

Нейрохимия, 2020, T. 37, № 3, стр. 271-279
Экзосомальный ангиогенин как возможный биомаркер при боковом амиотрофическом склерозе
М. В. Иванова 1, Е. О. Чеканова 1, Б. В. Белугин 2, И. В. Должикова 2, И. Л. Тутыхина 2, М. Н. Захарова 1
1 ФГБНУ “Научный центр неврологии”
Москва, Россия
2 ФГБУ “НИЦЭМ им. Н.Ф. Гамалеи” Минздрава России
Москва, Россия
Поступила в редакцию 06.01.2020
После доработки 08.01.2020
Принята к публикации 09.01.2020
Аннотация
Экзосомы представляют собой внеклеточные везикулы, обеспечивающие один из механизмов межклеточной коммуникации. Считается, что экзосомы могут быть носителями биологически активных молекул, связанных с прогрессированием и распространением патологического процесса при боковом амиотрофическом склерозе (БАС) – фатальном нейродегенеративном заболевании, при котором происходит гибель двигательных нейронов. Ангиогенин известен своим возможным значением в патогенезе (БАС). В работе проведен анализ содержания ангиогенина в плазме крови, ЦСЖ и полученных из них экзосомальных фракциях при БАС. В исследование были включены 30 пациентов с БАС и 26 соответсвующих им по полу и возрасту здоровых добровольцев. Относительное количество ангиогенина в исходной плазме и ЦСЖ оказалось значительно более высоким, чем в их экзосомальных фракциях (р < 0.05). При сопоставлении содержания ангиогенина как в плазме, так и в экзосомальной фракции, у пациентов с БАС были выявлены более низкие уровни, по сравнению со здоровыми добровольцами (р < 0.05), что может указывать на протективную роль ангиогенина. Определялась тенденция к выявлению больших концентраций ангиогенина в плазме и ее экзосомальной фракции у пациентов с лучшим функциональным состоянием. Таким образом, снижение уровня ангиогенина в плазме и в экзосомальной фракции плазмы может быть маркером прогрессирования заболевания, однако эти данные нуждаются в подтверждении в более крупной выборке.
ВВЕДЕНИЕ
Ангиогенин впервые был выделен в 1985 г. из культуральной среды линии клеток человеческой аденокарциномы кишечника НТ-29, где был обнаружен в связи со своей ангиогенной активностью. Зрелый ангиогенин представляет собой основный одноцепочечный белок, состоящий из 123 аминокислот и имеющий молекулярную массу около 14.4 кДа [1]. С момента окрытия ангиогенина были выявлены и другие его функции, такие как участие в нейрогенезе [2], нейропротекция [3], иммунорегуляция [4]. Ангиогенин относится к суперсемейству рибонуклеаз [1], способен к транслокации в ядро и активации транскрипции рибосомальной РНК [5], может стимулировать деградацию базальной мембраны [6], активировать сигнальную трансдукцию [5, 7], что связано с секрецией ряда биологически активных факторов. Показана также роль ангиогенина в патогенезе некоторых заболеваний, среди которых опухоли [8], болезнь Паркинсона [9], боковой амиотрофический склероз (БАС).
БАС представляет собой фатальное прогрессирующее нейродегенеративное заболевание с избирательным поражением двигательных нейронов. Заболевание имеет высокую гетерогенность в отношении темпов прогрессирования, но в среднем приводящее к гибели в течение 3–5 лет [10]. Патогенетические механизмы, лежащие в основе БАС, несмотря на активное изучение, во многом не ясны. К ним относят мутации, нарушение процессинга РНК, образование агрегатов патологических белков, дисфункцию митохондрий, окислительный стресс, воспаление и др., однако значение каждого из них до конца не определено [10]. По этой причине к настоящему моменту не существует эффективных методов лечения БАС, не разработаны биомаркеры для выявления предрасположенности, ранней диагностики и прогнозирования течения заболевания.
О возможной роли ангиогенина при БАС стало известно с 2004 г., когда у пациентов впервые были выявлены мутации в гене ANG, приводящие к ингибированию функции ангиогенина [11]. Мутации в гене ANG описаны как при семейных, так и при спорадических формах БАС. В результате мутаций описано снижение рибонуклеазной активности, способности ангиогенина к его транслокации в ядро, нарушение формирования стресс-гранул и снижение ангиогенной активности [12].
