

Нейрохимия, 2020, T. 37, № 3, стр. 249-256
Классическая модель индуцированного 6-гидроксидофамином паркинсонизма у крыс характеризуется односторонним повреждением структур мезолимбической системы мозга
А. В. Ставровская 1, Д. С. Бережной 1, Д. Н. Воронков 1, С. Л. Стволинский 1, Д. А. Абаимов 1, А. С. Ольшанский 1, А. С. Гущина 1, Н. Г. Ямщикова 1, Т. Н. Федорова 1
1 ФГБНУ “Научный центр неврологии”
Москва, Россия
Поступила в редакцию 28.10.2019
После доработки 24.03.2020
Принята к публикации 21.04.2020
Аннотация
Вопрос локализации повреждений в мозге при введении грызунам нейротоксина 6-гидроксидофамина (6-ГДА) является существенным для понимания механизмов развития паркинсонизма. В работе представлены результаты поведенческих тестов, иммуногистохимического исследования и исследования уровня дофамина в различных зонах стриатума на 25-й день после унилатерального интранигрального введения 6-ГДА крысам. Была обнаружена гибель нейронов не только в компактной части черной субстанции, но и в латеральном ядре вентральной тегментальной области (ВТО). Детальный анализ уровня дофамина показал снижение на 90% в дорсальном и на 73% в вентральном стриатуме, но только со стороны, ипсилатеральной введению токсина. Обнаруженные изменения ВТО подтверждают результаты морфологических исследований post mortem при болезни Паркинсона. Ранее выявленные нарушения эмоционального состояния у крыс с 6-ГДА-индуцированной моделью болезни Паркинсона могут быть связаны с показанным в настоящей работе повреждением вентральной тегментальной области мозга.
ВВЕДЕНИЕ
Болезнь Паркинсона (БП) – нейродегенеративное заболевание, характеризующееся постепенным нарастанием экстрапирамидной моторной симптоматики – брадикинезии, ригидности и тремора покоя, что связано с прогрессирующей гибелью дофаминергических нейронов черной субстанции среднего мозга [1]. Паркинсонический синдром характерен также для заболеваний группы “паркинсонизм-плюс” к которым относят прогрессирующий надъядерный паралич, мультисистемную атрофию, деменцию с тельцами Леви [1]. Выделяют, кроме того, вторичные формы – лекарственный, сосудистый, постинфекционный паркинсонизм и т.д.
В настоящее время накоплены многочисленные подтверждения того, что при БП нейродегенерация связана с патологической агрегацией в нейронах белка альфа-синуклеина и носит мультисистемный характер, затрагивая и периферические отделы нервной системы [2]. Моторная симптоматика при БП проявляется лишь после снижения уровня дофамина в нигростриарном пути на 60%, что соответствует гибели 50–60% нейронов компактной части черной субстанции (ЧСк) [3], однако немоторные нарушения во многих случаях опережают манифестацию двигательных расстройств на 5–10 и более лет [4]. Возникновение этих нарушений объясняется тем, что наряду с дегенеративными изменениями нигростриатной системы мозга при паркинсонизме имеет место также вовлечение мезолимбической дофаминергической системы и некоторых других нейротрансмиттерных систем.
