

Нейрохимия, 2019, T. 36, № 3, стр. 226-238
Влияние производных 3-оксипиридина и янтарной кислоты на активность моноаминоксидаз в коре головного мозга крыс с аллоксановым диабетом
И. А. Волчегорский 1, А. И. Синицкий 1, И. Ю. Мирошниченко 1, Л. М. Рассохина 1
1 ФГБОУ ВО “Южно-Уральский государственный медицинский университет”
Министерства здравоохранения Российской Федерации
Челябинск, Россия
Поступила в редакцию 12.09.2018
После доработки 30.10.2018
Принята к публикации 02.11.2018
Аннотация
Изучено влияние оригинальных отечественных производных 3-оксипиридина и янтарной кислоты (эмоксипина, реамберина и мексидола) на динамику активности моноаминоксидаз (МАО-А и МАО-Б) в сопоставлении с уровнем биогенных аминов (серотонина и дофамина) в коре головного мозга на протяжении первых двух нед. аллоксанового диабета у крыс. Установлено, что курсовое введение производных 3-оксипиридина (эмоксипина и мексидола) животным с аллоксановым диабетом в дозах, эквивалентных терапевтическому диапазону для человека, предотвращало нарастание кортикальной активности МАО-А и параллельно развивающийся дефицит дофамина. По выраженности этих эффектов эмоксипин и мексидол не уступали α-липоевой кислоте, которая использовалась как препарат сравнения. Изолированное производное янтарной кислоты (реамберин) уменьшало активность МАО-А в неокортексе крыс с аллоксановым диабетом, но вызывало транзиторное снижение коркового содержания серотонина и дофамина на раннем этапе исследования и не корригировало дефицит дофамина к концу периода наблюдения. Курсовое введение всех изученных препаратов вызывало увеличение кортикальной активности МАО-Б, не связанное с параллельными сдвигами уровня моноаминов в коре мозга крыс с аллоксановым диабетом. Изменения активности кортикальных МАО и содержания моноаминов в коре мозга под действием изученных препаратов не зависели от их влияния на уровень циркулирующего кортикостерона и гипергликемию при аллоксановом диабете у крыс.
ВВЕДЕНИЕ
Моноаминоксидаза [МАО; амин: кислород оксидоредуктаза (дезаминирующая), (содержащая флавин); КФ 1.4.3.4] является маркерным ферментом наружной мембраны митохондрий, играющим ключевую роль в катаболизме моноаминов-нейротрансмиттеров [1–3]. Невзирая на широкую распространенность МАО в различных органах и тканях [2], особое внимание привлекает ее роль в регуляции эмоций и поведения [1, 2]. Показатели церебральной МАО-активности отражают особенности характера, склонность к аддикциям, а также предрасположенность к импульсивным реакциям и агрессивным действиям [4]. В первую очередь это касается хлоргилин-ингибируемой А-формы фермента (МАО-А), экспрессируемой в катехоламинергических нейронах головного мозга, и осуществляющей окислительное дезаминирование норадреналина и серотонина [1, 3, 5]. Депренил-ингибируемая форма МАО (МАО-Б), сосредоточенная преимущественно в серотонинергических нейронах и астроцитах, дезаминирует β-фенилэтиламин, рассматривающийся в качестве “эндогенного амфетамина” [1, 2]. Обе формы фермента участвуют в катаболизме дофамина, который дезаминируется преимущественно МАО-А у грызунов и МАО-Б у приматов (включая человека) [1, 2]. Независимо от формы фермента и структурного разнообразия его субстратов, одним из облигатных продуктов МАО-реакции является H2O2 [1], играющая важную роль в регуляции чувствительности к инсулину [6]. При этом важно подчеркнуть, что инсулин подавляет экспрессию генов МАО-А и МАО-Б в нервной ткани [7], уравновешивая противоположное действие глюкокортикостероидных гормонов (ГКГ) [1, 8]. Абсолютная или относительная недостаточность инсулина и/или длительная гиперпродукция ГКГ в результате хронического стресса [7] вызывают нарушение баланса эндокринной регуляции экспрессии генов МАО и приводят к прогрессирующему нарастанию активности этого фермента в головном мозге. Такая ситуация складывается при сахарном диабете (СД) 1-го и 2-го типа, а также при болезни Альцгеймера, которая характеризуется церебральной инсулинорезистентностью и аргументированно рассматривается как СД 3-его типа [7]. Прогрессирующее нарастание МАО-активности головного мозга при этих заболеваниях считается важной причиной нарушений моноаминергической (прежде всего серотонин- и дофаминергической) нейропередачи, обусловливающих развитие депрессии [7]. Стоит добавить, что чрезмерное усиление МАО-зависимой продукции H2O2 вызывает оксидативный стресс (ОС) [2, 9, 10], играющий существенную роль в патогенезе диабетической энцефалопатии [11].
Оригинальные отечественные производные 3‑оксипиридина и янтарной кислоты (эмоксипин, реамберин и мексидол), продемонстрировавшие высокую клиническую эффективность в комплексном лечении диабетических нейропатий [12], вызывают антидепрессивный эффект как у людей, больных СД [13], так и у животных с экспериментальным СД [14]. Эти лекарственные средства (ЛС) проявляют инсулинпотенцирующую и антиглюкокортикоидную активность в экспериментах in vivo [15, 16], а также обладают МАО-модулирующим действием in vitro [17]. Важно подчеркнуть, что направленность МАО-модулирующего действия отдельных производных 3-оксипиридина и янтарной кислоты определяет характер их влияния на ОС in vitro [17]. До настоящего времени остается неизученным влияние эмоксипина, реамберина и мексидола на церебральную активность МАО-А и МАО-Б при экспериментальном СД.
Представленная статья посвящена изучению влияния производных 3-оксипиридина и янтарной кислоты на динамику активности моноаминоксидаз в коре головного мозга крыс с аллоксановым диабетом. Данный отдел головного мозга был выбран для исследования в связи с тем, что он содержит значительную часть аксональных терминалей аминергических нейронов стволовой локализации [18] и относится к числу тех церебральных структур, которые наиболее уязвимы к диабетическому поражению [11]. Полученные данные были сопоставлены с уровнем циркулирующего кортикостерона и изменениями кортикального содержания серотонина и дофамина, дефицит которых считается важнейшей причиной развития депрессии [6]. Дополнительно были исследованы соответствующие эффекты α-липоевой кислоты (α-ЛК), которая ранее использовалась как препарат сравнения при изучении антидепрессивного и церебропротекторного действия производных 3-оксипиридина и янтарной кислоты у крыс с аллоксановым диабетом [19–21].
МЕТОДЫ ИССЛЕДОВАНИЯ
Исследование было проведено на 956 половозрелых беспородных крысах обоего пола, массой 180–250 г. Организация работы соответствовала отечественным и международным этическим нормам, регламентирующим эксперименты на животных [22]. СД моделировали путем внутрибрюшинного введения аллоксана моногидрата (“ДИАЭМ”, Россия) в дозе 163 мг/кг. Животные группы “интактный контроль” получали эквиобъемное количество 0.9% раствора NaCl.
