

Нейрохимия, 2020, T. 37, № 4, стр. 291-310
Молекулярно-клеточные механизмы регуляции состояния покоя и деления стволовых клеток гиппокампа
Д. И. Мальцев 1, О. В. Подгорный 2, 3
1 Федеральный центр мозга и нейротехнологий ФМБА России
Москва, Россия
2 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
3 Институт биологии развития им. Н.К. Кольцова РАН
Москва, Россия
Поступила в редакцию 18.04.2020
После доработки 28.04.2020
Принята к публикации 30.04.2020
Аннотация
Нейрогенез в зубчатой извилине гиппокампе происходит на протяжении всей жизни млекопитающих, в том числе и у человека. Считается, что новорожденные нейроны в процессе интеграции обеспечивают высокий уровень пластичности гиппокампальной нервной сети. Это необходимо для когнитивной функции мозга, памяти и формирования эмоций. Нейрогенез поддерживается пулом стволовых клеток, расположенных в субгранулярной зоне зубчатой извилины. Эти клетки бόльшую часть жизни пребывают в состоянии митотического покоя. С возрастом запас стволовых клеток постепенно уменьшается в результате их деления и необратимой дифференцировки в астроциты. Кинетика расходования запаса стволовых клеток определяется балансом между их покоем и делением. Вопрос о том, какие молекулярные механизмы регулируют этот баланс, является одним из ключевых в биологии стволовых клеток мозга. Разобраться в этом вопросе важно не только для того, чтобы понять фундаментальные принципы регуляции нейрогенеза, но и, чтобы разрабатывать новые стратегии использования внутреннего резерва стволовых клеток для коррекции дефицита пластичности гиппокампа при разнообразных патологиях мозга и старении. К настоящему моменту уже накопилось достаточно много новых и интересных данных о многообразии молекулярных механизмов, вовлеченных в регуляцию покоя и деления стволовых клеток. В предлагаемом обзоре мы систематизируем накопленные знания о том, какие сигнальные молекулы и клеточные рецепторы вовлечены в поддержание состояния покоя стволовых клеток, как происходит переключение состояния покоя на пролиферацию на уровне транскрипции генов, каково значение компонентов ниши (нервных связей, макро- и микроглии, внеклеточного матрикса) в регулировании баланса между покоем и делением стволовых клеток, а также в поддержании и истощении их пула.
ПОСТНАТАЛЬНЫЙ НЕЙРОГЕНЕЗ В ЗУБЧАТОЙ ИЗВИЛИНЕ ГИППОКАМПА И ЕГО ЗНАЧЕНИЕ ДЛЯ ФУНКЦИОНИРОВАНИЯ МОЗГА
Нейрогенез, т.е. рождение и функциональная интеграция новых нейронов в существующие нервные сети, продолжается на протяжении всей жизни в зубчатой извилине гиппокампа большинства млекопитающих [1–4], включая человека [5–10]. Гиппокампальный нейрогенез во взрослом мозге впервые был обнаружен и описан американским нейробиологом Джозефом Альтманом в 60-х гг. прошлого столетия [11–13]. Однако окончательное признание это явление получило лишь в середине–конце 90-х годов прошлого столетия [2], в результате чего была пересмотрена господствующая догма о том, что вместе с окончанием развития мозг утрачивает способность к продукции новых нервных клеток. Начиная с этого времени, постнатальный нейрогенез в гиппокампе вызывает большой интерес у исследователей в связи с его особой значимостью для когнитивной функции мозга, памяти и формирования эмоций в норме и при разнообразных патологиях [14–16].
Сегодня считается, что новорожденные нейроны обеспечивают высокий уровень пластичности гиппокампальной нервной сети. Это достигается в критический период созревания и интеграции новорожденных нейронов (между 4-ой и 6-ой неделями после окончания делений), когда они активно устанавливают синаптические связи и имеют электрофизиологические свойства, отличные от зрелых гранулярных нейронов. В частности, новорожденные нейроны обладают более низким порогом возбудимости, и в них легче индуцировать долговременную потенциацию (т.е. изменение силы синаптической передачи – один из основных механизмов синаптической пластичности). Такая пластичность, как полагают многие исследователи, необходима для формирования эпизодической памяти и генерирования нормальной реакции организма на стресс. Так, у животных с абляцией нейрогенеза развиваются признаки глубокой депрессии и нарушается восстановление уровня кортикостерона (гормона, регулирующего функционирование различных систем организма при стрессе) после однократного действия стрессирующего фактора [17]. Кроме того, у таких животных наблюдается когнитивный дефицит при некоторых видах гиппокамп-зависимого обучения и ухудшение формирования долговременной потенциации именно в зубчатой извилине [18]. С другой стороны – хемогенетическая активация новорожденных нейронов оказывает антидепрессивный эффект [19]. Функционирование новорожденных нервных клеток именно в период созревания играет ведущую роль в различении паттерна (в англ. яз. лит-ре: pattern separation), т.е. способности различать похожие опыт, события или ощущения, что важно для формирования эпизодической памяти [20]. Искусственное увеличение продукции новых нейронов улучшает эту функцию зубчатой извилины [21].
