servatius R.J., Pang K.C. // J. Neurosci. 2014. V. 34. № 2. P. 506–514.
Zhang H., Lin S.C., Nicolelis M.A. // J. Neurophysiol. 2011. V. 106. № 5. P. 2749–2763.
Simon A.P., Poindessous-Jazat F., Dutar P., Epelbaum J., Bassant M.H. // J. Neurosci. 2006. V. 26. № 35. P. 9038–9046.
Borhegyi Z., Varga V., Szilágyi N., Fabo D., Freund T.F. // J. Neurosci. 2004. V. 24. № 39. P. 8470–8479.
Freund T.F., Antal M. // Nature. 1988. V. 336. № 6195. P. 170–173.
McQuiston A.R. // Front. Synaptic Neurosci. 2014. V. 6. Article 20.
Toth K., Freund T.F., Miles R. // J. Physiol. 1997. V. 500 (Pt. 2). P. 463–474.
Goutagny R., Manseau F., Jackson J., Danik M., Williams S. // Hippocampus. 2008. V. 18. № 6. P. 531–535.
Hangya B., Borhegyi Z., Szilagyi N., Freund T.F., Varga V. // J. Neurosci. 2009. V. 29. № 25. P. 8094–8102.
Blasco-Ibáñez J.M., Freund T.F. // Eur. J. Neurosci. 1995. V. 7. № 10. P. 2170–2180.
Unal G., Crump M.G., Viney T.J., Éltes T., Katona L., Klausberger T., Somogyi P. // Brain Struct. Funct. 2018 V. 223. № 5. P. 2409–2432.
Joshi A., Salib M., Viney T.J., Dupret D., Somogyi P. // Neuron. 2017. V. 96. № 6. P. 1342–1357.
Tsanov M. // Prog Brain Res. 2015. V. 219. P. 103–120.
Lee M.G., Chrobak J.J., Sik A., Wiley R.G., Buzsaki G. // Neuroscience. 1994. V. 62. № 4. P. 1033–1047.
Bassant M.H., Simon A., Poindessous-Jazat F., Csaba Z., Epelbaum J., Dournaud P. // J. Neurosci. 2005. V. 25. № 8. P. 2032–2041.
Apartis E., Poindessous-Jazat F.R., Lamour Y.A., Bassant M.H. // J. Neurophysiol. 1998. V. 79. № 4. P. 1633–1642.
Cobb S.R., Buhl E.H., Halasy K., Paulsen O., Somogyi P. // Nature. 1995. V. 378. № 6552. P. 75–78.
Tsubokawa H., Ross W.N. // J. Neurosci. 1997. V. 17. № 15. P. 5782–5791.
Fernández de Sevilla D., Buño W. // J. Neurosci. 2010. V. 30. № 33. P. 11032–11042
Giessel A.J., Sabatini B.L. // Neuron. 2010. V. 68. № 5. P. 936–947.
Bell L.A., Bell K.A., McQuiston A.R. // J. Physiol. 2015. V. 593. № 1. P. 197–215.
Hasselmo M.E., Sarter M. // Neuropsychopharmacology. 2011. V. 36. № 1. P. 52–73.
Nagode D.A., Tang A.H., Karson M.A., Klugmann M., Alger B.E. // PLoS One. 2011. V. 6. № 11. e27691.
Nagode D.A., Tang A.H., Yang K., Alger B.E. // J. Physiol. 2014. V. 592. № 1. P. 103–123.
Силькис И.Г. // Журн. высш. нерв. деят. им. И.П. Павлова 2002. Т. 52. № 4. С. 392–405.
Силькис И.Г. // Успехи Физиол. Наук 2002. Т. 33. № 1. С. 40–57.
Steffensen S.C., Jones M.D., Hales K., Allison D.W. // Hippocampus. 2006. V. 16. № 12. P. 1080–1090.
Lawrence J.J., Grinspan Z.M., Statland J.M., McBain C.J. // J. Physiol. 2006. V. 571. (Pt. 3). P. 555–562.
Gołebiewski H., Eckersdorf B., Konopacki J. // Neuroreport. 1993. V. 4. № 12. P. 1323–1236.
Силькис И.Г. // Журн. высш. нерв. деят. им. И.П. Павлова 1995. Т. 45. № 1. С. 18–28
Scanziani M. // Neuron. 2000. V. 25. № 3. P. 673–681.