Через 72 ч после индукции СД крыс, получивших инъекцию аллоксана, равномерно распределяли на подгруппы экспериментальной терапии и контроля (“аллоксановый диабет – контроль”). Выбор данного срока для начала экспериментальной терапии обусловлен 4-фазным характером токсикодинамики аллоксана, вызывающего некротическую деструкцию β-эндокриноцитов и абсолютный дефицит инсулина в пределах 12–48 ч после введения [23]. Для надежного воспроизведения аллоксанового диабета период, предшествующий введению изученных ЛС, пролонгировали на сутки от максимальной латентности развития абсолютного дефицита инсулина. Изученные ЛС применяли в рамках трех схем введения – однократное введение без заместительной инсулинотерапии, 7- и 14-кратное введение на фоне заместительной инсулинотерапии. Первая инъекция ЛС всегда выполнялась через 3-е сут после введения аллоксана. Во всех случаях изучаемые препараты вводили внутрибрюшинно, один раз в сутки. Каждое ЛС применяли в 3-х дозах, экстраполированных из разовых дозировок терапевтического диапазона для человека с учетом различий в величинах относительной площади поверхности тела [24]. Во всех случаях минимальной дозой изучаемого диапазона являлась 1/2 от расчетного эквивалента средней терапевтической дозы (ЭСТД). В качестве максимальной дозировки использовался удвоенный ЭСТД. Эмоксипин (2-этил-6-метил-3-оксипиридина гидрохлорид, Московский эндокринный завод) использовали в дозах 6.25, 12.5 и 25 мг/кг. Мексидол (2-этил-6-метил-3-оксипиридина сукцинат, ООО “НПК “Фармасофт”, Россия) применяли в дозах 12.5, 25 и 50 мг/кг. Исследованные дозы 1.5% раствора реамберина (N-(1-дезокси-D-глюцитол-1-ил)-N-метиламмония натрия сукцинат, ООО “НТФФ “Полисан”, Санкт-Петербург) составили 12.5, 25 и 50 мл/кг. α-ЛК (берлитион, “Berlin-Chemie”, Германия) вводили в дозах 25, 50 и 100 мг/кг. Все дозировки изучаемых ЛС вводили в конечном объеме 50 мл/кг (при необходимости препараты разводили в 0.9% растворе NaCl). Животные контрольных подгрупп (“интактный контроль” и “аллоксановый диабет – контроль”) получали изотонический раствор NaCl в том же объеме. При 7- и 14-кратном применении изученных препаратов всем крысам с экспериментальным СД проводили базисную инсулинотерапию, начиная с 4 дня после инъекции аллоксана. С этой целью животным раз в сутки подкожно вводили 3 ед./кг инсулина аспарта двухфазного (НовоМикс 30 Пенфилл, “Novo nordisk”, Дания). В экспериментах с курсовым (7-и и 14-и дневным) введением изученных препаратов крысы подгруппы “интактный контроль” вместо инсулина получали 0.9% раствор NaCl в соответствующем объеме. В экспериментах с однократным применением изученных ЛС заместительную инсулинотерапию не проводили.
Сроки получения биологического материала соответствовали дизайну ранее выполненного исследования, в котором последовательно оценивались психотропные эффекты изучаемых препаратов и их влияние на клеточный состав церебральных структур при аллоксановом диабете у крыс [25]. Через 48 часов после заключительной инъекции изучаемых препаратов животных наркотизировали диэтиловым эфиром, декапитировали, получали кровь и быстро извлекали головной мозг. Из полученных образцов крови выделяли сыворотку, в которой регистрировали концентрацию кортикостерона экстракционно-флуориметрическим методом [26, 27]. Образцы головного мозга отмывали в охлажденном 100 мМ калий-фосфатном буфере (рН 7.6), осушали фильтровальной бумагой и помещали на охлажденные льдом чашки Петри. Все манипуляции с образцами нервной ткани проводили при температуре 0–4°С. Кору головного мозга выделяли по технологии, описанной Glowinski, Iversen [28], взвешивали и гомогенизировали в буфере вышеприведенного состава (соотношение 1 : 20 – вес/объем) с помощью гомогенизатора Поттера–Эвельгейма. Полученный гомогенат разделяли на три порции, одна из которых предназначалась для определения активности МАО, а две другие для регистрации содержания дофамина и серотонина.
Аликвоту первой порции гомогената (0.9 мл) при тщательном перемешивании разбавляли в 1.5 раза 0.9 М раствором сахарозы, приготовленным на 100 мМ калий-фосфатном буфере (рН 7.6), с целью обеспечения концентрации сахарозы (0.3 М), требующейся для дальнейших этапов работы [29]. Полученные образцы замораживали и хранили в герметично закрытых пластиковых пробирках при –80°С (морозильная камера Thermo Forma 905, Thermo Fisher Scientific, США). После размораживания из гомогенатов выделяли МАО-содержащую фракцию субмитохондриальных частиц [29]. На первом этапе этого процесса гомогенаты центрифугировали при 1824 g в течение 10 мин. Затем полученные супернатанты центрифугировали при 12 768 g в течение 35 мин (Hitachi Himac CT 15RE с ротором T15A62, Япония). Осадок ресуспендировали в 250 мкл раствора 0.3 М сахарозы, наслаивали на 2.0 мл 1.2 М раствора сахарозы, приготовленной на 100 мМ калий-фосфатном буфере (рН 7.6) и вновь центрифугировали при 12 687 g в течение 40 мин. После однократной промывки в буфере вышеуказанного состава, очищенные МАО-содержащие белковые преципитаты суспендировали в 1.0 мл того же буферного раствора, замораживали и хранили при –80°С вплоть до определения МАО-активности. После заключительного размораживания пробы разделяли на две порции, в одной из которых определяли активность МАО-А (с серотонин-гидрохлоридом (H9523, Sigma–Aldrich) в качестве субстрата), а в другой регистрировали активность МАО-Б (с солянокислым бензиламином (B5136, Sigma–Aldrich) как субстратом). В обоих случаях регистрацию МАО-активности проводили спектрофотометрическим методом, основанном на дериватизации альдегидных продуктов МАО-реакции 2,4-динитрофенилгидразином (2,4-ДФНГ) [29]. Этап дериватизации 5-гидроксииндолуксусного альдегида и бензальдегида выполнялся с минимальными модификациями, которые касались снижения концентрации 2,4-ДФНГ до 0.1 М в реактиве для фиксации альдегидов и уменьшения содержания тритона Х-100 до 3 г/л в реактиве для повышения хромогенности ДНФГ – производных. Оптическую плотность регистрировали на спектрофотометре СФ-56 (ОКБ “Спектр”, Россия). Активность ферментов выражали в единицах прироста оптической плотности 2,4-ДФНГ-дериватов альдегидных продуктов МАО-реакции за 1 мин инкубации в расчете на 1 г нервной ткани (ед.о.п./мин г ткани). Расчет ферментной активности на вес ткани (а не на количество белка в пробе) обусловлен ранее установленными существенными изменениями содержания белка в субмитохондриальной фракции нервной ткани крыс с аллоксановым диабетом.
Вторую и третью порции исходного гомогената депротеинировали равными объемами 0.8 М хлорной кислоты, содержащей 0.2% ЭДТА (для определения дофамина) и 20% трихлоруксусной кислоты (для определения серотонина), тщательно перемешивали в течение 10 мин и центрифугировали при 800 g в течение 20 мин при 4°С. Полученные супернатанты использовались для определения содержания изучаемых моноаминов. Уровень дофамина регистрировали флуориметрически после его предварительной адсорбции из депротеинатов на окиси алюминия с последующим элюированием уксусной кислотой и окислением йодом [30–32]. Содержание серотонина определяли модифицированным экстракционно-флуориметрическим методом по реакции с о-фталевым альдегидом [31, 33]. Флуоресценцию полученных образцов оценивали, с помощью анализатора “Флюорат-02-АБЛФ-Т” (ГК “Люмэкс”, Россия). Показатели содержания моноаминов выражали в микрограммах на грамм нервной ткани.