Многочисленные исследования показывают, что гиппокампальный нейрогенез чувствителен к разнообразным физиологическим и патологическим стимулам. Так, например, физическая активность (бег в колесе), содержание животных в обогащенной среде (в клетке с разнообразными лабиринтами, лесенками и пр.), введение антидепрессантов, как, например, ингибиторов обратного захвата серотонина, антидепрессивная терапия (электроконвульсивная терапия или глубокая стимуляция мозга) и некоторые виды гиппокамп-зависимого обучения увеличивают число новых нейронов в зубчатой извилине, улучшая при этом показатели в поведенческих тестах [22–28]. С другой стороны хронический стресс, нейродегенеративные (болезни Альцгеймера и Паркинсона) и нейропсихические (шизофрения, глубокая депрессия) заболевания, радиоактивное облучение, химиотерапия, травматическое повреждение мозга и старение всегда сопровождаются сниженным уровнем гиппокампального нейрогенеза [2, 8, 16, 29–35]. В то же время инсульт, наоборот, повышает уровень продукции новых нейронов в зубчатой извилине гиппокампа, но при этом новорожденные нейроны имеют аномальную морфологию и локализацию [36].
Еще целый ряд исследований указывает на необходимость новых нейронов в зубчатой извилине для достижения позитивных эффектов при терапии когнитивных нарушений или депрессии различной этиологии [26, 37]. Это, в частности, привело к созданию так называемой нейрогенной теории депрессии, в которой причиной развития депрессии считается недостаток продукции новых нейронов в зубчатой извилине [38], а восстановление уровня гиппокампального нейрогенеза необходимо, чтобы обратить депрессивное состояние. Хотя нейрогенная теория депрессии имеет много противников, сегодня уже ни у кого не вызывает сомнение наличие тесной связи между постнатальным гиппокампальным нейрогенезом, депрессией и антидепрессантами.
Старение мозга во многих случаях сопряжено со снижением когнитивной функции и/или развитием депрессии [39]. При этом хорошо известно, что и гипокампальный нейрогенез значительно снижается с возрастом [2, 9]. Хотя прямой связи между падением базового уровня продукции новых нейронов и развитием деменции или депрессии в старости не прослеживается, оказалось, что некоторые способы терапии когнитивных нарушений и депрессивных расстройств, обусловленных возрастными изменениями в мозге, на животных моделях в большей или меньшей степени повышают продукцию новых нейронов в стареющем мозге [40, 41].
Если говорить о нейрогенезе в цифрах, то рождение сотен нервных клеток в зрелом (но еще не стареющем) мозге в день в масштабах зубчатой извилины гиппокампа может показаться несущественным явлением (у грызунов зубчатая извилина содержит порядка миллиона гранулярных нейронов, у человека – порядка 10 миллионов гранулярных нейронов [42]. Однако по некоторым данным у человека за всю жизнь замещается примерно треть всех гранулярных нейронов [9], а у грызунов – около 10% [43, 44]. Эти цифры представляются уже вполне значимыми.
Все эти многочисленные факты, полученные не только при исследовании животных моделей, но и при анализе клинических образцов, показывают всю важность нейрогенеза и новорожденных нейронов как в функционировании нормального мозга, так и в патогенезе разнообразных когнитивных и эмоциональных расстройств. Поэтому, понимание молекулярных механизмов и клеточных процессов функционирования нейрогенного каскада имеет крайне важное значение для разработки эффективных видов терапии когнитивных нарушений и депрессивных расстройств различной этиологии, в том числе и при старении мозга.
НЕЙРОГЕННЫЙ КАСКАД И ЖИЗНЕННЫЙ ЦИКЛ СТВОЛОВЫХ КЛЕТОК В ГИППОКАМПЕ
Начало новым гранулярным нейронам дают стволовые клетки, тела которых расположены в субгранулярной зоне между гранулярным слоем и хилусом зубчатой извилины гиппокампа. Эти стволовые клетки часто называют клетками В типа [45] или клетками типа 1 [46, 47]. Они имеют апикальный отросток, который распространяется через всю толщу гранулярного слоя и заканчивается многочисленными ветвлениями в первой трети молекулярного слоя [48–51]. Морфологически эти стволовые клетки очень напоминают клетки радиальной глии, которые являются предшественниками всех основных типов клеток мозга и существуют только в период эмбрионального развития. В этой связи в англоязычной литературе их часто называют radial glia-like (stem) cells или RGL (stem) cells, т.е. (стволовые) клетки, похожие на радиальную глию (далее этот тип клеток мы будем называть RGL клетки (рис. 1)) [48, 52–54]. RGL клетки экспрессируют такие гены как nestin (ген-маркер нейроэктодермальных стволовых клеток, его экспрессия показана для мультипотентных клеток-предшественников разнообразных органов и тканей в развитии и в зрелом организме) и Sox2 (транскрипционный фактор, экспрессия которого важна для самообновления и поддержания клеток-предшественников нейронов и глии в развивающемся и зрелом мозге) и синтезируют белки, характерные для глиальных клеток GFAP (глиальный фибриллярный кислый белок, ) и BLBP (белок мозга, связывающий липиды) [45, 48, 49, 55, 56].
Рис. 1.