Экзосомы представляют собой класс внеклеточных везикул, по данным разных исследователей имеющих размеры 30–200 нм [13, 14]. Экзосомы имеют характерные особенности белкового, липидного и нуклеотидного состава, на основании которого возможно их распознавание. Среди молекул-маркеров экзосом описаны флотиллины, тетраспанины (CD63, CD81), белки теплового шока, некоторые липиды и др. [15]. Основной функцией экзосом является обеспечение коммуникации различных типов клеток, часто в отдаленных участках организма за счет транспорта биологически активных молекул к клеткам-мишеням. Показана роль экзосом в развитии патологического процесса при многих заболеваниях, в том числе болезнях центральной нервной системы, среди которых, в первую очередь, онкологические и нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона и БАС. При БАС в экспериментах на клеточных линиях, животных моделях было показано, что экзосомы могут участвовать в распространении патологических белков, способствуя развитию их нейротоксического эффекта [16]. В то же время выявлялись свидетельства возможной протективной роли экзосом при БАС, связанной с очищением клеток от белков с нарушенной упаковкой и переносом протективных факторов [17].
Предположительно существенная роль экзосом в патогенезе БАС позволяет их рассматривать в качестве возможных носителей биомаркеров прогрессирования и распространения патологического процесса при БАС. В настоящей работе представлено исследование ангиогенина в биологических жидкостях пациентов с БАС (плазма и ЦСЖ), и полученных из них экзосомальных фракциях, проанализированы взаимосвязи определяемых уровней ангиогенина с клиническими характеристиками заболевания.
МЕТОДЫ ИССЛЕДОВАНИЯ
Пациенты. В исследование были включены 30 пациентов с БАС с вероятным или достоверным диагнозом согласно критериям El Escorial [18]. среди которых 6 пациентов с семейными формами и/или с мутациями в генах, ассоциированных с БАС, и 24 пациента со спорадическими формами заболевания. Среди вошедших в группу исследования пациентов с БАС было 16 мужчин и 14 женщин. Медиана возраста составила 55.5 лет [48; 65]. Группу здоровых добровольцев составили 26 практически здоровых человек, соответствующих по полу и возрасту группе пациентов: 12 мужчин и 14 женщин, медиана возраста 56 лет [45; 60 ] .
Забор и хранение биологического материала. У всех пациентов и здоровых добровольцев производился забор крови из кубитальной вены в пробирки с цитратом, у 8 пациентов с БАС дополнительно производился забор цереброспинальной жидкости (ЦСЖ). Кровь центрифугировали при темпепатуре 4°С в течение 10 мин на скорости 3000 об./мин, отбирали плазму и центрифугировали последнюю в течение 15 мин при ускорении 3000 g. ЦСЖ центрифугировали при температуре 4°С и ускорении 13000 g в течение 30 мин. Подготовленный материал хранили в виде аликвот при температуре –80°С.
Выделение экзосом. Выделение экзосом проводили с помощью набора реактивов ExoEasy Maxi Kit (Qiagen). Данный набор используется для быстрой очистки и выделения экстраклеточных везикул из плазмы, из сыворотки и среды из-под клеточных культур. Принцип работы набора основан на аффинном связывании сорбентом колонки мембран внеклеточных везикул, находящихся в материале. Выделение экзосом проводили согласно инструкции производителя. Образец плазмы объемом 4 мл размораживали и смешивали с буфером для нанесения на колонку в равных объемах. Держали при комнатной температуре 5 мин. Затем наносили на колонку, центрифугировали при 500 g 2 мин, промывали сорбент буфером для промывки объемом 10 мл центрифугированием при скорости 5000 g 10 мин. На этапе элюирования колонку с сорбентом переносили с чистую пробирку, наносили на сорбент элюирующий буфер объемом 500 мкл инкубировали при комнатной температуре 1 мин и центрифугировали при 500 g 10 мин. Собранный элюат повторно наносили на колонку и центрифугировали при 5000 g 10 мин. Препараты экзосом сохраняли в виде аликвот по 100 мкл при –80°С.