Müller C., Remy S. // Cell Tissue Res. 2017. Dec. 18. [Epub ahead of print]
Smythe J.W., Colom L.V., Bland B.H. // Neurosci. Biobehav. Rev. 1992. V. 16. № 3. P. 289–308.
Colom L.V., Castaneda M.T., Reyna T., Hernandez S., Garrido-Sanabria E. // Synapse. 2005. V. 58. № 3. P. 151–164.
Robinson J., Manseau F., Ducharme G., Amilhon B., Vigneault E., El Mestikawy S., Williams S. // J. Neurosci. 2016. V. 36. № 10. P. 3016–3023.
Manns I.D., Alonso A., Jones B.E. // J. Neurophysiol. 2003 V. 89. № 2. P. 1057–1066.
Huh C.Y., Goutagny R., Williams S. // J. Neurosci. 2010. V. 30. № 47. P. 15951–15961.
Leão R.N., Targino Z.H., Colom L.V., Fisahn A. // J. Neurophysiol. 2015. V. 113. № 3. P. 971–980.
Асташева Е.В., Кичигина В.Ф. // Журн. высш. нерв. деят. им. И.П. Павлова, 2009. Т. 59 № 6. С. 742–749.
Colom L.V., Castaneda M.T., Aleman D., Touhami A. // Neurosci Lett. 2013. V. 541. P. 54–57.
Liu W., Kumar A., Alreja M. // Brain Res. 1998. V. 805. № 1–2. P. 220–233.
Tsanov M., Mamad O. // In: Society for Neuroscience Meeting, 2014. Abstarct, 463.24.
Manseau F., Danik M., Williams S. // J. Physiol. 2005. V. 566. P. 865–884.
Wu M., Newton S.S., Atkins J.B., Xu C., Duman R.S., Alreja M. // J. Pharmacol. Exp. Ther. 2003. V. 307. P. 535–543.
Wu M., Shanabrough M., Leranth C., Alreja M.C. // J. Neurosci. 2000. V. 20. P. 3900–3908.
Chapman C.A., Lacaille J.C. // J. Neurosci. 1999. V. 19. P. 8637–8645.
Zheng F, Khanna S. // Neuroscience. 2001. V. 103. № 4. P. 985–998.
Mamad O., McNamara H.M., Reilly R.B., Tsanov M. // Front. Behav. Neurosci. 2015. V. 9. Article 166.
Stewart M., Fox S.E. // J. Neurophysiol. 1989. V. 61. № 5. P. 982–993.
Stewart M., Fox S.E. // Exp. Brain Res. 1989. V. 77. № 3. P. 507–516.
Stewart M., Fox S.E. // Brain Res. 1989. V. 500. № 1–2. P. 55–60.
Matulewicz P., Kuśmierczak M., Orzeł-Gryglewska J., Jurkowlaniec E. // Brain Res. Bull. 2013. V. 96. P. 10–18.
Vertes R.P., Colom L.V., Fortin W.J., Bland B.H. // Exp. Brain Res. 1993. V. 96. № 3. P. 419–429.
Simon C., Kezunovic N., Ye M., Hyde J., Hayar A., Williams D.K., Garcia-Rill E. // J. Neurophysiol. 2010. V. 104. № 1. P. 463–474.
Kinney G.G., Vogel G.W., Feng P. // Brain Res. 1998. V. 809. № 2. P. 307–313.
Nowacka A., Jurkowlaniec E., Trojniar W. // Brain Res. Bull. 2002. V. 58. № 4. P. 377–384.
Stoiljkovic M., Kelley C., Nagy D., Hajós M. // Biochem. Pharmacol. 2015. V. 97. № 4. P. 445–453.
Muzur A. // J. Theor. Biol. 2005. V. 233. № 1. P. 103–118.
Takano Y., Hanada Y. // Neurosci. Lett. 2009. V. 455. № 1. P. 65–69.
Nuñez A., De la Roza C., Rodrigo-Angulo M.L., Buño W., Reinoso-Suárez F. // Brain Res. 1997. V. 754. № 1–2. P. 1–11.
Pötter-Nerger M., Reese R., Steigerwald F., Heiden J.A., Herzog J., Moll C.K.E., Hamel W., Ramirez-Pasos U., Falk D., Mehdorn M., Gerloff C., Deuschl G., Volkmann J. // Front. Hum. Neurosci. 2017. V. 11. Article 436.