Нейрогенный каскад в зубчатой извилине гиппокампа и молекулярные маркеры основных этапов рождения новых гранулярных нейронов. Обозначения: П. RGL – покоящаяся RGL клетка (стволовая клетка), Д. RGL – делящаяся RGL клетка (стволовая клетка), ТА КП – транзиторно-амплифицирующиеся клетки-предшественники, НБ – нейробласт, МНБ – мигрирующий нейробласт, НН – новорожденный нейрон, ГН– гранулярный нейрон, МС – молекулярный слой, ГС – гранулярный слой, СГЗ – субгранулярная зона. Стрелками с прерывистой линией обозначены возможные (1) самообновление RGL клеток посредством симметричных делений и (2) чередование покоящегося и пролиферативного состояния RGL клеток.
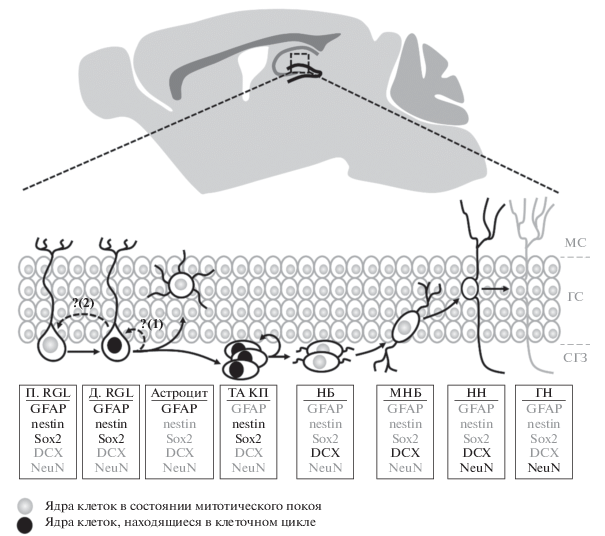
К настоящему моменту в литературе обсуждаются несколько моделей нейрогенеза, описывающих жизненный цикл стволовых клеток и последовательность клеточных событий, которые превращают их в новые нейроны гранулярного слоя (более подробно см. обзор [57]). Основные отличия моделей заключаются в описании способов деления стволовых клеток. Одни модели утверждают, что стволовые клетки в норме имеют неограниченный пролиферативный потенциал и регулярно подвергаются симметричным делениям, что обеспечивает поддержание их числа на протяжении всей жизни путем самовоспроизведения [48, 58, 59]. Другие – исключают такие явление [49, 60]. Третьи – допускают наличие симметричных делений в присутствии какого-либо физиологического или патологического стимула [51, 61]. Некоторые модели описывают чередование покоящегося и пролиферативного состояния RGL клеток или существование врéменной популяции активно делящихся RGL клеток, которая восполняется за счет входа в клеточный цикл покоящихся стволовых клеток [47, 48, 62]. Иные модели говорят о том, что гиппокампальные стволовые клетки являются стволовыми клетками однократного применения, т.е., однажды выйдя из состояния покоя, стволовые клетки подвергаются ограниченному числу делений с последующей необратимой дифференцировкой в астроциты [49, 63]. Наконец существуют модели, которые допускают наличие неких более ранних предшественников, которые дают начало RGL клеткам [56].
Если дебаты относительно способов деления RGL клеток все еще продолжаются в научной литературе, то дальнейшие этапы нейрогенного каскада, как правило, не подвергаются сомнениям (риc. 1). RGL клетки путем асимметричных делений генерируют потомков, которые уже совсем не похожи на материнские клетки [45, 48, 49, 62, 63]. В частности, они лишены апикального отростка и не синтезируют белков, характерных для глиальных клеток GFAP и BLBP [45, 49]. При этом они продолжают экспрессировать гены nestin и Sox2 и подвергаются ограниченному числу симметричных делений, увеличивая таким образом свою собственную популяцию. В литературе этот тип клеток называют транзиторно-амплифицирующимися нейральными предшественниками (transit amplifying neural progenitors) или клетками D типа (по некоторым другим классификациям эти клетки разделяют на клетки типов 2a и 2b) [15, 45, 46, 49]. Бóльшая часть этих клеток вскоре после рождения подвергается апоптозу или уничтожается клетками микроглии [64]. Оставшиеся жизнеспособными транзитороно-амплифицирующиеся клетки превращаются в даблкортин-экспрессирующие нейробласты (клетки типа 3), которые локально мигрируют и через последовательные стадии затем дифференцируются в зрелые гранулярные нейроны [46], интегрируясь в существующие нервные сети зубчатой извилины [4]. Весь нейрогенный цикл от RGL клетки до функционального гранулярного нейрона у грызунов занимает примерно 4 недели. Еще через две недели после этого отличить по морфологическим, молекулярным и электрофизиологическим признакам новорожденные гранулярные нейроны от тех, которые существуют с момента рождения, невозможно.