Электронная микроскопия. Аликвоту препарата экзосом фиксировали в равном объеме буфера Ито–Карновского – смеси тринитрофенола и альдегида – в течение ночи при температуре 4°С. Затем наносили материал на никелевые сетки (SPI, 30 Mesh). На каплю (20–30 мкл) фиксированных экзосом устанавливали сетку и инкубировали в течение часа при комнатной температуре. Затем сетку снимали и удаляли избыток жидкости фильтровальной бумагой. Негативное контрастирование проводили раствором 2% уранил-ацетата в течение 1 мин, избыток раствора удаляли и сетки высушивали. Контрастирование уранилацетатом позволяет окрасить белки, в результате чего окружение экзосомы или везикулы, связанной с белками, приобретает большую электронную плотность. Исследование препарата осуществляли на просвечивающем электронном микроскопе Jeol JEM-100SX при напряжении 80 кВ.
Детекция маркерных белков экзосом. Для подтверждения наличия экзосом в материале, полученном с помощью набора реактивов ExoEasy Maxi Kit (Qiagen), было проведено исследование таких наиболее распространенных маркеров экзосом, как CD63, CD81 и Flotillin-1. Определение маркеров экзосом проводилось методом вестерн-блот. В качестве положительного контроля были использованы стандарты экзосом Exosome Standards (Human Plasma) (NBP2-49838, Novus Biologicals), Purified Exosomes from Human CSF (Healthy Donors) (EXOP-530A-1, SBI), рекомбинантные белки-маркеры экзосом CD63 (11271-H08H, Sino Biological Inc.), CD81 (14244-H07H, Sino Biological Inc.), Flotillin-1 (H00010211-Q01-10ug Novus Biologicals). Электрофорез образцов проводили на гелях Mini-PROTEAN TGX Stain-Free Precast Gels 4–20% при напряжении 250 В в течение 15 мин. Проводился полусухой перенос белков на нитроцеллюлозную мембрану в системе для быстрого блоттинга Trans Turbo Blot (25 В, 1.3А, 7 мин (режим “mixeo”)). После переноса белков производили блокировку мембраны 4% раствором обезжиренного молока на фосфатно-солевом буфере (PBS) с добавлением детергента Tween 20 (0.1%) в течение часа. Затем проводили инкубацию с первичными антителами (mouse anti-Human CD63 sc-5275, mouse anti-Human CD81 sc-166029, mouse anti-Human Flotillin-1 sc-74566, SANTA CRUZ BIOTECHNOLOGY, INC.) в разведении 1 : 100 в течение 2 ч. После промывки осуществляли инкубацию со вторичными антителами, специфичными к антителам мыши (ECL Anti-mouse IgG, peroxidase-linked whole antibody (from sheep) NXA931, Amersham) в разведении 1 : 5000. Затем проводили детекцию с использованием набора для хемилюминесцентной детекции Clarity™ Western ECL Substrate (1705060, Bio-Rad). Чтобы исключить неспецифическое связывание вторичных антител с образцами и человеческими иммуноглобулинами, адсорбированными на мембране, был проведен вестерн-блот без инкубации с первичными антителами.
Определение концентрации ангиогенина. Определение ангиогенина проводили с помощью коммерческого набора Human Angiogenin Immunoassay (DAN00, R&D Systems) для количественного ИФА согласно инструкции производителя. В результате проведения вычислений исходные данные оптической плотности преобразовывались в концентрации ангиогенина в проанализированных в образцах, выраженные в пг/мл. Для сопоставления полученных данных проводили расчет количества ангиогенина по отношению к общему количеству белка в образце, результат выражали в пг/мг. Измерение концентрации белка в образцах производили с помощью прибора Nanodrop 2000 (Thermo Fisher Scientific).