Учитывая известную роль нарушений углеводного обмена в развитии диабетической энцефалопатии [11], в параллельной серии экспериментов оценивали влияние исследуемых препаратов на выраженность гипергликемии у крыс с аллоксановым диабетом (набор реагентов “Новоглюк-К, М (500)”, ЗАО “Вектор-Бест”, Россия). Уровень гликемии регистрировали на фоне предшествующей депривации пищи в течение 24 ч при сохранении свободного доступа к воде. По своему дизайну дополнительная серия экспериментов соответствовала основной серии, в которой изучалось влияние препаратов на активность МАО и содержание моноаминов в коре головного мозга крыс с экспериментальным сахарным диабетом.
Статистический анализ выполнен с использованием пакета прикладных компьютерных программ SPSS-17.0. Данные обработаны методами дескриптивной статистики и представлены в виде медианы (Me) и диапазона между “нижним” (LQ, 25 процентиль) и “верхним” (UQ, 75 процентиль) квартилями. О достоверности межгрупповых различий судили по U-критерию Манна–Уитни. Оценку взаимосвязи между стандартизованными по соответствующим контролям результатами экспериментальных серий проводили путем расчета коэффициентов корреляции Спирмена (rs). Проверка статистических гипотез выполнялась при критическом уровне значимости р = 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
В результате проведенного исследования было установлено, что развитие экспериментального СД у крыс сопровождается быстрым нарастанием активности МАО в коре головного мозга (табл. 1–4). Прежде всего, это касается МАО-А, медианная активность которой в группе “аллоксановый диабет – контроль” без заместительной инсулинотерапии в 1.68 раза превысила соответствующий показатель “интактного контроля” уже через 5 сут после введения аллоксана. По-видимому, прирост активности данного фермента при аллоксановом диабете, также как параллельно развивающаяся гипергликемия (табл. 4), обусловлен абсолютным дефицитом инсулина. Важно заметить, что заместительная инсулинотерапия в период с 4-го по 10-й день после инъекции аллоксана, оказалась недостаточной для коррекции гипергликемии (табл. 4), но привела к временной нормализации активности МАО-А в коре мозга на 12-ые сут эксперимента (табл. 1). Это согласуется с представлениями об ингибирующем влиянии инсулина на экспрессию генов МАО [7]. Вместе с тем, несмотря на продолжение заместительной инсулинотерапии в период с 11-го по 17‑й день после введения аллоксана, активность кортикальной МАО-А в группе “аллоксановый диабет-контроль” повторно возросла и к 19-м сут эксперимента достигла 437.5% от соответствующего показателя “интактного контроля” (табл. 1). Полученные данные отражают формирование вторичной инсулинорезистентности [35], прогрессирование которой в динамике аллоксанового диабета привело к неэффективности использованной дозы инсулина как в отношении коррекции гипергликемии (табл. 4), так и предотвращения роста активности МАО-А в коре головного мозга (табл. 1). Стоит добавить, что на фоне абсолютного дефицита инсулина [23], отягощенного инсулинорезистентностью [35], у контрольных крыс с аллоксановым диабетом уровень циркулирующего кортикостерона соответствовал значениям “интактного контроля” на всех сроках эксперимента (табл. 3). Не исключено, что подобное нарушение эндокринного равновесия ведет к несбалансированному усилению не только гипергликемизирующего действия эндогенного ГКГ (кортикостерона), но и его стимулирующего влияния на экспрессию гена МАО-А [1] с последующим нарастанием активности этого фермента в коре головного мозга (табл. 1). Похожая, но менее выраженная динамика была отмечена для кортикальной МАО-Б, активность которой в группе “аллоксановый диабет – контроль” увеличивалась в 1.56 раза относительно “интактного контроля” только к 19 сут эксперимента (табл. 1). Важно подчеркнуть, что максимальное нарастание кортикальной МАО-активности к концу эксперимента сопровождалось более чем трехкратным снижением содержания дофамина в неокортексе крыс с аллоксановым диабетом (табл. 2).
Таблица 1.
Влияние производных 3-оксипиридина и янтарной кислоты на активность моноаминоксидаз в коре головного мозга у крыс с аллоксановым диабетом [Me (LQ-UQ)]
| Группа | Активность моноаминоксидаз, ед.о.п./ г ткани мин | |||||
|---|---|---|---|---|---|---|
| МАО-А | МАО-Б | |||||
| Кратность введения ЛС | 1-кратное | 7-кратное | 14-кратное | 1-кратное | 7-кратное | 14-кратное |
| Интактный контроль | 0.19 (0.15–0.25) (n = 10) |
0.17 (0.07–0.29) (n = 10) |
0.08 (0.03–0.27) (n = 10) |
0.38 (0.30–0.53) (n = 10) |
0.50 (0.43–0.53) (n = 10) |
0.27 (0.18–0.39) (n = 10) |
| Аллоксановый диабет – контроль | 0.32* (0.26–0.36) (n = 10) |
0.17 (0.09–0.27) (n = 10) |
0.35* (0.27–0.39) (n = 11) |
0.36 (0.16–0.53) (n = 10) |
0.43 (0.39–0.46) (n = 10) |
0.42* (0.35–0.53) (n = 11) |
| Эмоксипин | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
0.18** (0.16–0.21) (n = 10) |
0.27 (0.22–0.37) (n = 10) |
0.10** (0.04–0.17) (n = 10) |
0.36 (0.27–0.46) (n = 10) |
0.50** (0.44–0.60) (n = 10) |
0.45 (0.36–0.63) (n = 10) |
| ЭСТД (12.5 мг/кг) |
0.20** (0.16–0.28) (n = 10) |
0.20 (0.11–0.24) (n = 10) |
0.20 (0.13–0.34) (n = 10) |
0.22 (0.19–0.44) (n = 10) |
0.48** (0.4–0.62) (n = 10) |
0.36 (0.30–0.46) (n = 10) |
| 2ЭСТД (25 мг/кг) |
0.25 (0.18–0.39) (n = 10) |
0.11 (0.06–0.23) (n = 10) |
0.13** (0.07–0.27) (n = 10) |
0.22 (0.13–0.38) (n = 10) |
0.52** (0.4–0.59) (n = 10) |
0.52 (0.38–0.58) (n = 10) |
| Реамберин | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
0.15** (0.12–0.28) (n = 10) |
0.16 (0.13–0.31) (n = 10) |
0.16** (0.14–0.27) (n = 10) |
0.35 (0.20–0.46) (n = 10) |
0.44 (0.39–0.49) (n = 10) |
0.37 (0.33–0.49) (n = 10) |
| ЭСТД (12.5 мг/кг) |
0.17 (0.09–0.34) (n = 10) |
0.26 (0.12–0.35) (n = 10) |
0.24** (0.17–0.29) (n = 10) |
0.33 (0.13–0.39) (n = 10) |
0.51 (0.37–0.56) (n = 10) |
0.41 (0.35–0.50) (n = 10) |
| 2ЭСТД (25 мг/кг) |
0.13** (0.10–0.16) (n = 10) |
0.18 (0.16–0.20) (n = 10) |
0.18** (0.12–0.27) (n = 10) |
0.34 (0.29–0.37) (n = 10) |
0.44 (0.38–0.46) (n = 10) |
0.55** (0.52–0.62) (n = 10) |
| Мексидол | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
0.13** (0.11–0.25) (n = 10) |
0.14 (0.09–0.31) (n = 10) |
0.07** (0.04–0.11) (n = 10) |
0.30 (0.27–0.34) (n = 10) |
0.48** (0.43–0.53) (n = 10) |
0.59** (0.54–0.60) (n = 10) |
| ЭСТД (12.5 мг/кг) |
0.17** (0.09–0.22) (n = 10) |
0.29 (0.10–0.35) (n = 10) |
0.06** (0.03–0.09) (n = 10) |
0.43 (0.39–0.52) (n = 10) |
0.59** (0.45–0.64) (n = 10) |
0.62** (0.52–0.74) (n = 10) |
| 2ЭСТД (25 мг/кг) |
0.16** (0.13–0.21) (n = 10) |
0.16 (0.10–0.30) (n = 10) |
0.14** (0.09–0.18) (n = 10) |
0.34 (0.20–0.36) (n = 10) |
0.56** (0.47–0.56) (n = 10) |
0.45 (0.43–0.58) (n = 10) |
| α-Липоевая кислота | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
0.19** (0.16–0.21) (n = 10) |
0.20 (0.15–0.26) (n = 11) |
0.38 (0.17–0.42) (n = 10) |
0.38 (0.36–0.48) (n = 10) |
0.47** (0.43–0.51) (n = 11) |
0.45 (0.41–0.48) (n = 10) |
| ЭСТД (12.5 мг/кг) |
0.11** (0.09–0.13) (n = 10) |
0.22 (0.20–0.29) (n = 10) |
0.16** (0.12–0.20) (n = 10) |
0.35 (0.33–0.39) (n = 10) |
0.38 (0.28–0.48) (n = 10) |
0.50 (0.46–0.54) (n = 10) |
| 2ЭСТД (25 мг/кг) |
0.16** (0.15–0.24) (n = 11) |
0.17 (0.14–0.20) (n = 10) |
0.14** (0.12–0.29) (n = 10) |
0.37 (0.32–0.39) (n = 11) |
0.43 (0.41–0.47) (n = 10) |
0.60** (0.47–0.70) (n = 10) |
Примечание: здесь и в табл. 2, 3, 4 различия достоверны (р < 0.05) по сравнению: * с группой “интактный контроль”; ** с группой “аллоксановый диабет – контроль”; показатели, по которым установлены достоверные различия, выделены полужирным шрифтом.