Zavala B., Tan H., Ashkan K., Foltynie T., Limousin P., Zrinzo L., Zaghloul K., Brown P. // Neuroimage. 2016. V. 137. P. 178–187.
Alam M., Heissler H.E., Schwabe K., Krauss J.K. // Exp. Neurol. 2012. V. 233. № 1. P. 233–242.
Sitti I., Acar G., Zisakis A.K., Özdemir M., Acar F., Burchiel K.J. // Stereotact. Funct. Neurosurg. 2016. V. 94. № 1. P. 54–59.
Steininger T.L., Rye D.B., Wainer B.H. // J. Comp. Neurol. 1992. V. 321. № 4. P. 515–543.
Sugimoto T., Hattori T. // Neuroscience. 1984. V. 11. № 4. P. 931–946.
Hallanger A.E., Wainer B.H. // J. Comp. Neurol. 1988. V. 274. № 4. P. 483–515.
Semba K., Fibiger H.C. // J. Comp. Neurol. 1992. V. 323. № 3. P. 387–410.
Hosseini N., Alaei H., Reisi P., Radahmadi M. // Brain Res. 2017. V. 1655. P. 122–127.
Kocsis B., Vertes R.P. // Hippocampus. 1997. V. 7. № 2. P. 204–214.
Żakowski W., Braszka Ł., Zawistowski P., Orzeł-Gryglewska J., Jurkowlaniec E. // Neurosci. Lett. 2017. V. 645. P. 19–24.
Ruan M., Young C.K., McNaughton N. // Front. Neural Circuits. 2017. V. 11. Article 62.
McNaughton N., Logan B., Panickar K.S., Kirk I.J., Pan W. X., Brown N. T., Heenan A. // Hippocampus. 1995. V. 5. № 6. P. 534–545.
Sharp P. E., Koester K. // Hippocampus. 2008. V. 18. № 9. P. 862–878.
Aranda L. // J. Chem. Neuroanat. 2016. V. 74. P. 11–17.
Maglóczky Z., Acsády L., Freund T.F. // Hippocampus. 1994. V. 4. № 3. P. 322–334.
Kirk I. J., Oddie S. D., Konopacki J., Bland B.H. // J. Neurosci. 1996. V. 16. № 17. P. 5547–5554.
Kocsis B., Kaminski M. // Hippocampus. 2006. V. 16. № 6. P. 531–540.
Chee S.S., Menard J.L., Dringenberg H.C. // J. Neurophysiol. 2015. V. 113. № 6. P. 1831–1841.
Kocsis B., Vertes R.P. // J. Neurosci. 1994. V. 14. № 11(Pt. 2). P. 7040–7052.
Kirk I.J. // Neurosci. Biobehav. Rev. 1998. V. 22. № 2. P. 291–302.
Vertes R.P., Kocsis B. // Neuroscience. 1997. V. 81. № 4. P. 893–926.
Leranth C., Carpi D., Buzsaki G., Kiss J. // Neuroscience. 1999. V. 88. № 3. P. 701–718.
Kowalczyk T., Bocian R., Caban B., Konopacki J. // Hippocampus. 2014. V. 24. № 1. P. 7–20.
Ariffin M.Z., Jiang F., Low C.M., Khanna S. // Hippocampus. 2010. V. 20. № 7. P. 852–865.
Pedersen N.P., Ferrari L., Venner A., Wang J.L., Abbott S.B.G., Vujovic N., Arrigoni E., Saper C.B., Fuller P.M. // Nat. Commun. 2017. V. 8. № 1. P. 1405.
Jiang F., Khanna S. // Hippocampus. 2006. V. 16. № 10. P. 891–905.
Nitsch R., Leranth C. // J. Comp. Neurol. 1996. V. 364. № 3. P. 425–438.
Ascoli G.A., Atkeson J.C. // Biosystems. 2005. V. 79. № 1–3. P. 173–181.
Leranth C., Kiss J. // J. Neurosci. 1996. V. 16. № 23. P. 7699–7710.
Borhegyi Z., Freund T.F. // Brain Res. Bull. 1998. V. 46. № 5. P. 453–459.
Gonzalo-Ruiz A., Morte L., Flecha J.M., Sanz J.M. // Anat. Embryol. 1999. V. 200. P. 377–392
Mercer A., Trigg H.L., Thomson A.M. // J. Neurosci. 2007. V. 27. № 27. P. 7329–7338.