Статистическая обработка данных. Статистическая обработка данных проводилась с помощью программы GraphPad Prism 8 (GraphPad Software, США). Данные по клиническим характеристикам пациентов и содержанию ангиогенина представлены в виде медиан [Ме] и межквартильного размаха [Q1; Q3], на графиках отображены медианы и 95% доверительный интервал. Нормальность распределения данных внутри одной группы определяли в тесте на нормальность Д’Агостина–Пирсона. Попарные сравнения между группами исследования проводили с помощью непараметрического критерия Манна–Уитни. Анализ корреляций уровней ангиогенина в различном материале с клиническими параметрами заболевания проводился с помощью коэффициента ранговой корреляции Спирмена. Статистически значимыми считались различия при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Клиническая характеристика пациентов с БАС. У 11 (36.7%) пациентов была бульбарная форма заболевания, у 9 (30%) – шейно-грудная, у 10 (33.3%) – пояснично-крестцовая. У основной части пациентов имелись клинические симптомы поражения двигательных нейронов на 2 или 3 анатомических уровнях (стадия 2 и 3): 11 и 13, соответственно, у 5 пациентов дополнительно к 3 уровням поражения имели место дыхательные нарушения, корректирующиеся проведением вентиляции легких (стадия 4В), у 1 пациента – выраженные нарушения глотания, требующие установки гастростомы (стадия 4А). Медиана длительности заболевания составила 24 мес. Медиана значений по шкале ALS-FRS-R, характеризующей функциональное состояние при БАС и включающей оценку речи, глотания, функции рук, ходьбы, дыхательной функции, равнялась 39 (максимальное значение, соответствующее норме, – 48 баллов). Медиана значений жизненной емкости легких (ЖЕЛ) составила 80%, индекса массы тела (ИМТ) – 26.2. В группу исследования вошли преимущественно пациенты с относительно быстрыми (снижение балла по шкале ALS-FRS-R на 8 и более единиц в год) или медленными темпами прогрессирования заболевания (снижение балла по шкале ALS-FRS-R на 3 и менее в год) примерно в равном соотношении. Более подробная характеристика групп исследования представлена в табл. 1.
Таблица 1.
Характеристика групп исследования
| Параметр | Пациенты с БАС (n = 40) | Здоровые добровольцы (n = 26) | р |
|---|---|---|---|
| Пол (М/Ж) | 16/14 | 12/14 | 0.593 |
| Возраст, лет (Me [Q1; Q3]) | 55.5 [48; 65] | 56 [45; 60 ] | 0.24 |
| Длительность заболевания, мес. (Me [Q1; Q3]) | 24 [13; 48 ] | – | |
| Форма заболевания, n (%): | – | ||
| 1. бульбарная | 13 (32.5%) | ||
| 2. шейно-грудная | 13 (32.5%) | ||
| 3. пояснично-крестцовая | 14 (35%) | ||
| Стадия заболевания, n (%): | – | ||
| 1 | 2 (5%) | ||
| 2 | 16 (40%) | ||
| 3 | 17 (43.5%) | ||
| 4A | 1 (2.5%) | ||
| 4B | 4 (10%) | ||
| Балл по шкале ALS-FRS-R (Me [Q1; Q3]) | 39 [35; 43 ] | – | |
| ЖЕЛ, % (Me [Q1; Q3]) | 80 [62.25; 109] | – | |
| ИМТ (Me [Q1; Q3]) | 26.2 [23.97; 29.66] | – | |
| Мутации, n: | – | ||
| 1. SOD1 G93A | 3 | ||
| 2. С9orf72, экспансия GGGGCC-повторов | 3 | ||
| 3. TARDBP – неизвестная мутация | 1 | ||
| 4. Семейный случай, мутация не выявлена | 2 |
Характеристика экзосомальной фракции
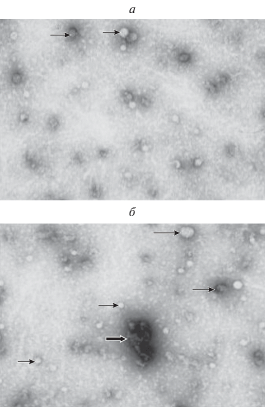
Трансмиссионная электронная микроскопия (ЭМ) была проведена для исследования экзосомальной фракции плазмы шести пациентов (3 здоровых добровольцев и 3 пациентов с диагнозом БАС), результаты оказались относительно однородными и представлены на рис. 1. Мембранные везикулы обладают сферической морфологией, характерной для экзосом. С помощью програмного обеспечения ImageJ (NIH) были проведены измерения размеров экзосом, представленных на снимках. Было выявлено, что диаметр везикул варьирует от 16 до 94 нм, средний диаметр составил 31.35 ± 11.45 нм. Существенных различий между препаратами экзосом, выделенных из плазмы здоровых добровольцев и больных с БАС, не определялось. В препаратах экзосом встречаются крупные везикулы – от 50 до 100 нм, также характерным является наличие в препаратах агрегатов экзосом и белков плазмы (рис. 1б).