Хотя токсические модели паркинсонизма на экспериментальных животных служили одним из первых инструментов исследования механизмов нарушений при БП, наряду с фармакологической блокадой синтеза и нейротрансмиссии дофамина [5], они остаются незаменимыми для оценки компенсаторных, пластических изменений в базальных ядрах, поиска причин повышенной уязвимости нигральных нейронов при БП, разработки стратегий нейропротекции и клеточной терапии при паркинсонизме. Классическими моделями является введение в различные отделы мозга нейротоксинов 6‑гидроксидофамина (6-ГДА) и 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МФТП), приводящих к гибели дофаминергических нейронов за счет индукции окислительных повреждений клетки и нарушения окислительного фосфорилирования [6]. В случае системного введения токсинов, например, МФТП, размер поражения значительно варьирует, что сказывается и на вариабельности проявления моторной симптоматики. При интракраниальном введении итоговый размер поражения и скорость его развития зависит от места введения токсина. Так, наиболее быструю и селективную гибель нейронов ЧСк с развитием характерной моторной симптоматики вызывает введение 6-ГДА непосредственно в средний мозг [7]. Чаще используют одностороннее интранигральное введение, при котором наблюдается снижение дофамина в мезостриарном дофаминергическом пути. Животные с таким повреждением не демонстрируют афагии и адипсии, в отличие от двусторонних разрушений ЧСк [8]. Кроме того, одностороннее введение токсина соответствует латерализации моторных нарушений в начальных стадиях БП [1]. В последнее время, учитывая возросший интерес к преклинической стадии БП, все больше внимания привлекают немоторные проявления паркинсонизма в данной модели [9, 10], связанные с нарушениями в мезолимбической и мезокортикальной дофаминергических системах. Подобные нейрохимические нарушения наблюдаются при введении 6-ГДА в стриатум, что сопровождается медленным развитием паркинсоноподобной симптоматики на протяжении 3 недель [10]. Для такого способа введения 6-ГДА детально описаны нейродегенеративные изменения как в ЧСк, так и ВТО [11]. Но в большинстве работ с интранигральным введением токсина, сделан акцент лишь на локальные изменениях в ЧСк и быстром развитии моторной симптоматики паркинсонизма [7, 12, 13]. Таким образом, в какой степени локальное введение 6-ГДА в ЧСк влияет на мезолимбическую дофаминергическую систему остается неясным. С учетом современных работ по гетерогенности популяций дофаминергических нейронов среднего мозга требуется более детальное изучение нейродегенерации в данной модели [14].
Вместе с тем, во многих работах ставится вопрос о локальности повреждений в 6-ГДА модели паркинсонизма. Так, были получены данные об изменении уровня дофамина в контралатеральном стриатуме [15, 12 ] при введении 6-ГДА в ЧСк, что рассматривают как свидетельство перекрестной дофаминергической иннервации и взаимодействия разных дофаминергических путей.
Поскольку нейродегенеративный процесс при БП у человека не ограничен черной субстанцией, важной для понимания механизмов данной патологии представляется оценка изменений не только в нигростриатной системе, но и в структурах, мезолимбической и мезокортикальной дофаминергических системам, формируемых нейронами ВТО [4]. В связи с этим представляет интерес изучение гибели дофаминергических нейронов в вентральной тегментальной области (ВТО) мозга при локальном введении 6-ГДА как в зоны ее иннервации – базальные ядра [16, 11 ], так и в ЧСк.
Цель исследования – охарактеризовать выраженность нейродегенеративного процесса в нигростриарной и мезолимбической дофаминовых системах и связанные с ними нарушения поведения при моделировании паркинсонизма с помощью одностороннего интранигрального введения 6-гидроксидофамина.
МАТЕРИАЛЫ И МЕТОДЫ
Эксперимент проведен на 18 аутбредных крысах самцах Wistar в возрасте 3.5 мес., с массой тела 300–350 г, которые содержались в стандартных контролируемых условиях вивария при 12-ч световом цикле.
Для получения модели паркинсонического синдрома животных помещали на раму лабораторного стереотаксиса (Stoelting Co., США), скальп надрезали и через просверленные в черепе отверстия в средний мозг справа вводили 6-OHDA в дозе 12 мкг в 3 мкл 0.05% раствора аскорбиновой кислоты в соответствии с координатами атласа мозга крыс (АР = –4.8; L = 1.9; V = 8.0) [17]. Ложнооперированным животным (n = 9) вводили растворитель в том же объеме. Для анестезии применяли золетил 100 в дозе 3 мг/100 г и ксиланит в дозе 3 мг/кг внутримышечно, для премедикации использовали атропин в дозе 0.04 мг/кг подкожно за 10–15 мин до введения ксиланита.
В течение 2 нед. животным давали оправиться от операции, на 14-й и 20-й день после введения токсина животных тестировали в “Открытом поле” (ОП). Установка для изучения изменения двигательной активности и ориентировочно-исследовательского поведения представляла собой короб 97 × 97 × 40 см из жесткого ПВХ (“Открытая наука”, Россия). Поведение крыс фиксировали в течение 3-х мин, последующий анализ данных проводили с помощью системы видео наблюдения Any-Maze (Stoelting Inc., США) с программным обеспечением.