Таблица 2.
Влияние производных 3-оксипиридина и янтарной кислоты на содержание серотонина и дофамина в коре головного мозга у крыс с аллоксановым диабетом [Me (LQ-UQ)]
| Группа | Содержание моноаминов, мкг / г ткани | |||||
|---|---|---|---|---|---|---|
| Серотонин | Дофамин | |||||
| Кратность введения ЛС | 1–кратное | 7–кратное | 14–кратное | 1–кратное | 7–кратное | 14–кратное |
| Интактный контроль | 2.99 (1.61–4.98) (n = 10) |
2.43 (1.14–5.92) (n = 10) |
3.81 (1.57–5.40) (n = 10) |
2.71 (2.07–6.04) (n = 10) |
1.89 (0.82–4.61) (n = 10) |
3.25 (1.54–5.41) (n = 10) |
| Аллоксановый диабет – контроль | 3.75 (2.80–5.24) (n = 10) |
2.83 (2.40–4.44) (n = 10) |
1.11 (0.74–4.95) (n = 11) |
3.43 (2.47–5.70) (n = 10) |
2.68 (1.70–4.80) (n = 10) |
1.01* (0.87–4.78) (n = 11) |
| Эмоксипин | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
3.17 (2.84–4.86) (n = 10) |
3.83 (1.49–3.88) (n = 10) |
3.13 (2.01–3.89) (n = 10) |
3.94 (1.93–5.60) (n = 10) |
3.48 (1.73–5.60) (n = 10) |
3.30 (2.37–3.96) (n = 10) |
| ЭСТД (12.5 мг/кг) |
3.27 (1.61–4.09) (n = 10) |
3.68 (1.52–5.82) (n = 10) |
2.13 (1.43–3.55) (n = 10) |
3.01 (1.61–3.96) (n = 10) |
4.03 (1.02–5.04) (n = 10) |
2.35 (1.07–3.07) (n = 10) |
| 2ЭСТД (25 мг/кг) |
3.54 (3.33–3.59) (n = 10) |
3.94 (2.31–4.18) (n = 10) |
4.41 (3.09–5.28) (n = 10) |
3.34 (3.09–4.77) (n = 10) |
3.82 (2.23–4.21) (n = 10) |
5.29** (3.42–5.83) (n = 10) |
| Реамберин | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
2.17** (1.73–4.32) (n = 10) |
2.57 (1.30–5.27) (n = 10) |
1.75 (1.24–3.71) (n = 10) |
1.97** (1.36–2.80) (n = 10) |
1.83 (1.13–6.02) (n = 10) |
2.05 (1.61–3.74) (n = 10) |
| ЭСТД (12.5 мг/кг) |
2.94 (1.40–4.74) (n = 10) |
2.32 (1.53–3.40) (n = 10) |
3.52 (2.99–4.48) (n = 10) |
3.38 (1.30–5.30) (n = 10) |
2.78 (1.23–3.30) (n = 10) |
3.71 (2.77–4.59) (n = 10) |
| 2ЭСТД (25 мг/кг) |
1.93** (0.80–2.98) (n = 10) |
5.25 (4.66–5.49) (n = 10) |
2.34 (0.86–4.04) (n = 10) |
2.85 (1.61–3.03) (n = 10) |
5.15 (1.79–6.16) (n = 10) |
3.10 (1.06–5.40) (n = 10) |
| Мексидол | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
2.92 (2.65–3.15) (n = 10) |
2.48 (2.00–6.29) (n = 10) |
4.67 (4.25–5.06) (n = 10) |
2.90 (2.22–3.31) (n = 10) |
2.85 (2.29–5.64) (n = 10) |
5.85** (5.26–6.44) (n = 10) |
| ЭСТД (12.5 мг/кг) |
3.56 (0.84–4.47) (n = 10) |
3.73 (1.49–4.65) (n = 10) |
4.16 (3.81–4.93) (n = 10) |
3.54 (1.89–4.50) (n = 10) |
4.48 (1.88–5.19) (n = 10) |
5.38** (1.58–6.20) (n = 10) |
| 2ЭСТД (25 мг/кг) |
3.30 (2.35–4.42) (n = 10) |
4.31 (2.86–5.31) (n = 10) |
4.50 (1.06–5.17) (n = 10) |
2.85 (2.47–4.29) (n = 10) |
4.58 (3.09–5.33) (n = 10) |
4.25 (1.49–4.56) (n = 10) |
| α-Липоевая кислота | ||||||
| 1/2ЭСТД (6.25 мг/кг) |
2.12** (1.85–2.52) (n = 10) |
0.81 (0.21–4.76) (n = 11) |
0.72 (0.70–1.03) (n = 10) |
2.24** (1.91–2.96) (n = 10) |
3.88 (0.24–6.35) (n = 11) |
1.26 (0.94–3.16) (n = 10) |
| ЭСТД (12.5 мг/кг) |
1.85** (1.47–4.29) (n = 10) |
4.23** (3.97–6.03) (n = 10) |
4.05 (0.53–5.23) (n = 10) |
3.01 (1.80–4.76) (n = 10) |
4.17 (2.99–4.67) (n = 10) |
3.86 (1.77–4.59) (n = 10) |
| 2ЭСТД (25 мг/кг) |
3.01 (1.86–3.81) (n = 11) |
2.78 (1.85–4.17) (n = 10) |
1.51 (1.31–2.88) (n = 10) |
2.87 (2.14–3.23) (n = 11) |
2.67 (1.87–3.58) (n = 10) |
1.80** (1.15–4.16) (n = 10) |
Таблица 3.