Рис. 1.
ЭМ экзосомальной фракции здоровых добровольцев и пациентов с БАС (а и б соответственно). Простыми стрелками указаны типичные для данного препарата везикулы, фигурными – агрегаты везикул и белков плазмы.
В образцах экзосом, выделенных из плазмы крови как пациентов с БАС, так и здоровых добровольцев, а также ЦСЖ, с помощью набора ExoEasy Maxi Kit были выявлены все три маркера экзосом (CD63, CD81, Flotillin-1). Таким образом, наличие экзосом в исследованном материале было подтверждено с помощью двух методов.
Определение ангиогенина в плазме, ЦСЖ и экзосомальных фракциях. Содержание ангиогенина в плазме у пациентов с БАС равнялось 6368.76 [5302.41; 7800.83] пг/мг белка, в экзосомальной фракции плазмы – 1869.22 [1235.59; 2373.60] пг/мг белка, что в 3.4 раза ниже. У здоровых добровольцев уровень ангиогенина в плазме составил 7657.30 [6592.24; 8569.09] пг/мг белка, в экзосомальной фракции плазмы – 2900.73 [2202.86; 3968.40] пг/мг белка, что в 2.6 раз ниже. Таким образом, относительное количество ангиогенина в исходной плазме оказалось значительно более высоким, чем в ее экзосомальной фракции, это значение как у пациентов, так и у здоровых добровольцев было статистически значимым (р < 0.0001) (рис. 2).
Рис. 2.
Уровень ангиогенина в плазме и ее экзосомальной фракции у пациентов с БАС и здоровых добровольцев. БАС – боковой амиотрофический склероз.
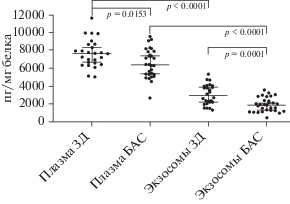
При сопоставлении содержания ангиогенина как в плазме, так и в экзосомальной фракции, у пациентов с БАС были выявлены более низкие уровни, по сравнению со здоровыми добровольцами, различия были статистически значимыми, что может указывать на протективную роль ангиогенина (рис. 2).
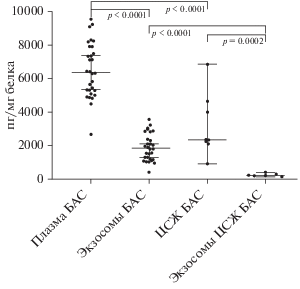
Содержание ангиогенина было оценено также в ЦСЖ пациентов с БАС, где оно составило 2365.19 [2219.35; 4175.06] пг/мг белка и ее экзосомальной фракции, где его уровень составил 223.50 [181.27; 266.21] пг/мг белка, что в 10.58 раз ниже. Это различие статистически значимо (р = 0.0002). Относительное количество ангиогенина в плазме у пациентов с БАС статистически выше, чем в ЦСЖ (р < 0.0001) (рис. 3). Тем не менее, во всем исследованном материале содержание ангиогенина было относительно высоким, что может говорить о том, что ангиогенин может переноситься как в составе внеклеточных везикул, так и вне их.
Рис. 3.
Уровень ангиогенина в плазме, ЦСЖ и экзосомальных фракциях у пациентов с БАС. ЦСЖ – цереброспинальная жидкость, БАС – боковой амиотрофический склероз.