На 25-й день эксперимента крыс декапитировали и извлекали мозг для дальнейшего биохимического и гистологического анализа. Мозг разделяли на льду и в соответствии с атласом [17] выделяли префронтальную кору, область покрышки среднего мозга, дорсальный и вентральный стриатум билатерально. У части животных (по 3 из каждой группы) блок мозга, содержащий средний мозг, фиксировали в 4% формалине на PBS для иммуногистохимического анализа на тирозингидроксилазу. Полученные образцы стриатума и коры замораживали в жидком азоте и использовали для последующего измерения уровня моноаминов с помощью метода ВЭЖХ/ЭД.
Для морфологического исследования были взяты 3 образца мозга из каждой группы. Образцы пропитывали средой O.C.T. (Tissue Tek, Sakura, США) и готовили серию фронтальных срезов толщиной 10 мкм (всего 36 срезов, взятых через 20 мкм) на криостате Cryo 3 (Tissue Tek, Sakura, США), продвигаясь в каудальном направлении от трека иглы (области введения токсина). Область исследования была выбрана таким образом, чтобы захватить большую часть парабрахиального пигментированного ядра и черной субстанции. Из полученной серии срезов для подсчета нейронов брали каждый шестой срез. Для выявления тирозингидроксилазы использовали кроличьи поликлональные антитела (1 : 500, Sigma, Germany) и вторичные антитела козы к иммуноглобулинам кролика, меченные флуорохромом CF488 (1 : 150, Sigma, Germany). Срезы изучали под флуоресцентным микроскопом Eclipse NiU (Nikon; Япония). При помощи программы ImageJ (Wayne Rasband (NIH); США) оценивали количество дофаминергических нейронов в дорсальной и латеральной областях ЧСк и в области парабрахиального ядра (латерального ядра ВТО) [14].
Дополнительно для оценки повреждения латерального ядра ВТО отдельной группе животных (n = 6) через две недели после введения 6-ГДА стереотаксичеcки вводили в стриатум (АР = 0.3; L = 3.0; V = 7.0) ретроградный трейсер DiI (10 мкл 0.5% р-р в диметилсульфоксиде (Sigma, Германия)). Половина животных получала инъекции в ипсилатеральный стриатум, а половина – в противоположное полушарие. Через две недели после введения DiI животных декапитировали, мозг фискировали как описано выше и на вибратоме (Microm HM560V, Thermo Fischer, США) готовили срезы толщиной 25 мкм.
В разных зонах стриатума определяли содержание дофамина (ДА) и его основных метаболитов 3,4-дигидроксифенилуксусной кислоты (ДОФУК) и гомованилиновой кислоты (ГВК). Ткань мозга гомогенизировали в 20 объемах 0.1N HClO4 с добавлением диоксибензиламина (0.5 нмоль/мл) в качестве внутреннего стандарта, затем центрифугировали при 10 000 g в течение 10 мин, для анализа использовали супернатант. Определение проводили методом высокоэффективной жидкостной хроматографии с электрохимической детекцией на хроматографе LC-304T (BAS, West Lafayette, США) с аналитической колонкой C18 (4 × 150 мм, 4 мкм, Phenomenex, США).
Все эксперименты проводились в соответствии с международными правилами по работе с лабораторными животными, c соблюдением биоэтических норм и возможным сокращением числа использованных животных [18]. Статистическую обработку проводили с использованием программы “Statistica 10.0”. Для определения статистической значимости различий использовали факторный дисперсионный анализ (ANOVA), для сравнения между группами использовали апостериорные тесты Тьюки (Tukey test) и t-критерий Стьюдента. Различия считались значимыми при p < 0.05. Результаты представлены в виде среднего значения ± ошибка среднего (M ± SEM) и среднего значения ± стандартное отклонение (M ± SD).
РЕЗУЛЬТАТЫ
Поведение. Сопоставление веса животных, получавших инъекцию 6-ГДА, с контрольными животными, выявило значимое снижение веса на 13-й день после операции (316.6 ± 35.5 г против 346.0 ± 21.3 г в контрольной группе, p < 0.05), которое нивелировалось к 19-му дню эксперимента (338.6 ± 23.9 г против 357.5 ± 21.7 г в контрольной группе). Тестирование в ОП позволило обнаружить изменения в поведении экспериментальных крыс относительно контрольных в оба исследованных срока после операции: на 14-й и 20-й день (табл. 1). Было показано, что введение токсина привело к значимому снижению длины пройденного в ОП пути (F(1,16) = 38.07; p < 0.01), уменьшению количества стоек (F(1,16) = 11.88; p < 0.05) и увеличению количества спонтанных вращений вправо (F(1,16) = 7.57; p < 0.05). Для животных экспериментальной группы была характерна латерализация отдельных двигательных действий, проявившаяся в увеличенном числе спонтанных вращений вправо по сравнению с числом вращений влево, что характерно для унилатерального разрушения дофаминергической иннервации [19]. Стоит отметить, что различия в количестве вращений в сторону повреждения были более выражены у экспериментальной группы при первом тестировании, а при втором значимых отличий по этому параметру уже не выявлялось (табл. 1).