Cholvin T., Hok V., Giorgi L., Chaillan F.A., Poucet B. // J. Neurosci. 2018. V. 38. № 1. P. 158–172.
Cassel J.C., Pereira de Vasconcelos A., Loureiro M., Cholvin T., Dalrymple-Alford J.C., Vertes R.P. // Prog. Neurobiol. 2013. V. 111. P. 34–52.
Van der Werf Y.D., Witter M.P., Groenewegen H.J. // Brain Res. Rev. 2002. V. 39. № 2–3. P. 107–140.
McKenna J.T., Vertes R.P. // J. Comp. Neurol. 2004. V. 480. № 2. P. 115–142.
Bokor H., Csáki A., Kocsis K., Kiss J. // Eur. J. Neurosci. 2002. V. 16. № 7. P. 1227–1239.
Vertes R.P., Hoover W.B., do Valle A.C., Sherman A., Rodriguez J.J. // J. Comp. Neurol. 2006. V. 499. № 5. P. 768–796.
Morales G.J., Ramcharan E.J., Sundararaman N., Morgera S.D., Vertes R.P. // Conf. Proc. IEEE Eng. Med. Biol. Soc. 2007. P. 2480–2484.
Levey A.I., Hallanger A.E., Wainer B.H. // J. Comp. Neurol. 1987. V. 257. № 3. P. 317–332.
Krout K.E., Belzer R.E., Loewy A.D. // J. Comp. Neurol. 2002. V. 448. № 1. P. 53–101.
Bernard R., Lydic R., Baghdoyan H.A. // J. Pharmacol. Exp. Ther. 2006. V. 317. № 1. P. 163–171.
Stoiljkovic M., Kelley C., Nagy D., Leventhal L., Hajós M. // Neuropharmacology. 2016. V. 110 (Pt. A). P. 102–108.
Lara-Vásquez A., Espinosa N., Durán E., Stockle M., Fuentealba P. // Sci. Rep. 2016. V. 6. P. 29807.
Heckers S., Geula C., Mesulam M.M. // J. Comp. Neurol. 1992. V. 325. № 1. P. 68–82.
Markello R.D., Spreng R.N., Luh W.M., Anderson A.K., De Rosa E. // Neuroimage. 2018. V. 173. P. 287–297.
Gengler S., Mallot H.A., Hölscher C. // Behav. Brain Res. 2005. V. 164. № 1. P. 73–82.
DeCoteau W.E., Thorn C., Gibson D.J., Courtemanche R., Mitra P., Kubota Y., Graybiel A.M. // Proc. Natl. Acad. Sci. USA. 2007. V. 104. № 13. P. 5644–5649.
Bohnen N.I., Grothe M.J., Ray N.J., Müller M.L.T.M., Teipel S.J. // Curr. Geriatr. Rep. 2018. V. 7. № 1. P. 1–11.
Gołebiewski H., Eckersdorf B., Błaszczyk M., Grabowski R., Konopacki J. // Neuroreport. 1994. V. 5. № 15. P. 1989–1992.
Hamam B.N., Sinai M., Poirier G., Chapman C.A. // Hippocampus. 2007. V. 17. № 2. P. 103–113.
Markevich V.A., Grigoryan G.A., Dawe G.S., Stephenson J.D. // Журн. высш. нерв. деят. им. И.П. Павлова. 2006. Т. 56. № 2. P. 257–263.
Dickson C.T., Kirk I.J., Oddie S.D., Bland B.H. // Hippocampus. 1995. V. 5. № 4. P. 306–319.
Newman E.L., Hasselmo M.E. // Front. Syst. Neurosci. 2014. V. 8. Article 193.
Gloveli T., Schmitz D., Empson R.M. Heinemann U. // J. Neurophysiol. 1997. V. 78. № 6. P. 3444–3449.
Dvorak-Carbone H., Schuman E.M. // J. Neurophysiol. 1999. V. 82. № 6. P. 3213–3222.
Newman E.L., Gillet S.N., Climer J.R., Hasselmo M.E. // J. Neurosci. 2013. V. 33. № 50. P. 19635–19646.
Barry C., Heys J.G., Hasselmo M.E. // Front. Neural. Circuits. 2012. V. 6. Article 5.
Hyman J.M., Wyble B.P., Goyal V., Rossi C.A., Hasselmo M.E. // J. Neurosci. 2003. V. 23. № 37. P. 11725–11731.