При сопоставлении данных ИФА ангиогенина с клиническими характеристиками заболевания, такими как возраст пациента, длительность заболевания, балл по шкале ALSFRS-R, форма, стадия заболевания, жизненная емкость легких (ЖЕЛ), индекс массы тела (ИМТ), отмечалась тенденция к выявлению больших концентраций ангиогенина в плазме и ее экзосомальной фракции у пациентов с более высокой оценкой по шкале ALSFRS-R, хотя обнаруженная взаимосвязь не достигла уровня статистической значимости (р = 0.06). Таким образом, снижение уровня ангиогенина в плазме и в экзосомальной фракции плазмы может быть маркером прогрессирования заболевания, однако эта гипотеза нуждается в подтверждении в более крупной выборке. Других взаимосвязей с клиническими характеристиками заболевания обнаружено не было. Ассоциаций между уровнями ангиогенина в плазме, ЦСЖ, экзосомальных фракциях и наличием генетически обусловленной формы заболевания или какой-либо конкретной мутации не обнаружено.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Снижение уровня ангиогенина, выявленное нами у пациентов с БАС, по сравнению со здоровыми добровольцами, и более выраженное у пациентов с худшим функциональным состоянием указывает на протективное значение ангиогенина при БАС. Отличием нашего исследования от других работ, в которых анализировались уровни ангиогенина при патологии, является дополнительное измерение уровня ангиогенина в экзосомальных фракциях. Полученные нами данные указывают на снижение уровня ангиогенина при БАС во всех исследованных образцах. Также отмечена тенденция к выявлению более низких уровней ангиогенина у пациентов, находящихся в худшем функциональном состоянии. Хотя эти данные нуждаются в подтверждении в более крупной выборке пациентов, наши результаты указывают на возможное значение ангиогенина как плазмы, так и ее экзосомальной фракции как маркера прогрессирования заболевания при БАС.
Полученные нами данные согласуются с представлениями о возможной роли ангиогенина при БАС, полученными на основании результатов работ, проведенных на клеточных линиях и животных моделях. Было показано, что ангиогенин в больших количествах экспрессируется во взрослых мотонейронах и может защищать культуры мотонейронов от различных повреждающих воздействий, имеющих место при БАС, таких как эксайтотоксичность, гипоксия, стресс эндоплазматического ретикулума [19]. Рекомбинантный ангиогенин оказывал нейропротективное действие на культуры мотонейронов, а его введение мышам с мутацией SOD1G93A – лабораторной модели БАС, что приводило к значительному увеличению длительности жизни и двигательных функций у данных животных [20]. Протективный эффект ангиогенина реализуется только при воздействии на мотонейроны диких форм белка, но не его мутантных вариантов, отмечающихся у некоторых пациентов с БАС. Культуры клеток P19 поглощают как дикую форму ангиогенина, так и его варианты, связанные с БАС, однако ангиогенин дикого типа способствует дифференцировке клеток Р19 и росту нейритов, а связанные с БАС формы не имеют такого эффекта. Ангиогенин дикого типа защищает нейроны от гипоксия-индуцированной клеточной гибели, эксайтотоксичности и сывороточного голодания, а варианты, связанные с БАС, не оказывают нейропротективного действия [3, 20, 21].
В работе, изучавшей влияние структуры ангиогенина дикого типа и 11 его вариантов, связанных с БАС, было показано, что структурные изменения ангиогенина обусловливают его отрицательное воздействие на выживаемость нейронов преимущественно за счет влияния мутаций на РНКазную активность и способность индуцировать образование стрессовых гранул в нейрональных клеточных линиях [12]. Ангиогенин дикого типа в условиях гипоксии или голодания клетки индуцирует образование стресс-гранул в результате отщепления от тРНК антикодоновой петли и образования тРНК-производных стресс-индуцированных РНК. Последние несут целый ряд функций, включая подавление трансляции и деградацию специфических мРНК. В целом, стресс-гранулы обладают нейропротективной функцией, позволяя клетке пережить неблагоприятные условия, подавляя метаболизм РНК, поэтому одним из механизмов дегенерации мотонейронов у пациентов с мутациями в гене Ang может быть именно нарушение формирования стресс гранул. В то же время необходимо отметить, что агрегаты других мутантных белков, таких как как TDP-43, колокализуются со стресс-гранулами, формируя патологические агрегаты [22].