Таблица 1.
Основные параметры поведения животных в тесте “открытое поле”. Приведены результаты post hoc равнений (LSD test). * значимые отличия от контрольной группы с p < 0.05; – отсутствие измерения в этот день
| Группа | Тестирование | Пройденный путь, м | Количество стоек | Количество вращений вправо | Количество вращений влево |
|---|---|---|---|---|---|
| Контроль | 14 день | 11.9 ± 2.2 | – | 3.3 ± 1.0 | 2.3 ± 1.0 |
| 20 день | 8.0 ± 1.2 | 11.9 ± 4.5 | 2.4 ± 1.1 | 1.8 ± 1.0 | |
| 6-ГДА | 14 день | 6.1 ± 1.6* | – | 5.1 ± 2.1* | 0.6 ± 0.8* |
| 20 день | 4.2 ± 2.5* | 4.2 ± 4.8* | 3.8 ± 2.5 | 0.3 ± 0.5* |
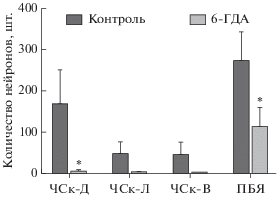
Гистология. Подсчет нейронов в компактной части черной субстанции показал значимое снижение плотности нейронов в ЧСк и парабрахиальном ядре ВТО (F(2,4) = 10.18; p = 0.046). При этом более выраженное снижение наблюдалось в дорсолатеральной части, включающей нейроны ЧСк (см. рис. 1, 2). Так, у животных, которым вводили 6-ГДА, на 23-й день полностью отсутствовали нейроны в дорсолатеральной части, а в медиальной части (ПБЯ) их количество снижалось только на 58%. Следовательно, токсическое воздействие 6-ГДА распространялось не только на дорсолатеральную часть ЧСк, куда вводился токсин, но и на структуры ВТО. Полученные результаты подтверждались качественной оценкой изменений на срезах черной субстанции после ипсилатерального введения в стриатум животных, получавших 6-ГДА ретроградного трейсера DiI (рис. 1) – наблюдалась потеря меченых DiI нейронов в парабрахиальном ядре ВТО и полное исчезновение метки в ЧСк.
Рис. 1.
Повреждение нейронов компактной части черной субстанции (ЧСк) и парабрахиального ядра вентрального поля покрышки среднего мозга (ПБЯ) при одностороннем интранигральном введении 6-ГДА. а, б – Иммуногистохимическое выявление тирозингидроксилазы (TH). в, г – Выявление ретроградно меченых нейронов при введении красителя DiI на контралатеральной повреждению стороне (в) и на стороне введения 6-ГДА (г). ЧСк – компактная часть черной субстанции, ЧСк-Л –латеральная область компактной части черной субстанции, ЧСк-В – вентральная область, ЧСк-Д – дорсальная область и ПБЯ – область парабрахиального пигментированного ядра ВТО. Справа – сторона введения 6-ГДА.
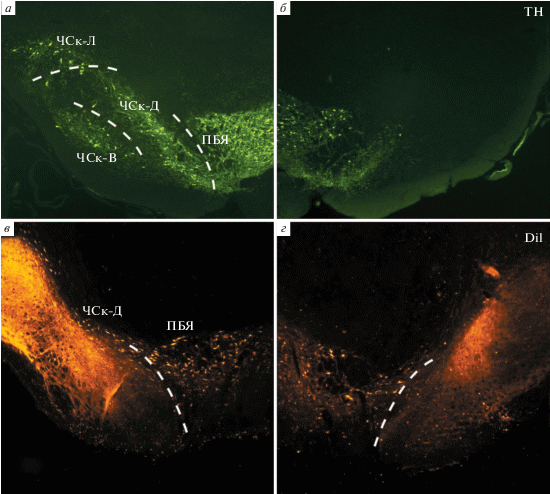
Рис. 2.