Влияние производных 3-оксипиридина и янтарной кислоты на уровнь кортикостерона в сыворотке у крыс с аллоксановым диабетом [Me (LQ-UQ)]
| Группа | Кортикостерон, нмоль/л | ||
|---|---|---|---|
| Кратность введения ЛС | 1-кратное | 7-кратное | 14-кратное |
| Интактный контроль | 206.5 (142.1–439.4) (n = 10) |
156.5 (145.3–230.9) (n = 10) |
247.2 (199.3–346.0) (n = 10) |
| Аллоксановый диабет – контроль | 200.8 (178.5–240.7) (n = 10) |
215.7 (172.3–249.8) (n = 10) |
286.6 (150.8–297.2) (n = 11) |
| Эмоксипин | |||
| 1/2ЭСТД (6.25 мг/кг) |
255.8 (137.7–320.2) (n = 10) |
188.3 (135.3–277.9) (n = 10) |
238.8 (131.4–382.5) (n = 10) |
| ЭСТД (12.5 мг/кг) |
210.8 (129.2–228.9) (n = 10) |
369.8 (198.1–510.5) (n = 10) |
236.9 (167.9–381.1) (n = 10) |
| 2ЭСТД (25 мг/кг) |
308.2 (160.7–473.1) (n = 10) |
192.8 (125.4–263.2) (n = 10) |
186.8 (110.9–256.7) (n = 10) |
| Реамберин | |||
| 1/2ЭСТД (6.25 мг/кг) |
178.5 (125.1–285.8) (n = 10) |
246.6 (177.1–317.1) (n = 10) |
176.2 (145.3–292.6) (n = 10) |
| ЭСТД (12.5 мг/кг) |
170.0 (146.8–211.3) (n = 10) |
185.9 (163.3–240.5) (n = 10) |
156.1 (144.0–185.3) (n = 10) |
| 2ЭСТД (25 мг/кг) |
186.4 (150.3–288.7) (n = 10) |
231.3 (142.7–274.7) (n = 10) |
163.8 (125.1–181.7) (n = 10) |
| Мексидол | |||
| 1/2ЭСТД (6.25 мг/кг) |
151.6** (141.0–187.7) (n = 10) |
385.9 (146.6–502.8) (n = 10) |
184.1 (177.1–586.6) (n = 10) |
| ЭСТД (12.5 мг/кг) |
189.6 (145.0–273.4) (n = 10) |
193.3 (183.0–266.2) (n = 10) |
341.1** (301.7–390.1) (n = 10) |
| 2ЭСТД (25 мг/кг) |
167.4 (124.3–285.6) (n = 10) |
177.1 (152.8–237.7) (n = 10) |
201.8 (154.7–243.1) (n = 10) |
| α-Липоевая кислота | |||
| 1/2ЭСТД (6.25 мг/кг) |
138.0** (118.9–192.1) (n = 10) |
156.7 (116.1–197.6) (n = 11) |
159.0 (137.8–167.6) (n = 10) |
| ЭСТД (12.5 мг/кг) |
143.7 (115.8–252.6) (n = 10) |
168.1** (145.2–185.9) (n = 10) |
414.8** (253.2–545.7) (n = 10) |
| 2ЭСТД (25 мг/кг) |
280.0 (164.4–288.5) (n = 11) |
144.9** (135.2–180.2) (n = 10) |
145.8** (123.4–162.0) (n = 10) |
Таблица 4.
Влияние производных 3-оксипиридина и янтарной кислоты на выраженность гликемии (ммоль/л) в сыворотке у крыс с аллоксановым диабетом [Me (LQ-UQ)]
| Группа, доза | Лекарственный препарат | |||
|---|---|---|---|---|
| Эмоксипин | Реамберин | Мексидол | α-Липоевая кислота |
|
| 1-кратное введение ЛС | ||||
| Интактный контроль | 4.3 (4.0–5.8) (n = 11) |
4.5 (3.5–5.6) (n = 10) |
4.1 (3.1–5.9) (n = 13) |
4.1 (3.1–5.9) (n = 13) |
| Аллоксановый диабет – контроль | 19.9* (12.0–25.2) (n = 11) |
20.1* (4.8–22.6) (n = 10) |
12.3* (3.2–14.1) (n = 11) |
12.3* (3.2–14.1) (n = 11) |
| 1/2ЭСТД | 12.2 (6.5–27.1) (n = 11) |
4.1 (2.2–10.1) (n = 10) |
17.9 (3.8–21.8) (n = 11) |
10.1 (4.9–23.4) (n = 12) |
| ЭСТД | 4.8 (3.8–21.4) (n = 11) |
5.9 (3.3–9.5) (n = 10) |
6.2 (4.6–9.9) (n = 11) |
9.2 (5.1–16.6) (n = 11) |
| 2ЭСТД | 4.8 (3.1–24.3) (n = 11) |
10.0 (3.6–18.9) (n = 10) |
8.2 (4.9–16.1) (n = 11) |
7.2 (4.1–24.1) (n = 10) |
| 7-кратное введение ЛС | ||||
| Интактный контроль | 4.8 (4.1–5.1) (n = 10) |
4.8 (3.6–5.7) (n = 10) |
4.8 (4.1–5.1) (n = 10) |
4.8 (3.6–5.7) (n = 10) |
| Аллоксановый диабет – контроль | 14.3* (7.7–15.9) (n = 11) |
15.3* (5.0–24.3) (n = 11) |
14.3* (7.7–15.9) (n = 11) |
15.3* (5.0–24.3) (n = 11) |
| 1/2ЭСТД | 6.5** (4.8–7.6) (n = 12) |
5.9 (5.3–7.7) (n = 11) |
7.2** (5.6–8.0) (n = 11) |
7.0 (5.7–9.2) (n = 13) |
| ЭСТД | 6.2** (5.7–8.0) (n = 10) |
11.9 (5.1–18.1) (n = 13) |
6.2** (5.3–7.1) (n = 10) |
7.3 (4.0–12.9) (n = 11) |
| 2ЭСТД | 6.0** (3.6–7.8) (n = 12) |
5.3 (4.5–17.6) (n = 11) |
6.8** (5.3–8.8) (n = 10) |
12.1 (6.0–19.2) (n = 12) |
| 14-кратное введение ЛС | ||||
| Интактный контроль | 5.9 (4.6–6.4) (n = 11) |
5.6 (5.4–6.1) (n = 10) |
5.9 (4.6–6.4) (n = 11) |
5.6 (5.4–6.1) (n = 10) |
| Аллоксановый диабет – контроль | 15.6* (11.5–17.2) (n = 10) |
15.5* (13.9–16.9) (n = 11) |
15.6* (11.5–17.2) (n = 10) |
15.5* (13.9–16.9) (n = 11) |
| 1/2ЭСТД | 7.0** (6.0–12.8) (n = 10) |
5.7 (4.9–7.2) (n = 10) |
5.3** (4.9–6.7) (n = 10) |
7.4** (6.4–8.4) (n = 10) |
| ЭСТД | 5.7** (5.0–6.9) (n = 10) |
7.6 (5.2–8.0) (n = 10) |
6.8** (5.2–9.1) (n = 10) |
6.7** (6.3–7.0) (n = 10) |
| 2ЭСТД | 5.8** (4.5–6.6) (n = 10) |
7.1 (6.3–8.2) (n = 10) |
6.5** (5.9–6.8) (n = 10) |
6.7** (5.7–7.5) (n = 11) |
Аналогичная, но статистически не значимая тенденция прослеживалась в отношении кортикального содержания серотонина. Полученные данные свидетельствуют о том, что нарастание активности МАО в коре головного мозга крыс с экспериментальным СД приводит к развитию дефицита дофамина, одновременно являющегося субстратом МАО-А и МАО-Б [1, 2]. Развивающаяся на таком фоне недостаточность дофаминергической нейропередачи в структурах переднего мозга считается узловым нейрохимическим механизмом патогенеза депрессии [36], проявления которой наблюдаются как у людей, страдающих СД [13, 37], так и у лабораторных грызунов с аллоксановым диабетом [14]. Следует заметить также, что наибольший прирост МАО-активности в неокортексе крыс с аллоксановым диабетом по срокам своего развития совпадал с максимальным накоплением липофусцина в первичной соматосенсорной коре и значительным уменьшением числа нейронов в ее поверхностных и глубоких слоях [19]. Это хорошо согласуется с представлениями о роли МАО в эскалации оксидативного поражения головного мозга при СД [6] и старении [10].