В отличие от наших данных, в нескольких исследованиях определялось повышение уровня ангиогенина в плазме крови у пациентов с БАС, которое расценивали как стрессовую реакцию, направленную на защиту гибнущих мотонейронов [23, 24]. Научной группой Skorupa было выдвинуто предположение о том, что ангиогенин, выделяемый мотонейронами под воздействием стресса, может служить сигналом призыва к помощи, адресованного астроцитам и эндотелиальным клеткам. В своих экспериментах они показали, что секреция ангиогенина мотонейронами в значительной степени активируется в ответ на стресс, а выделяемый ими ангиогенин поглощается почти исключительно астроглией, модифицируя в ней профиль РНК. Поступление ангиогенина в астроциты также было необходимым условием для защиты стресс-индуцированного повреждения мотонейронов с помощью ангиогенина [25].
В ряде других исследований повышения уровня ангиогенина в биологических жидкостях у пациентов с БАС выявлено не было. В работе Ilzecka различий концентраций ангиогенина в ЦСЖ между пациентами с БАС и контрольной группой выявлено не было [26]. В другом недавно опубликованном исследовании статистически значимых различий уровня ангиогенина в ЦСЖ у пациентов с БАС и здоровых добровольцев выявлено не было [27]. В работе McLaughlin и соавт. было показано статистически значимое снижение концентраций ангиогенина в сыворотке и плазме крови, а также ЦСЖ, по сравнению с контрольной группой [28]. В мета-анализе, проанализировавшем уровни ангиогенина в сыворотке крови у пациентов с нейродегенеративными заболевания (в основном с БАС), в сравнении со здоровыми добровольцами, статистически значимых различий между группами выявлено не было [29]. Ограничением нашего и многих других исследований, вошедших в последний мета-анализ, является относительно небольшой размер выборки. Как было показано ранее, уровень ангиогенина может варьировать в зависмости от целого ряда факторов, таких как возраст, пол, размеры тела, генетические особенности [30]. Таким образом, вопрос о значении ангиогенина в качестве биомаркера при БАС требует дополнительного изучения с включением в исследование более крупной выборки пациентов.
ЗАКЛЮЧЕНИЕ
Исследование показало, что уровни ангиогенина относительно высоки как в плазме, так и в ЦСЖ пациентов с БАС, так и в экзосомальной фракции. Тем не менее его содержание значительно выше в исходной плазме и ЦСЖ. У пациентов с БАС содержание ангиогенина в плазме и ее экзосомальной фракции понижено, по сравнению со здоровыми людьми. Обнаружена тенденция к взаимосвязи более низких уровней ангиогенина в плазме и ее экзосомальной фракции с худшим функциональным состоянием пациентов с БАС. Полученные данные указывают на возможную протективную роль ангиогенина при БАС, механизмы реализации этого эффекта нуждаются в дополнительном изучении. Полученные результаты создают основу для возможного использования ангиогенина в качестве маркера прогрессирования патологического процесса при БАС, однако этот вопрос также требует подтверждения в более крупных исследовниях.
Список литературы
Fett J.W., Strydom D.J., Lobb R.R., Alderman E.M., Bethune J.L., Riordan J.F., Vallee B.L. // Biochemistry. 1985. V. 24. P. 5480–5486.
Subramanian V., Feng Y. // Hum. Mol. Genet. 2007. V. 16. P. 1445–1453.
Subramanian V., Crabtree B., Acharya K.R. // Hum. Mol. Genet. 2008. V. 17. P. 130–149.
Hooper L.V., Stappenbeck T.S., Hong C.V., Gordon J.I. // Nat. Immunol. 2003. V. 4. P. 269–273.
Xu Z., Monti D.M., Hu G. // Biochem. Biophys. Res. Commun. 2001. V. 285. P. 909–914.
Soncin F. // Proc. Natl. Acad. Sci. USA. 1992. V. 89. P. 2232–2236.
Liu S., Yu D., Xu Z.P., Riordan J.F., Hu G.F. // Biochem. Biophys. Res. Commun. 2001. V. 287. P. 305–310.
Li S., Hu M.G., Sun Y., Yoshioka N., Ibaragi S., Sheng J., Sun G. // Mol. Cancer Res. 2013. V. 11. P. 1203–1214
Steidinger T.U., Slone S.R., Ding H., Standaert D.G, Yacoubian T.A. // PLoS One. 2013. V. 8. P. e56092
Захарова М.Н., Брылев Л.В., Авдюнина И.А., Лысогорская Е.В., Воробьева А.А., Иванова М.В., Червяков А.В., Васильев А.В., Бакулин И.С. // Неврология. Национальное руководство. Под ред. Гусева Е.И., Коновалова А.Н., Скворцовой В.И. М.: ГЭОТАР-Медиа, 2018. С. 644–662.