Подсчет количества нейронов в разных отделах покрышки среднего мозга на 23-й день после введения 6-ГДА. * p < 0.05 по сравнению с группой контроля по критерию Стьюдента. Остальные обозначения, как на рис. 1.
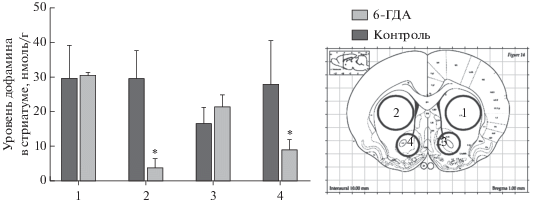
Катехоламины. Анализ содержания дофамина и его основных метаболитов – 3,4-диоксифенил-уксусной кислоты (ДОФУК) и гомованилиновой кислоты (ГВК) – в различных областях стриатума выявил их значимое снижение только со стороны, ипсилатеральной по отношению к введению токсина в черную субстанцию (F(3,12) = 47.89; p < 0.001 для дорсальной части; F(3,11) = 3.57; p < 0.05 для вентральной). С контралатеральной стороны изменений метаболизма дофамина обнаружено не было (F(2,13) = 1.31; p = 0.29 для дорсальной части; F(2,13) = 2.80; p = 0.09 для вентральной) (табл. 2). Выявленные изменения были более выраженными в дорсальной части ипсилатерального стриатума (рис. 3): снижение уровня дофамина на 88% (p < 0.001), ДОФУК на 88% (p < 0.001), ГВК на 80% (p < 0.001). В вентральной части, включающей прилежащее ядро, было обнаружено значимое снижение уровня дофамина на 65% (p < 0.01) но ДОФУК (62% от контрольных значений, р = 0.07) и ГВК (88% от контрольных значений, р = 0.59) по отношению к значениям контрольной группы не изменялись.
Таблица 2.
Изменение содержания дофамина и его основных метаболитов в разных областях стриатума животных на 23 день после введения 6-ГДА: 1 – левый дорсальный, 2 – правый дорсальный, 3 – левый вентральный, 4 – правый вентральный, * p < 0.05 по сравнению с группой контроля по критерию Стьюдента
| Группа | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| ДА контроль | 29.4 ± 9.6 | 29.3 ± 8.2 | 16.3 ± 7.0 | 27.7 ± 12.7 |
| ДА 6-ГДА | 30.2 ± 2.8 | 3.1 ± 3.5* | 21.1 ± 5.3 | 9.7 ± 5.2* |
| ДОФУК контроль | 9.2 ± 1.3 | 11.0 ± 1.9 | 14.9 ± 3.5 | 13.7 ± 3.1 |
| ДОФУК 6-ГДА | 10.5 ± 1.8 | 1.2 ± 2.0* | 17.4 ± 1.8 | 8.5 ± 6.8 |
| ГВК контроль | 2.3 ± 0.3 | 2.5 ± 0.5 | 2.6 ± 0.7 | 2.9 ± 0.6 |
| ГВК 6-ГДА | 2.7 ± 0.3 | 0.5 ± 0.8* | 3.7 ± 0.7 | 2.5 ± 1.7 |
Рис. 3.
Измерение содержания дофамина в разных областях стриатума животных на 23-й день после введения 6-ГДА. Сбоку схематично предcтавлено выделение зон стриатума по атласу (AP = +1 мм; ([17]): 1 – левый дорсальный, 2 – правый дорсальный, 3 – левый вентральный, 4 – правый вентральный, * p < 0.05 по сравнению с группой контроля по критерию Стьюдента.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
6-ГДА – гидроксилированный аналог естественного нейротрансмиттера дофамина – один из самых распространенных нейротоксинов, используемых в моделях дегенерации центральных катехоламинергических проекций [6]. Несмотря на более чем 50-летнюю историю изучения 6-ГДА [8], вопросы локальности повреждений, временной динамики развития патологии и роли различных патологических каскадов в развитии 6-ГДА-индуцированных повреждений остаются нерешенными [20, 21]. Так, в большинстве работ исследователи концентрируются на гибели нейронов в компактной части черной субстанции и оценивают повреждения в отдаленные сроки после введения токсинов [22], что не позволяет оценить динамику развития нейродегенерации. В качестве основных механизмов токсичности 6-ГДА предполагают его аутоокисление и ферментативное окисление моноаминоксидазами, что ведет к образованию хинонов, перекиси водорода, суперкосид-аниона и гидроксильных радикалов [6, 23]. Интересно, что в физиологических условиях в результате окисления дофамина образуется хиноновая, окисленная форма 6-ГДА, что послужило поводом для гипотезы о роли эндогенного 6-ГДА в патогенезе БП [24]. Большинство авторов называют апоптоз основным механизмом гибели нейронов ЧСк при введении 6-ГДА, однако показаны и другие типы гибели клеток – некроз и аутофагия [20, 25 ], а их соотношение вероятно зависит от активации механизмов нейровоспаления.