Оказалось, что разные физиологические и патологические стимулы действуют на различные этапы нейрогенеза. Так, например, физические нагрузки (бег в колесе), антидепрессанты, относящиеся к классу ингибиторов обратного захвата серотонина, и глубокая стимуляция мозга увеличивают число делящихся транзиторно-амплифицирующихся клеток, при этом не влияя на деление RGL клеток [23, 49]. Содержание животных в обогащенной среде, но без доступа к беговому колесу, повышает выживаемость новорожденных нейробластов, не влияя при этом на пролиферацию клеток в зубчатой извилине [27]. Электроконвульсивная терапия [65], индуцированная каинатом эпилептическая активность [51] и введение мемантина (мемaнтин – неконкурентный низкоаффинный потенциал-зависимый блокатор ионного канала NMDA-рецептора к глутамату; он применяется для улучшения повседневной деятельности у пациентов, страдающих болезнью Альцгеймера) [61] стимулируют деления RGL клеток, существенно увеличивая нейрогенез в зубчатой извилине.
БАЛАНС МЕЖДУ ПОКОЕМ И ДЕЛЕНИЕМ – КЛЮЧЕВОЙ МЕХАНИЗМ ПОДДЕРЖАНИЯ ПУЛА RGL КЛЕТОК И НЕЙРОГЕНЕЗА НА ПРОТЯЖЕНИИ ВСЕЙ ЖИЗНИ ОРГАНИЗМА
Продолжительное (т.е. существенно превышающее длительность одного клеточного цикла) пребывание в состоянии покоя (G0-фазе клеточного цикла) является одним из характерных признаков соматических стволовых клеток многих органов и тканей таких как, например, система кроветворения, поперечно-полосатая мускулатура или волосяные фолликулы [66–69]. Даже в кишечнике, ткани с очень интенсивным самообновлением, существует субпопуляция, так называемых, +4 стволовых клеток, которые делятся редко и восполняют пул активно делящихся стволовых клеток в криптах [70, 71]. Сегодня считается, что пребывание в состоянии покоя защищает геномы стволовых клеток от неизбежного накопления потенциально опасных, вызывающих рост опухолей мутаций, которые возникают в результате активной репликации ДНК. В отличие от митотического покоя терминально-дифференцированных или стареющих клеток покой стволовых клеток обратим, и под действием внутренних и/или внешних факторов покоящиеся стволовые клетки входят в клеточный цикл и делятся. Более того, покоящиеся стволовые клетки пребывают не в некоем пассивном или “спящем” состоянии, а, наоборот, они активно воспринимают разнообразные стимулы среды микроокружения за счет функционирования разнообразных клеточных сигнальных систем [66].
Несмотря на различия в описании жизненного цикла RGL клеток, все предложенные к настоящему моменту клеточные модели гиппокампального нейрогенеза сходятся в том, что большая часть RGL клеток длительно пребывает в состоянии покоя. Баланс между покоем и делением RGL клеток определяет поддержание их пула на протяжении всей жизни и темп рождения новых нервных клеток. Нарушение этого баланса, даже врéменное, может существенно изменить нейрогенез в оставшийся период жизни организма. Так, например, гиперактивность нейронов гиппокампа, вызванная введением каиновой кислоты, агониста каинатных рецепторов к глутамату, приводит к существенным изменениям в продукции новых нейронов и в работе нейрогенного каскада [51]. Вызванная гиперактивность нейронов стимулирует выход RGL клеток из состояния покоя и их деление. При невысокой дозе (0.74 мМ) каиновой кислоты (при такой дозе развивается эпилептиформная активность мозга) работа нейрогенного каскада в целом сохраняется, а стимуляция деления покоящихся RGL клеток приводит к увеличению числа новорожденных нейронов. В то же время при повышенной дозе (1 мМ) каиновой кислоты (такая доза используется для моделирования мезиальной височной эпилепсии) активация de novo деления покоящихся RGL клеток не приводит к увеличению продукции новых нервных клеток. Наоборот, гиперактивность гиппокампальных нейронов приводит к тому, что начинают доминировать симметричные деления RGL клеток. Потомки, которые получаются в результате таких симметричных делений в последствии необратимо дифференцируются астроциты с молекулярными и морфологическими признаками реактивной глии. В обоих случаях активация деления покоящихся RGL клеток вызывала ускоренное истощение пула стволовых клеток, что в свою очередь приводило существенному снижению нейрогенеза в оставшийся период жизни модельных животных.
Поиск и идентификация молекулярных механизмов, которые управляют покоем и пролиферацией RGL клеток в зубчатой извилине, имеет важное фундаментальное и практическое значение. Во-первых, необходимо объяснить – каким образом регулируется постепенный расход RGL клеток на протяжении всей жизни в условиях физиологической нормы. Иными словами, необходимо идентифицировать механизмы, которые определяют, когда те или иные RGL клетки должны выйти из состояния покоя и начать делиться в нормальном мозге. Идентификация таких механизмов позволит дополнить существующие модели гиппокампального нейрогенеза новыми фундаментальными знаниями. Во-вторых, важно понять – какие сигнальные пути оказываются задействованы при избыточной активации покоящихся RGL клеток под действием тех или иных патологических или физиологических стимулов. Определение таких сигнальных путей позволит установить потенциальные молекулярные мишени для предотвращения преждевременного истощения пула RGL клеток и, следовательно, сохранения нейрогенеза на уровне физиологической нормы. Кроме того, возможность искусственно управлять делением и покоем стволовых клеток открывает новые перспективы к использованию внутренних резервов стволовых клеток для коррекции дефицита пластичности при различных патологиях мозга.