Greenway M.J., Alexander M.D., Ennis S., Traynor B.J., Corr B., Frost E., Green A., Hardiman O. // Neurology. 2004. V. 63. P. 1936–1938.
Thiyagarajan N., Ferguson R., Subramanian V., Acharya K.R. // Nat. Commun. 2012. V. 3. P. 1121.
Thery C., Boussac M., Veron P., Ricciardi–Castagnoli P., Raposo G., Garin J., Amigorena S. // J. Immunol. 2001. V. 166. P. 7309–7318.
Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. // Nat. Biotechnol. 2011. V. 29. P. 341–345.
Subra C., Grand D., Laulagnier K., Stella A., Lambeau G., Paillasse M., De Medina P., Monsarrat B., Perret B., Silvente-Poirot S., Poirot M., Record M. // J. Lipid Research. 2010. V. 51. P. 2105–2120.
Park S., Ahn E.S., Kim Y. // Cell Biol. Int. 2015. V. 39. P. 379–387.
Iguchi Y., Eid L., Parent M., Soucy G., Bareil C., Riku Y., Kawai K., Takagi S., Yoshida M., Katsuno M., Sobue G., Julien J. // Brain. 2016. V. 139. P. 3187–3201.
Ludolph A., Drory V., Hardiman O., Nakano I., Ravits J., Robberecht W., Shefner J. // Amyotroph Lateral Scler Frontotemporal Degener. 2015. V. 16. P. 291–292.
Greenway M.J., Andersen P.M., Russ C., Ennis S., Cashman S., Donaghy C. // Nat. Genet. 2006. V. 38. P. 411–413.
Kieran D., Sebastia J., Greenway M.J., King M.A., Connaughton D., Concannon C.G., Fenner B. // J. Neurosci. 2008. V. 28. P. 14056–14061.
Sebastia J., Kieran D., Breen B., King M.A., Netteland D.F., Joyce D., Fitzpatrick S.F. // Cell Death Differ. 2009. V. 16. P. 1238–1247.
Parker S.J., Meyerowitz J., James J.L., Liddell J.R., Crouch P.J., Kanninen K.M., White A.R. // Neurochem. Int. 2012. V. 60. P. 415–24.
Cronin S., Greenway M.J., Ennis S., Kieran D., Green A., Prehn J.H., Hardiman O. // Neurology. 2006. V. 67. P. 1833–1836.
van Es M.A., Veldink J.H., Schelhaas H.J., Bloem B.R., Sodaar P., van Nuenen B.F., Verbeek M. // J. Neurol. Neurosurg. Psychiatry. 2014. V. 85. P. 1439–1440.
Skorupa A., King M.A., Aparicio I.M., Düssmann H., Coughlan K., Breen B. // J. Neurosci. 2012. V. 32. P. 5024–5038.
Ilzecka J. // Acta Clin. Croat. 2008. V. 47. P. 77–79.
Morelli C., Tiloca C., Colombrita C., Zambon A., Soranna D., Lafronza A., Solca F., Carelli L., Poletti B., Doretti A., Verde F., Maderna L., Ticozzi N., Ratti A., Silani V. // Amyotroph. Lateral Scler. Frontotemporal Degener. 2019. V. 18. P. 1–7.
McLaughlin R.L., Phukan J., McCormack W., Lynch D.S., Greenway M., Cronin S., Saunders J., Slowik A., Tomik B., Andersen P.M., Bradley D.J., Jakeman P., Hardiman O. // PLoS One. 2010. V. 5. e15402.
Yu D., Cai Y., Zhou W., Sheng J., Xu Z. // Dis. Markers. 2018. V. 2018. P. 1984718.
Pantsulaia I., Trofimov S., Kobyliansky E., Livshits G. // Clinical Endocrinology. 2006. V. 64. P. 271–279.
Дополнительные материалы отсутствуют.