Несмотря на быстрое действие 6-ГДА, характеризующееся гибелью нейронов в течение 12 ч после введения в черную субстанцию и снижением дофамина в течение 2–3 дней [26], динамика развития моторной симптоматики в данной модели оказывается значительно более медленной. При этом, на 3–4 неделю приходятся основные функциональные перестройки в сети базальных ганглиев, выраженные в усилении активности ретикулярной части черной субстанции и бледного шара. Известно, что двигательная активность также достигает минимума к 3–4 нед. после операции [27, 28]. В данном исследовании было показано, что на этих сроках горизонтальная и вертикальная активность у животных опытной группы существенно ниже контрольных значений, но у животных не обнаруживалось птоза, тремора, или выраженной ригидности, характерных для поздних стадий паркинсонизма [5]. Ранее, на модели введении 6-ГДА в область ЧСк нами была проведена оценка не только двигательной дисфункции, но и психоэмоционального состояния животных в тесте “Сужающаяся дорожка” с присвоением баллов по шкале невротизации [29, 30]. Было показано наличие “нестандартной” поведенческой активности крыс с моделью БП, в том числе стереотипных движений головой, интенсивного обнюхивания и лизания установки, стереотипных жевательных движений, длительных замираний и др. Такие животные либо вовсе отказывались от поступательного движения, либо преодолевали расстояние до укрытия за значительно более длительное время, чем контрольные. Полученные данные свидетельствовали о развитии у экспериментальных крыс неврозо-подобного состояния. В соответствие с этим, представляло интерес охарактеризовать выраженность нарушений дофаминергической системы мозга крыс именно на сроке 21–25 дней после токсического повреждения ЧСк.
Морфологический анализ выявил не только полное исчезновение дофаминовых нейронов в ЧСк, но и значительное снижение их количества в парабрахиальном ядре ВТО, примыкающем к дорсальному рогу ЧСк [14]. Это совпадает с литературными данными по другим способам введения 6-ГДА [11], где также наблюдается гибель нейронов ВТО при выраженном повреждении ЧСк (>70%). Можно предположить, что, поскольку гибель нейронов в области введения 6-ГДА сопровождается активацией микроглии, это ведет к усилению нейродегенеративного процесса, распространяющегося и на относительно удаленные от места введения токсина области. К сожалению, для характеристики уязвимости различных зон покрышки к 6-ГДА-индуцированным повреждениям недостаточно анализа в конечной точке и необходим более детальный анализ ранних стадий развития патологии.
Полученные данные по уровню дофамина и его метаболитов в разных областях стриатума свидетельствуют о том, что в правом дорсальном стриатуме, судя по уровню дофамина, наблюдается полное исчезновение дофаминергической иннервации. Интересно, что нарушения иннервации проявляются и в вентральном стриатуме справа, что дополнительно подтверждает повреждение ВТО, проецирующейся в данную область [14].
Нейроанатомически обнаруживается до 5% нейронов черной субстанции (в том числе – дофаминергических), проецирующихся в контралатеральный стриатум [31]. Недавно было продемонстрировано высвобождение дофамина в контралатеральном стриатуме перекрестными нигростриарными проекциями в ответ на стимуляцию черной субстанции [15]. Предполагают, что контралатеральные нигростриарные проекции участвуют в пластических перестройках в базальных ядрах при повреждении ЧС в эксперименте и при болезни Паркинсона [15]. В то же время, локальное повреждение области покрышки среднего мозга справа в нашем эксперименте значимо не влияло на метаболизм дофамина с контралатеральной стороны: уровень дофамина и ДОФУК не отличается значимо от контрольных животных. Очевидно, что перекрестная дофамин-ергическая иннервация практически не сказывалась на проявлении нейропатологии в данной модели. В других исследованиях также было показано, что при гибели нейронов более 75% последующей реиннервации стриатума не наблюдается [32].