Покой и деление RGL клеток регулируется как внутренними факторами, так и сигналами, поступающими извне. В частности, пребывание RGL клеток в покоящемся или пролиферативном состоянии зависит от транскрипционной активности целого ряда генов и клеточного метаболизма [72–76]. Кроме того, жизнь RGL клеток происходит в тесном взаимодействии c компонентами “стволовой” ниши [77]: астроцитами, гранулярными нейронами, парвальбумин-позитивными тормозными интернейронами, клетками микроглии, кровеносными сосудами и внеклеточным матриксом. Все эти компоненты “стволовой” ниши влияют на покой и деление RGL клеток и на нейрогенез в целом через контактные взаимодействия или высвобождение растворимых факторов (ростовых факторов, нейротрофинов, цитокинов, морфогенов, гормонов и т.д.) и малых молекул (нейротрасмиттеров) [77–81]. Ниже мы рассмотрим последние достижения в изучении внутренних механизмов и внешних сигналов, которые регулируют покой и деление RGL клеток зубчатой извилины гиппокампа.
АВТОНОМНАЯ РЕГУЛЯЦИЯ ПОКОЯ И ДЕЛЕНИЯ RGL КЛЕТОК ЗУБЧАТОЙ ИЗВИЛИНЫ ГИППОКАМПА
Выход RGL клеток из состояния покоя и их вхождение в клеточный цикл очевидно сопровождается существенными изменениями в транскрипционной программе. Поэтому одним из наиболее информативных подходов к тому, чтобы понять – какие внутренние механизмы регулируют пребывание RGL клеток в состоянии покоя или деления, является определение и сравнение транскрипционных профилей покоящихся и делящихся RGL клеток. Благодаря созданию репортерных линий мышей, у которых стволовые клетки “подсвечены” флуоресцентными белками, экспрессия генов которых в свою очередь находится под контролем таких промоторов, как nestin, LPA1 (ген рецептора лизофосфатидной кислоты 1, он также как и ген nestin экспрессируется в RGL клетках и транзиторно-амплифицирующихся клетках) и Hopx (ген-маркер RGL клеток зрелого гиппокампа), появилась возможность выделять RGL клетки с помощью флуоресцентно-активируемой сортировки [82–84] для последующего РНК-секвенирования популяций или даже одиночных RGL клеток. Разделение популяции RGL клеток на покоящуюся и пролиферирующую фракции проводили либо путем сортировки с использованием дополнительно флуоресцентно-меченного лиганда EGFR (рецептора эпидермального фактора роста) [84], либо путем взятия образцов разного возраста с различным содержанием делящихся и покоящихся RGL клеток [82], либо с помощью биоинформатического группирования транскрипционных профилей одиночных RGL клеток [83]. Интересно, что полученные такими разными способами наборы данных о транскрипции генов в покоящихся и делящихся RGL клетках привели различные исследовательские группы к похожим выводам и позволили предположить динамику молекулярных изменений клеток при переходе от покоящегося к пролиферативному состоянию. Так, например, в противоположность некоторым ожиданиям, покоящиеся RGL клетки в отличие от активированных RGL клеток имеют более высокий уровень экспрессии генов, относящихся к таким категориям генной онтологии, как межклеточная и внутриклеточная коммуникация [82, 83]. В частности, в покоящихся RGL клетках на высоком уровне экспрессируются гены, связанные с такими видами межклеточной коммуникации, как Notch-, BMP-, wnt-, MAPK-сигнальные пути, нейротрофины, инсулин, различные межклеточные контакты и нейротрансмиттеры глутамат и ГАМК. При этом функционирование Notch- и BMP-сигнальных путей необходимо для удержания RGL клеток в состоянии покоя [79, 85], тогда как wnt- и MAPK-сигнальные пути связаны с активацией пролиферации в зубчатой извилине гиппокампа [86, 87]. Внутриклеточная коммуникация в покоящихся RGL клетках обеспечивается продукцией белков разнообразных метаболических путей, связанных с глутатионом и биосинтезом некоторых классов липидов и жирных кислот. Эти данные в явном виде подтверждают, что “покой” RGL клеток – это функциональное состояние, при котором они активно воспринимают сигналы микроокружения и при необходимости могут ответить на внешние стимулы. С другой стороны, в покоящихся RGL клетках подавлена экспрессия генов, связанных с реорганизацией хроматина, регуляцией процессов транскрипции и трансляции, прогрессированием клеток через различные фазы клеточного цикла и синтезом субъединиц рибосом, имеющих цитоплазматическую локализацию [82, 83]. Активация же покоящихся RGL клеток сопровождается увеличением уровня экспрессии этих генов и одновременным снижением в уровне экспрессии генов, вовлеченных во внутри- и межклеточную сигнализацию. Интересно, что рост уровня экспрессии генов, кодирующих субъединицы рибосом, предшествует изменениям в уровне экспрессии генов, продукты которых участвуют управлении продвижением клетки по различным фазам клеточного цикла. Более того, было показано, что избыточная экспрессия гена, кодирующего транскрипционный фактор REST (в англ. яз. лит-ре: repressor element 1-silencing transcription factor) повышает уровень биосинтеза рибосомальных белков и активирует деление покоящихся RGL клеток [88]. Похожую картину изменения в уровне экспрессии генов, относящихся к различным категориям генной онтологии, при переходе стволовых клеток из покоящегося в пролиферативное состояние обнаружили при анализе транскриптомов отдельных клеток, выделенных из субвентрикулярной области боковых желудочков – зоны, где также происходит постнатальный нейрогенез [89].