Производные 3-оксипиридина и янтарной кислоты оказали существенное влияние на показатели МАО-активности и содержание моноаминов в коре головного мозга крыс с аллоксановым диабетом (табл. 1, 2). Все изученные препараты снижали активность кортикальной МАО-А на 38–66% по сравнению с группой “аллоксановый диабет – контроль” уже при однократном введении до начала заместительной инсулинотерапии. Изолированное производное 3-оксипиридина (эмоксипин) вызывало такой эффект при использовании в относительно низких дозах (1/2ЭСТД и ЭСТД). Изолированное производное янтарной кислоты (реамберин) снижало активность кортикальной МАО-А в минимальной (1/2ЭСТД) и максимальной (2ЭСТД) дозах. Мексидол, одновременно являющийся производным 3-оксипиридина и янтарной кислоты, подавлял кортикальную МАО-А во всех изученных дозах. Препарат сравнения (α‑ЛК), также как мексидол, снижал активность МАО-А коры мозга во всем диапазоне применяемых доз. Ни одно из изученных ЛС при однократном введении не повлияло на активность МАО-Б в неокортексе крыс с аллоксановым диабетом.
Направленность изменений кортикального уровня моноаминов у крыс с аллоксановым диабетом при однократном введении изученных препаратов не соответствовала их влиянию на МАО-активность коры мозга (табл. 2). Как видно, невзирая на существенное снижение активности МАО-А под действием всех исследуемых ЛС (табл. 1), разовое введение реамберина (в дозах 1/2ЭСТД и 2ЭСТД) и α-ЛК (в дозах 1/2ЭСТД и ЭСТД) вызвало парадоксальное уменьшение кортикального содержания серотонина на 42–50% по сравнению с группой “аллоксановый диабет – контроль” (табл. 2). Кроме того, однократное применение этих же препаратов в минимальных дозах на 36–43% снизило содержание дофамина в коре мозга крыс с экспериментальным СД. Не исключено, что уменьшение содержания серотонина и дофамина в коре мозга больных животных под действием реамберина и α-ЛК связано с вероятным моноамин-либераторным действием этих ЛС и/или их способностью подавлять обратный нейрональный захват моноаминов из синаптической щели. Правомерность этого предположения иллюстрируется сопоставимостью антидепрессивной активности реамберина и α-ЛК [20] с соответствующим действием амитриптилина, ингибирующего обратный захват серотонина и норадреналина [18].
Семидневное введение изучаемых препаратов на фоне заместительной инсулинотерапии не оказало никакого влияния на кортикальную активность МАО-А при экспериментальном СД, но в большинстве случаев привело к увеличению активности МАО-Б на 9–37% по сравнению с группой “аллоксановый диабет – контроль” (табл. 1). Это касается производных 3-оксипиридина (эмоксипина и мексидола), увеличивавших кортикальную активность МАО-Б у больных животных при введении во всех использованных дозах. Такой же эффект оказывал препарат сравнения (α-ЛК) в минимальной дозе. Реамберин при введении в средней дозе вызывал аналогичную тенденцию, которая не достигала уровня статистической значимости. Полученные результаты могут быть связаны с индуцируемой инсулином продукцией H2O2 [38], которая увеличивает церебральную активность МАО-Б, не влияя на активность МАО-А или снижая ее [39]. Не исключено, что инсулинпотенцирующее действие изученных ЛС [15] на фоне 7-и дневного введения инсулина способствует повышению Nox4-зависимой продукции перекиси водорода, оптимальное накопление которой необходимо для редокс-амплификации эффектов данного гормона [6]. Вполне возможно, что слабо выраженный прирост кортикальной активности МАО-Б под действием изученных препаратов вносит определенный вклад в развитие их нейропротекторного эффекта при аллоксановом диабете [19, 21]. Такая возможность тоже связана с инсулиномиметическим действием H2O2, в условиях, когда этот субстрат-независимый продукт МАО-реакции накапливается в диапазоне относительно низких (физиологических) концентраций [38]. Вместе с тем необходимо подчеркнуть, что супероптимальная продукция H2O2 в результате чрезмерного прироста МАО-активности является важным фактором развития ОС, инсулинорезистентности и нейронального апоптоза [6, 10, 38].
Введение изученных ЛС на протяжении 7 дней практически не оказало влияния на содержание моноаминов в коре головного мозга крыс с аллоксановым диабетом. Единственное исключение составила α-ЛК, недельное применение которой в средней дозе привело к нарастанию кортикального уровня серотонина в 1.5 раза по сравнению с группой “аллоксановый диабет – контроль” (табл. 2). Данный сдвиг не зависел от показателей корковой МАО-активности, которая не менялась в результате семидневного введения α-ЛК в дозе ЭСТД (табл. 1).
Двухнедельный курс введения изученных препаратов на фоне заместительной инсулинотерапии вызвал разнонаправленные изменения активности МАО-А и МАО-Б в коре мозга животных с экспериментальным СД (табл. 1). Все исследованные ЛС снижали активность кортикальной МАО-А в 1.46–5.83 раза в сравнении с группой “аллоксановый диабет – контроль”. Сукцинат-содержащие ЛС (реамберин и мексидол) оказывали такое действие во всем диапазоне применяемых доз, изолированное производное 3-оксипиридина (эмоксипин) – в минимальной и максимальной дозах, а препарат сравнения (α-ЛК) – в относительно высоких дозах (ЭСТД и 2ЭСТД). Одновременно, все препараты кроме эмоксипина увеличивали кортикальную активность МАО-Б на 31–48% по сравнению с группой “аллоксановый диабет – контроль”. Реамберин и α-ЛК вызывали такой эффект в максимальной дозе, а мексидол в относительно низких дозировках (1/2ЭСТД и ЭСТД). По-видимому, изменения кортикальной активности МАО-Б при 14-дневном применении изученных ЛС у крыс с аллоксановым диабетом отражают эскалацию тех сдвигов, которые начали развиваться при 7-дневном использовании соответствующих препаратов и были подробно обсуждены выше.
14-дневное применение большинства изученных ЛС при аллоксановом диабете существенно (в 1.78-5.79 раза) увеличило уровень дофамина в коре мозга больных животных (табл. 2). Это касается эмоксипина и α-ЛК, вводимых в максимальной дозе, а также мексидола, используемого в относительно низких дозах (1/2ЭСТД и ЭСТД). Реамберин не оказывал такого действия.
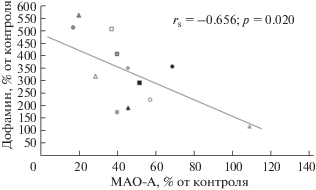
Стандартизация полученных данных по соответствующим контролям и последующий корреляционный анализ продемонстрировали обратную зависимость между влиянием препаратов на содержание дофамина и активность МАО-А в коре головного мозга крыс с аллоксановым диабетом. Как видно (рис. 1), нарастание кортикального уровня дофамина в результате двухнедельного введения изученных ЛС в значительной степени обусловлено их ингибирующим влиянием на МАО-А. При этом ни один из исследуемых препаратов не повлиял на кортикальный уровень серотонина (табл. 2), являющегося наиболее специфичным субстратом МАО-А.