Сопутствующая активация микроглии также проявляется наиболее активно в первую неделю и к 3-й неделе уже сходит на нет [13]. В данном исследовании мы наблюдаем конечный результат совершившегося патологического процесса. Тем не менее, обнаруженные изменения ВТО в 6-ГДА-индуцированной модели заслуживают особого внимания, поскольку подтверждают результаты морфологических исследований post mortem при болезни Паркинсона [33]. Подобные нарушения, как правило, рассматривают как одну из причин немоторной симптоматики БП [34]. Таким образом, ранее выявленные нарушения эмоционального состояния у крыс с 6-ГДА-индуцированной моделью БП [9, 29, 30] могут быть связаны с показанным в настоящей работе повреждением вентрально-тегментальной области мозга.
ВЫВОДЫ
1. Одностороннее введение 12 мкг 6-ГДА в черную субстанцию через 3 недели приводило к гибели дофаминовых нейронов ЧСк и резкому падению содержания дофамина на 88% в дорсальном ипсилатеральном стриатуме, а также снижению общей двигательной активности у экспериментальных животных.
2. Гибель дофаминовых нейронов под действием 6-ГДА отмечалась также в вентральной тегментальной области, но в меньшей степени (снижение числа нейронов в парабрахиальном ядре ВТО на 58%). Уровень дофамина в вентральном стриатуме на стороне повреждения снижался на 65%.
3. Введение 6-ГДА в ЧСк повреждает структуры нигростриарной и мезолимбической дофаминовых систем мозга ипсилатерально. При этом участия межполушарных (перекрестных) нигростриарных дофаминергических проекций в восполнении дефицита медиатора на исследуемом сроке не выявлено.
Список литературы
Шток В.Н., Левин О.С. // Клиническая синдромологическая классификация экстрапирамидных расстройств. М.: МедПресс-Информ, 2014. 112 с.
Иллариошкин С.Н., Власенко А.Г., Федотова Е.Ю. // Анналы клин. и эксперим. неврологии. 2013. Т. 7. № 2. С. 39–50.
Braak H., Ghebremedhin E., Rub U., Bratzke H., Del Tredici K. // Cell Tissue Res. 2004. V. 318. № 1. P. 121–134.
Chaudhuri K.R., Healy D.G., Schapira A.H. // Lancet Neurol. 2006. V. 5. № 3. P. 235–245.
Jagmag S.A., Tripathi N., Shukla S.D., Maiti S., Khurana S. // Front. Neurosci. 2016. V. 9. № 503. P. 1–13.
Schober A. // Cell Tissue Res. 2004. V. 318. № 1. P. 215–224.
Jeon B.S., Jackson-Lewis V., Burke R.E. // Neurodegeneration. 1995. V. 4. № 2. P. 131–137.
Ungerstedt U. // Eur. J. Pharmacol. 1968. V. 5. № 1. P. 107–110.
Branchi I., D’Andrea I., Armida M., Cassano T., Pezzola A., Potenza R.L., Morgese M.G., Popoli P., Alleva E. // J. Neurosci. Res. 2008. V. 86. № 9. P. 2050–2061.
Tadaiesky M.T., Dombrowski P.A., Figueiredo C.P., Cargnin-Ferreira E., Da Cunha C., Takahashi R.N. // Neuroscience. 2008. V. 156. № 4. P. 830–840.
Healy-Stoffel M., Omar Ahmad S., Stanford J.A., Levant B. // Brain Res. 2014. V. 1574. P. 113–119.
Schwarting R.K.W., Huston J.P. // Progress in Neurobiology. 1996. V. 49. № 3. P. 215–266. https://doi.org/10.1016/S0301-0082(96)00015-9
Walsh S. Finn D.P., Dowd E. // Neuroscience. 2011. V. 175. P. 251–261.
Vogt Weisenhorn D.M., Giesert F., Wurst W. // J. Neurochem. 2016. V. 139. S. 1. P. 8–26.