Факторы, управляющие прохождением клетки через различные фазы клеточного цикла, очевидно являются одними из ключевых внутренних регуляторов покоя и деления RGL клеток зубчатой извилины гиппокампа. К таким факторам относятся прежде всего циклины, циклин-зависимые киназы и их ингибиторы. Биосинтез и деградация циклинов и циклин-зависимых киназ строго привязаны к определенным временным промежуткам на шкале клеточного цикла. Ингибиторы циклин-зависимых киназ управляют их активностью путем связывания и выступают в качестве негативных регуляторов пролиферации. Циклины D типа (D1, D2 и D3) в отличие от других семейств циклинов появляются первыми при активации деления покоящихся клеток, а их экспрессия зависит от присутствия митогенов [90, 91]. Они образуют комплексы с циклин-зависимыми киназами CDK4 и CDK6, таким образом активируя их [92–94]. Это обеспечивает продвижение клеток через контрольную точку (в англ. яз. лит-ре: G1 checkpoint или restriction checkpoint) в поздней G1 фазе клеточного цикла, подготавливая клетки к вступлению в фазу синтеза (S-фазу). Циклины D рассматриваются в качестве основных посредников между внешними стимулами и молекулярным аппаратом клетки, который управляет клеточным циклом. Среди трех циклинов семейства циклин D2, как оказалось, имеет наиболее важное значение для постнатального нейрогенеза как в гиппокампе, так и в субвентрикулярной зоне боковых желудочков [95]. Во-первых, только циклин D2 экспрессируется в делящихся клетках зубчатой извилины гиппокампа. Во-вторых, у животных с нокаутом именно циклина D2, но не D1 постнатальный нейрогенез в гиппокампе был многократно снижен в первые 30 дней после рождения, а позднее практически совсем отсутствовал [95, 96]. Кроме того, нокаутные по циклину D2 животные, в отличие от мышей дикого типа, не показывали увеличения пролиферации в зубчатой извилине в ответ на стимул в виде содержания их в условиях “обогащенной” среды [95]. Этот результат показывает значимость циклина D2 для передачи внешних стимулов молекулярному аппарату, управляющему клеточным циклом стволовых и транзиторно-амплифицирующихся клеток. Хотя в этих работах и не был проведен детальный анализ роли циклина D2 в активации покоящихся стволовых клеток, очевидно, что столь значимые эффекты недостатка продукции циклина D2 на нейрогенез у нокаутных животных не могут происходить без нарушения баланса между покоем и делением RGL клеток. Кроме того, у нокаутных животных обнаруживаются дефекты в развитии гранулярного слоя зубчатой извилины [96]. Это наблюдение в совокупности с почти полностью отсутствующим гиппокампальным нейрогенезом примерно после 30-го постнатального дня может также указывать на то, что функционирование циклина D2 в развивающемся мозге может быть важно для формирования и поддержания той субпопуляции клеток-предшественников эмбриональной зубчатой извилины, которая в постнатальный период должна стать пулом покоящихся RGL клеток.
Хотя нокаут гена, кодирующего циклин D1, не влияет на гиппокампальный нейрогенез [95], оказалось, что избыточная одновременная экспрессия генов, кодирующих циклин D1 и циклин-зависимую киназу СDK4, приводит к увеличению пролиферации в зубчатой извилине и нейрогенеза в целом [97]. Более того, было показано, что этот подход позволяет значительно увеличить продукцию новых нейронов в стареющем мозге, улучшая при этом показатели в тестах на пространственное и контекст-зависимое обучение [98]. Количественный анализ клеток различных этапов нейрогенного каскада показал увеличение числа RGL клеток у животных с избыточной экспрессией генов, кодирующих циклин D1 и CDK4, по сравнению с контролем [97]. Вместе эти работы показывают, что избыточная продукция циклина D1 и CDK4 обращает возрастной расход RGL клеток через стимуляцию симметричных делений.
Ингибиторы циклин-зависимых киназ p16, относящийся к семейству Ink4/ARF, и p21, p27 и p57, относящиеся к семейству Cip/Kip, оказывают влияние на пролиферацию и на нейрогенез в целом в зубчатой извилине гиппокампа [73, 99–101]. У животных, нокаутных по генам, кодирующим эти белки, пролиферация и нейрогенез в зубчатой извилине повышены. Избыточная же экспрессия генов ингибиторов циклин-зависимых киназ подавляет деление стволовых и транзиторно-амплифицирующихся клеток. Более того, было показано, что белки p27 и p57 присутствуют в ядрах большинства RGL клеток [73, 102]. Подавление экспрессии генов этих белков приводило к увеличению доли делящихся RGL клеток в зубчатой извилине и росту продукции новорожденных нейронов. В то же время в случае белка p57 было показано, что у животных с кондиционным нокаутом (экспрессия гена выключалась только в RGL клетках в определенное экспериментатором время) сначала наблюдалось увеличение числа делящихся клеток и рост продукции новых нейронов, тогда как с увеличением времени, в течение которого экспрессия гена этого белка была подавлена, нейрогенез снижался, а пул RGL клеток истощался. Результаты этих исследований демонстрируют ключевую роль внутриклеточных регуляторов клеточного цикла в поддержании баланса между покоем и делением RGL клеток. Более того, ингибиторы циклин-зависимых киназ могут играть ключевую роль в выделении субпопуляции нейральных клеток-предшественников эмбрионального мозга, которые затем станут покоящимися RGL клетками взрослого гиппокампа, иными словами, они могут управлять сменой эмбрионального нейрогенеза на взрослый нейрогенз в зубчатой извилине гиппокампа [103].