Рис. 1.
Зависимость между влиянием изученных препаратов на содержание дофамина и активность МАО-А в коре головного мозга крыс с аллоксановым диабетом. Примечание: 1) треугольником обозначены 1/2ЭСТД, кругом – ЭСТД, квадратом – 2ЭСТД; 2) серая заливка - α-липоевая кислота; черная заливка – реамберин; серая заливка с очерченными контурами – мексидол; прозрачная заливка с очерченными контурами – эмоксипин.
Изменения МАО-активности и содержания моноаминов в коре мозга под действием изученных ЛС в целом не зависели от их влияния на концентрацию кортикостерона в сыворотке крови крыс с аллоксановым диабетом. Эмоксипин и реамберин, обладающие выраженным МАО-модулирующим действием (табл. 1), не оказали значимого влияния на концентрацию циркулирующего кортикостерона. Лишь мексидол и препарат сравнения (α-ЛК) вызвали значимые изменения уровня этого ГКГ (табл. 3). В обоих случаях данный эффект зависел от дозы и кратности введения ЛС. Однократное применение мексидола и α-ЛК в минимальной дозе снижало концентрацию циркулирующего кортикостерона на 25–31% в сравнении с группой “аллоксановый диабет– контроль”. Аналогичный эффект подобной выраженности наблюдался при 7-кратном введении относительно высоких доз α-ЛК (ЭСТД и 2ЭСТД). Двухнедельное применение α-ЛК в максимальной дозе приводило к практически двукратному снижению уровня циркулирующего кортикостерона. 14-дневное применение мексидола и α-ЛК в дозе ЭСТД вызвало противоположный эффект, проявившийся нарастанием концентрации кортикостерона на 19–45% по сравнению с группой “аллоксановый диабет – контроль”. Полученные данные укладываются в рамки представлений об антистрессорном действии мексидола [40], которое согласуется с его умеренно выраженной седативной активностью [41, 42]. α-ЛК оказывает еще более выраженное седативное действие [41], позволяющее предположить наличие антистрессорной активности у данного ЛС. Не исключено, что уменьшение концентрации кортикостерона при соответствующих режимах введения мексидола и α-ЛК обусловлено нарастанием порога стресс-реакции и снижением функциональной активности гипоталамо-гипофизарно-адренокортикальной оси (ГГАО) в результате развития седативного эффекта. Вместе с тем, мексидол и α-ЛК наряду с седативным действием обладают антиглюкокортикоидной активностью [16], которая при длительном введении этих препаратов может вызвать нарушение регуляции ГГАО по механизму “длинной петли отрицательной обратной связи” и привести к нарастанию уровня циркулирующих ГКГ.
Подобный механизм может лежать в основе прироста концентрации кортикостерона в крови крыс с экспериментальным СД при двухнедельном введении мексидола и α-ЛК в дозе ЭСТД.
Влияние изученных ЛС на активность кортикальных МАО и содержание моноаминов в коре мозга не зависело от гликемических эффектов соответствующих препаратов при экспериментальном СД. Большинство изученных препаратов значимо корригировали гипергликемию у крыс с аллоксановым диабетом (табл. 4). Чаще всего данный эффект развивался при курсовом введении изучаемых ЛС во всех использованных дозах на фоне заместительной инсулинотерапии. Это касается производных 3-оксипиридина (эмоксипина и мексидола), которые снижали гипергликемию в 1.99–2.94 раза при 7- и 14-дневном введении, а также α-ЛК, которая уменьшала гипергликемию в 2.1–2.3 раза только при двухнедельном применении. Исключение составил реамберин, применение которого вызывало тенденции аналогичной направленности, которые не достигали уровня статистической значимости.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Совокупность полученных данных свидетельствует о том, что развитие аллоксанового диабета у крыс сопровождается выраженным нарастанием МАО-активности в коре их головного мозга на протяжении всего эксперимента. В первую очередь это связано с динамикой кортикальной активности МАО-А, которая возрастала на 68% на начальном этапе эксперимента и, после транзиторной нормализации повторно увеличивалась более чем в 4 раза на его завершающем этапе. Менее выраженный и относительное поздний сдвиг аналогичной направленности был отмечен для кортикальной МАО-Б, активность которой увеличивалась на 56% только к концу эксперимента. Одновременное и наиболее выраженное нарастание активности МАО-А и МАО-Б в конце периода наблюдения сопровождалось более чем трехкратным снижением содержания дофамина в неокортексе крыс с аллоксановым диабетом. Оригинальные отечественные производные 3-оксипиридина и янтарной кислоты (эмоксипин, реамберин и мексидол) и α-ЛК, используемая в качестве препарата сравнения, эффективно корригировали прирост кортикальной активности МАО-А у крыс с аллоксановым диабетом. Введение этих препаратов в дозах, эквивалентных терапевтическому диапазону для человека, вызывало снижение кортикальной активности МАО-А на 38–66% при однократном и на 32–83% при 14-кратном применении. На заключительном сроке наблюдения изученные лекарственные средства корригировали дефицит дофамина в неокортексе животных с аллоксановым диабетом, увеличивая уровень этого моноамина в достоверном соответствии со снижением корковой активности МАО-А. В отношении коррекции кортикального дефицита дофамина при аллоксановом диабете наиболее эффективными оказались производные 3-оксипиридина (эмоксипин и мексидол). По обсуждаемому действию эти ЛС не уступали препарату сравнения (α-ЛК) и превосходили изолированное производное янтарной кислоты (реамберин). Важно добавить, что на начальном этапе исследования эмоксипин и мексидол, в отличие от реамберина и α-ЛК, не вызывали транзиторного снижения уровня дофамина и серотонина в коре головного мозга крыс с аллоксановым диабетом. Обсуждаемая динамика коркового содержания моноаминов не зависела от влияния изучаемых препаратов на активность МАО-Б, которая возрастала на 9–37% при 7-кратном введении эмоксипина, мексидола и α-ЛК и на 31–48% при 14-кратном применении реамберина, мексидола и α-ЛК. Изменения МАО-активности и содержания моноаминов в коре мозга под действием изученных препаратов не зависели от их влияния на выраженность гипергликемии и содержание кортикостерона в крови крыс с аллоксановым диабетом. Полученные результаты хорошо согласуются с известным церебропротекторным и антидепрессивным действием изученных препаратов [19, 20], а также соответствуют данным об их МАО-модулирующей активности in vitro [17].
ЗАКЛЮЧЕНИЕ
Курсовое введение производных 3-оксипиридина (эмоксипина и мексидола) в дозах, эквивалентных терапевтическому диапазону для человека, предотвращает нарастание активности МАО-А и параллельно развивающийся дефицит дофамина в коре головного мозга в динамике первых двух недель аллоксанового диабета у крыс. По выраженности этих эффектов эмоксипин и мексидол не уступают α-ЛК. Изолированное производное янтарной кислоты (реамберин) уменьшает активность МАО-А в неокортексе животных с аллоксановым диабетом, но вызывает транзиторное снижение содержания серотонина и дофамина на раннем этапе исследования и не корригирует дефицит дофамина к концу периода наблюдения. Курсовое введение всех изученных препаратов вызывает увеличение кортикальной активности МАО-Б, не связанной с параллельными сдвигами уровня моноаминов в коре мозга крыс с аллоксановым диабетом. Изменения активности кортикальных МАО и содержания моноаминов в коре мозга под действием изученных препаратов не зависят от их влияния на уровень циркулирующего кортикостерона и гипергликемию при аллоксановом диабете у крыс.