Fox M.E., Mikhailova M.A., Bass C.E., Takmakov P., Gainetdinov R.R., Budygin E.A., Wightmanet R.M. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. № 25. P. 6985–6990. https://doi.org/10.1073/pnas.1603629113
Sauer H., Oertel W.H. // Neuroscience. 1994. V. 59. № 2. P. 401–415.
Paxinos G., Watson C. // The Rat Brain in Stereotaxic Coordinates: Hard Cover Edition. 6th Edition. Elsevier, 2006. P. 456.
Каркищенко Н.Н., Грачев С.В. // Руководство по лабораторным животным и альтернативным моделям в биомедицинских технологиях. М.: Профиль, 2010. 346 с.
Dunnett S.B., Iversen S.D. // Neuropharmacology. 1982. V. 21. № 9. P. 899–908.
Hernandez-Baltazar D., Zavala-Flores L.M., Villanueva-Olivo A. // Neurologia. 2017. V. 32. № 8. P. 533–539.
Hoglinger G.U., Carrard G., Michel P.P., Medja F., Lombes A., Ruberg M., Friguet B., Hirsch E.C. // J. Neurochem. 2003. V. 86. № 5. P. 1297–1307.
Ferro M.M., Bellissimo M.I., Anselmo-Franci J.A., Angellucci M.E., Canteras N.S., Da Cunha C. // J. Neurosci. Methods. 2005. V. 148. № 1. P. 78–87.
Glinka Y.Y., Youdim M.B. // Eur. J. Pharmacol. 1995. V. 292. № 3–4. P. 329–332.
Jellinger K., Linert L., Kienzl E., Herlinger E., Youdim M. // J. Neural Transmission. Sup. 1995. V. 46. P. 297–314.
Hernandez-Baltazar D., Mendoza-Garrido M., Martinez-Fong D. // PLoS ONE. 2013. V. 8. № 8. e70951. https://doi.org/10.1317/journal.pone.0070951
Deumens R., Blokland A., Prickaerts J. // Exp. Neurol. 2002. V. 175. № 2. P. 303–317.
Ставровская А.В., Воронков Д.Н., Ямщикова Н.Г., Ольшанский А.С., Худоерков Р.М., Хаспеков Л.Г., Иллариошкин С.Н. // Анналы неврологии. 2015. Т. 9. № 2. С. 28–32.
Ставровская А.В., Конорова И.Л., Иллариошкин С.Н., Федорова Т.Н., Стельмашук Е.В., Ямщикова Н.Г., Стволинский С.Л., Генрихс Е.Е., Коновалова Е.В. // Технологии моделирования заболеваний нервной системы. В кн.: Неврология ХХI в.: диагностические, лечебные и исследовательские технологии: Руководство для врачей. В 3-х т. / Под ред. Пирадова М.А., Иллариошкина С.Н., Танашян М.М. Т. III. М.: ООО “АТМО”, 2015. С. 73–133.
Ставровская А.В., Ольшанский А.С., Ямщикова Н.Г., Гущина А.С. // Вестник Российской Военно-медицинской академии. 2018. Т. 3. № 63. С. 104–106.
Бережной Д.С., Федорова Т.Н., Куликова О.И., Ставровская А.В., Абаимов Д.А., Гущина А.С., Ольшанский А.С., Воронков Д.Н., Стволинский С.Л. // Вестник РГМУ. 2019. № 5. С. 49–56. https://doi.org/10.24075/vrgmu.2019.060
Lieu C.A., Subramanian T. // Brain Res. Bull. 2012. V. 87. № 1. P. 1–9. https://doi.org/10.1016/j.brainresbull.2011.09.013
Stanic D., Finkelstein D. I., Bourke D.W., Drago J., Horne M.K. // Eur. J. Neurosci. 2003. V. 18. № 5. P. 1175–1188.
Mythri R.B., Venkateshappa C., Harish G., Mahadevan A., Muthane U.B., Yasha T.C., Srinivas Bharath M.M., Shankar S.K. // Neurochem. Res. 2011. V. 36. № 8. P. 1452–1463.
Bruck A., Kurki T., Kaasinen V., Vahlberg T., Rinne J.O. // J. Neurol. Neurosurg. Psychiatry. 2004. V. 75. № 10. P. 1467–1469.
Дополнительные материалы отсутствуют.