Следующая категория внутренних регуляторов покоя и деления RGL клеток – это факторы транскрипции. Дифференциальная экспрессия целого ряда генов, связанных с регуляцией транскрипции, была обнаружена при анализе транскриптомов одиночных покоящихся и делящихся RGL клеток. Так клетки с высоким уровнем экспрессии генов Sox9, Id2 и Id3 образовывали кластер покоящихся RGL клеток, тогда как экспрессия генов Egr1, Fos, Sox4, Sox11 и Ascl1 была ассоциирована с кластером делящихся RGL клеток. Среди этих факторов транскрипции Ascl1 (в англ. яз. лит-ре: achaete-scute family bHLH transcription factor 1) заслуживает особого внимания, поскольку сравнение транскриптомов активированных RGL клеток с результатами секвенирования участков, с которыми связывается Ascl1 (участки были определены путем иммунопреципитации хроматина), показало, что около 17% последовательностей, транскрибируемых в активированных RGL клеток, находятся под прямым контролем этого фактора транскрипции [72, 83]. Экспериментальные исследования роли Ascl1 в регуляции нейрогенеза с использованием кондиционного нокаутирования гена этого фактора транскрипции в RGL клетках зубчатой извилины гиппокампа показали, что покоящиеся RGL клетки в принципе не могут выйти из состояния покоя в его отсутствие даже при воздействии внешних про-нейрогенных факторов [72]. При этом гиппокампальный нейрогенез полностью блокируется. Было также обнаружено, что Asсl1 связывается с промоторной областью гена Ccnd2, кодирующего циклин D2, а у животных с кондиционным нокаутом гена Ascl1 экспрессия гена Ccnd2 оказывается подавлена. Эти результаты показывают, что Ascl1 напрямую контролирует клеточный цикл RGL клеток в зубчатой извилине гиппокампа. Ascl1 сегодня рассматривается в качестве одного из главных интеграторов внешних и внутренних сигналов, который запускает транскрипционную программу активации покоящихся RGL клеток [72], а экспрессия гена этого транскрипционного фактора является предвестником скорого начала деления покоящихся RGL клеток [63]. Интересно, что избыточная продукция Ascl1 в делящихся клетках путем доставки ретровирусного вектора с геном Ascl1, находящимся под промотором CAG, изменяет судьбу потомков стволовых клеток – они начинают дифференцироваться преимущественно в олигодендроциты, а не в гранулярные нейроны как обычно [104].
Активность Ascl1 регулируется как на уровне экспрессии его гена, так и на посттрансляционном уровне. Так, было показано, что экспрессия гена Ascl1 модулируется транскрипционным фактором Hes1 – эффектором Notch-сигнального пути и репрессором транскрипции [105, 106]. Для того, чтобы проследить – как между собой связаны экспрессии генов Ascl1 и Hes1 в покоящихся и делящихся RGL клетках, были получены трансгенные мыши, в которых активность транскрипции этих генов детектировалась с помощью флуоресцентных белков или люциферазы. Оказалось, что экспрессия гена Hes1 осциллирует как в делящихся, так и в покоящихся RGL клетках. Но в последних высота пиков и провалов в уровне экспрессии больше, чем высота пиков в делящихся RGL клетках. При этом уровень экспрессии Ascl1 в покоящихся RGL клетках остается постоянно низким, а в делящихся – колеблется в противофазе осцилляциям уровня экспрессии Hes1. Таким образом, общий высокий уровень экспрессии Hes1 подавляет экспрессию Ascl1, заставляя RGL клетки пребывать в состоянии покоя. Инактивация же Hes1 приводит к врéменному усилению пролиферации и продукции новых нейронов в зубчатой извилине гиппокампа с последующим истощением пула RGL клеток и преждевременным прекращением нейрогенеза.
Скрининг с помощью масс-спектрометрии белков, которые ко-иммунопреципитируются с фактором транскрипции Ascl1, показал взаимодействие Ascl1 c Е3-убиквитинлигазой Huwe1 [62]. Мутации в этом белке, как было показано, могут быть причиной некоторых психических заболеваний и умственной отсталости [107]. Оказалось, что Huwe1 вызывает убиквитинопосредованный распад белка Ascl1 в делящихся RGL клетках, предотвращая, таким образом, накопление циклинов D и вызывая возврат делящихся RGL клеток в состояние покоя [62]. Прямой контроль циклинов D, а также многочисленные данные о том, что экспрессия Ascl1 управляется эффекторами нескольких важных для постнатального нейрогенеза сигнальных путей таких, как Notch, Sonic Hedgehog и BMP (от англ. яз. bone morphogenic protein) [105, 108, 109], подтверждают, что фактор транскрипции Ascl1 выступает в роли, своего рода, интегратора внутренних и внешних сигналов, управляя активацией покоящихся RGL клеток.