Croxson P.L., Browning P.G., Gaffan D., Baxter M.G. // J. Neurosci. 2012. V. 32. № 40. P. 13787–13795.
Vann S.D., Nelson A.J. // Prog. Brain Res. 2015. V. 219. P. 163–185.
Hata M., Kazui H., Tanaka T., Ishii R., Canuet L., Pascual-Marqui R.D., Aoki Y., Ikeda S., Kanemoto H., Yoshiyama K., Iwase M., Takeda M. // Clin. Neurophysiol. 2016. V. 127. № 2. P. 1269–1278.
Sanchez-Alavez M., Robledo P., Wills D.N., Havstad J., Ehlers C.L. // Brain Res. 2014. V. 1559. P. 11–25.
Başar E., Güntekin B., Tülay E., Yener G.G. // Brain Res. 2010. V. 1357. P. 79–90.
Yener G.G., Başar E. // Suppl. Clin. Neurophysiol. 2013. V. 62. P. 237–273.
Tanninen S.E., Nouriziabari B., Morrissey M.D., Bakir R., Dayton R.D., Klein R.L., Takehara-Nishiuchi K. // Neurobiol. Aging. 2017. V. 58. P. 151–162.
Braak H., Braak E. // Acta Neuropathol. 1991. V. 81. № 3. P. 261–268.
Ishino H., Otsuki S. // Folia Psychiatr. Neurol. Jpn. 1975. V. 29. № 2. P. 179–187.
Orr G., Rao G., Houston F.P., McNaughton B.L., Barnes C.A. // Hippocampus. 2001. V. 11. № 6. P. 647–654.
Федотова И.Р., Узаков Ш.С., Фролов А.А., Маркевич В.А. // Журн. высш. нерв. деят. им. И.П. Павлова. 2010. Т. 60. № 4. С. 486–492.
Leung L.S., Shen B., Rajakumar N., Ma J. // J. Neurosci. 2003. V. 23. № 28. P. 9297–9304.
Isaac J.T., Buchanan K.A., Muller R.U., Mellor J.R. // J. Neurosci. 2009. V. 29. № 21. P. 6840–6850.
Doralp S., Leung L.S. // Neurobiol. Learn. Mem. 2008. V. 90. № 2. P. 382–388.
Lebois E.P., Trimper J.B., Hu C., Levey A.I., Manns J.R. // ACS Chem. Neurosci. 2016. V. 7. № 10. P. 1393–1405.
Gu Z., Yakel J.L. // Neuron. 2011. V. 71. № 1. P. 155–165.
Douchamps V., Jeewajee A., Blundell P., Burgess N., Lever C. // J. Neurosci. 2013. V. 33. № 20. P. 8689–8704.
Villarreal D.M., Gross A.L., Derrick B.E. // J. Neurosci. 2007. V. 27. № 49. P. 13457–13467.
Easton A., Douchamps V., Eacott M., Lever C. // Neuropsychologia. 2012. V. 50. № 13. P. 3156–3168.
Rispoli V., Ragusa S., Nisticò R., Marra R., Russo E., Leo A., Felicitá V., Rotiroti D. // J. Alzheimers Dis. 2013. V. 35. № 4. P. 833–846.
Woodnorth M.A., McNaughton N. // Cogn. Affect Behav. Neurosci. 2002. V. 2. № 1. P. 76–83.
Ruan M., Young C.K., McNaughton N. // Hippocampus. 2011. V. 21. № 10. P. 1074–1081.
Sharp P. E., Turner-Williams S. // J. Neurophysiol. 2005. V. 94. № 3. 1920–1927.
Cholvin T., Loureiro M., Cassel R., Cosquer B., Geiger K., De Sa Nogueira D., Raingard H., Robelin L., Kelche C., Pereira de Vasconcelos A., Cassel J.C. // J. Neurosci. 2013. V. 33. № 20. P. 8772–8783.
Cassel J.C., Pereira de Vasconcelos A. // Prog. Brain Res. 2015. V. 219. P. 145–161.
Loureiro M., Cholvin T., Lopez J., Merienne N., Latreche A., Cosquer B., Geiger K., Kelche C., Cassel J.C., Pereira de Vasconcelos A. // J. Neurosci. 2012. V. 32. № 29. P. 9947–9959.
Hembrook J.R., Mair R.G. // Hippocampus. 2011. V. 21. № 8. P. 815–826.
Дополнительные материалы отсутствуют.