ИСТОЧНИК ФИНАНСИРОВАНИЯ
Работа выполнена в рамках государственного задания на осуществление научных исследований и разработок “Фармакофизиология и биохимическая фармакология производных 3-оксипиридина и янтарной кислоты” (№ государственной регистрации: АААА-А18-118021890008-4. Дата регистрации: 18.02.2018 г.).
Список литературы
Duncan J.W., Johnson S., Ou X.M. // Drug Disc. Ther. 2012. V. 6. № 3. P. 112–122.
Bortolato M., Chen K., Shih J.C. // Adv. Drug Del. Rev. 2008. V. 60. № 13–14. P. 1527–1533.
Горкин В.З. // Аминоксидазы и их значение в медицине. М.: Медицина, 1981. 335 с.
Harro J., Oreland L. // Progr. Neuro-Psychopharmacol. Biol. Psychiatry. 2016. V. 69. P. 101–111.
Youdim M.B.H., Edmondson D., Tipton K.F. // Nat. Rev. Neurosci. 2006. V. 7. № 4. P. 295.
Goldstein B.J., Mahadev K., Wu X. // Diabetes. 2005. V. 54. №2. P.311-321.
Kleinridders A., Cai W., Cappellucci L., Ghazarian A., Collins W.R., Vienberg S.G., Pothos E.N., Kahn C.R. // Proc. Natl. Acad. Sci. USA. 2015. V. 12. № 11. P. 3463–3468.
Chen K., Ou X.M., Wu J.B., Shih J.C. // Mol. Pharmacol. 2011. V. 79. № 2. P. 308–317.
Duncan J.W., Zhang X., Wang N., Johnson S., Harris S., Udemgba C., Ou X.M., Youdim M.B.H., Stockmeier C.A., Wang J.M. // Neuropharmacology. 2016. V. 105. P. 329–340.
Волчегорский И.А., Шемяков С.Е., Турыгин В.В., Малиновская Н.В. // Бюл. эксп. биол. мед. 2001. Т. 132. № 8. С. 174–177.
Kodl C.T., Seaquist E.R. // Endocr. Rev. 2008. V. 29. № 4. P. 494–511.
Волчегорский И.А., Москвичева М.Г., Чащина Е.Н. // Журн. неврол. и психиатр. им. C.C. Корсакова. 2005. Т. 105. № 2. С. 41–45.
Волчегорский И.А., Местер Н.В. // Клин. мед. 2007. Т. 85. № 2. С. 40–45.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М., Файзуллин Р.М., Малкин М.П., Пряхина К.Е., Калугина А.В. // Эксп. клин. фармакол. 2014. Т. 77. № 4. С. 14–20.
Волчегорский И.А., Рассохина Л.М., Мирошниченко И.Ю., Местер К.М., Новоселов П.Н., Астахова Т.В. // Бюл. эксп. биол. мед. 2010. Т. 150. № 9. С. 295–301.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М., Файзуллин Р.М., Пряхина К.Е. // Рос. физиол. журн. им. И.М. Сеченова. 2016. Т. 102. № 11. С. 1312–1322.
Волчегорский И.А., Синицкий А.И., Мирошниченко И.Ю., Рассохина Л.М. // Хим.-фарм. журн. 2018. Т. 52. № 1. С. 3–7.
Nestler E.J., Hyman S.E., Malenka R.C. // Molecular Neuropharmacology: a Foundation for Clinical Neuroscience. N.Y.: McGraw-Hill, 2001. 539 p.
Волчегорский И.А., Рассохина Л.М., Мирошниченко И.Ю. // Журн. неврол. психиатр. им. C.C. Корсакова. 2013. Т. 113. № 6. С. 50–61.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М., Малкин М.П., Файзуллин Р.М., Пряхина К.Е., Калугина А.В. // Журн. неврол. психиатр. им. C.C. Корсакова. 2015. Т. 115. № 2. С. 48–52.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М., Файзуллин Р.М. // Журн. неврол. психиатр. им. C.C. Корсакова. 2016. Т. 116. № 6. С. 53–59.
Миронов А.Н., Бунятян Н.Д., Васильев А.Н., Верстакова О.Л., Журавлева М.В., Лепахин В.К., Коробов Н.В., Меркулов В.А., Орехов С.Н., Сакаева И.В., Утешев Д.Б., Яворский А.Н. // Руководство по проведению доклинических исследований лекарственных средств. М.: Гриф и К, 2012. 944 с.
Lenzen S. // Diabetologia. 2008. V. 51. P. 216–226.
Волчегорский И.А., Долгушин И.И., Колесников О.Л., Цейликман В.Э. // Экспериментальное моделирование и лабораторная оценка адаптационных реакций организма. Челябинск: Издательство ЧГПУ, 2000. 167 с.
Рассохина Л.М. Автореф. дисc. … докт. мед. наук. Челябинск: ЮУГМУ, 2014. 48 с.
De Moor P., Osinski P., Deckx R., Steeno O. // Clinica Chimica Acta. 1962. V. 7. № 4. P. 475–480.
Балашов Ю.Г. // Физиол. журн. СССР. 1990. № 12. С. 280–283.
Glowinski J., Iversen L.L. // J. Neurochem. 1966. V. 13. P. 655–669.
Huang G., Zhu F., Chen Y., Chen S., Liu Z., Li X., Gan L., Zhang L., Yu Y. // Anal. Biochem. 2016. V. 512. P. 18–25.
Камышников В.С. // Справочник по клинико-биохимическим исследованиям и лабораторной диагностике. М.: МЕДпресс-информ, 2009. 896 с.
Колб В.Г., Камышников В.С. // Справочник по клинической химии. Минск: Беларусь, 1982. 366 с.
Матлина Э.Ш., Меньшиков В.В. // Клиническая биохимия катехоламинов. М.: Медицина, 1967. 304 с.
Лобода Е.Б., Макаров Ю.П. // Лаб. дело. 1974. № 4. С. 219–221.
Волчегорский И.А., Рассохина Л.М., Мирошниченко И.Ю. // Рос. физиол. журн. им. И.М. Сеченова. 2013. Т. 99. № 4. С. 491–500.
Волчегорский И.А., Колесников О.Л., Цейликман В.Э., Колесникова А.А., Владимиров А.Ю. // Пробл. эндокринол. 1997. Т. 43. № 2. С. 38–41.
Lavergne F., Jay T.M. // Front. Neurosci. 2010. V. 4. P. 192.
Волчегорский И.А., Москвичева М.Г., Чащина Е.Н. // Клин. мед. 2004. Т. 82. № 11. С. 31–35.
Iwakami S., Misu H., Takeda T., Sugimori M., Matsugo S., Kaneko S., Takamura T. // PLoS One. 2011. V. 6. № 11. P. e27401.
Konradi C., Riederer P., Youdim M.B.H. // J. Neur. Trans. Suppl. 1986. V. 22. P. 61–73.
Воронина Т.А. // Фарматека. 2009. № 6. С. 35–38.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М. // Рос. физиол. журн. им. И.М. Сеченова. 2017. Т. 103. № 7. С. 755–767.
Волчегорский И.А., Мирошниченко И.Ю., Рассохина Л.М., Файзуллин Р.М. // Эксп. клин. фармакол. 2013. Т. 76. № 7. С. 6–10.
Дополнительные материалы отсутствуют.