Восприимчивость покоящихся RGL клеток к внешним стимулам сильно зависит от функционирования внутриклеточных компонентов различных сигнальных каскадов. Так, например, было показано, что избыточная продукция Notch2 (одного из рецепторов Notch-сигнального пути, функционирование которого необходимо для переключении пролиферации прогениторных клеток на их дифференцировку в развивающемся мозге) в RGL клетках снижает пролиферацию в зубчатой извилине и продукцию нейробластов, сохраняя при этом пул покоящихся RGL клеток [110]. Подавление же экспрессии гена Notch2 в RGL клетках наоборот приводило врéменному увеличению пролиферации RGL клеток и нейрогенеза с последующим существенным снижением продукции новых нейронов и истощением пула покоящихся RGL клеток. В этой экспериментальной системе нокдаун гена Id4, кодирующего один из ингибиторов ДНК-связывающих белков и являющегося компонентом Notch2 сигнального пути, восстанавливает пролиферативную активность RGL клеток, которая была подавлена избыточной продукцией Notch2 [110]. Этот результат показывает, что Id4 важен для Notch-зависимого удержания RGL клеток в состоянии покоя. Аналогичном образом поддержание покоя RGL клеток зубчатой извилины гиппокампа обеспечивают транскрипционные факторы RBPJκ, еще один эффектор Notch-сигнального пути [85], и Smad4, эффектор канонического BMP-сигнального пути [79].
Похожий эффект на пул стволовых клеток был обнаружен при кондиционном нокауте гена Notch1, кодирующего еще один рецептор Notch-сигнального пути [111]. Оказалось, что через 60 дней после подавления экспрессии гена Notch1 в RGL клетках, число таких клеток значительно снижено по сравнению с контрольными животными дикого типа. Сами авторы работы объясняют свои результаты тем, что Notch1-зависимый сигнальный путь необходим для того, чтобы RGL клетки самообновлялись через симметричные деления. В свете современных представлений о жизненном цикле RGL клеток можно предположить, что, скорее всего, Notch1-зависимый сигнальный путь важен для удержания RGL клеток в состоянии покоя нежели для самообновления.
Регуляция транскрипционной программы при переходе RGL клеток из покоящегося в пролиферативное состояние и обратно осуществляется также с помощью эпигенетических механизмов. Так, например, было показано, что функционирование фактора CHD7 (в англ. яз. лит-ре: chromodomain–helicase-DNAbinding protein 7), который относится к семейству факторов, ремоделирующих хроматин, необходимо для поддержания пула покоящихся RGL клеток [74]. Влияя на структуру и доступность ДНК, факторы, ремоделирующие хроматин, регулируют транскрипционную программу в клетках, управляя, таким образом, многими физиологическими процессами. С использованием кондиционного нокаута гена Chd7 в RGL клетках модельных животных было показано, что недостаток синтеза CHD7 приводит к врéменному усилению пролиферации и продукции новых нейронов в зубчатой извилине гиппокампа на протяжении, по крайней мере, 3 месяцев после индукции нокаута в RGL клетках. Однако, затем через 8 месяцев это приводит к истощению пула RGL клеток и существенному снижению нейрогенеза по сравнению с контрольными животными. Интересно, что ген этого фактора экспрессируется делящимися, но не покоящимися RGL клетками. В этой связи авторы предположили, что CHD7 важен для возврата делящихся RGL клеток обратно в состояние покоя [74] и, таким образом, для поддержания пула RGL клеток на протяжении всей жизни.
Помимо Huwe1-зависимого распада транскрипционного фактора Ascl1 посттрансляционный контроль других белков в RGL клетках играет важную роль в регуляции баланса между их покоем и делением. Так, изменения гиппокампального нейрогенеза были обнаружены у мышей с нокаутом по гену Dnajc5, который кодирует кошаперон CSP-α (в англ. яз. лит-ре: cochaperone cysteine string protein-α) [112]. Этот кошаперон относится к семейству белков теплового шока HSP40. Он стабилизирует белок SNAP25 (от англ. Soluble NSF Attachment Protein 25 kDa), являющийся мембранным белком аксональных терминалей и входящий в состав комплекса SNARE (от англ. Soluble NSF Attachment Receptor; функционирование этого комплекса обеспечивает накопление везикул, содержащих нейротрасмиттер, в аксональной терминали и их слияние с плазматической мембраной, обеспечивая таким образом синаптическую передачу нервного возбуждения). Кондиционное нокаутирование гена Dnajc5 в RGL клетках приводило к усилению их пролиферации с последующим истощением пула покоящихся RGL клеток. Кроме того, было показано, что гиперактивация покоящихся RGL клеток у животных, у которых отсутствует кошаперон CSP-α, опосредована активацией mTORC1-сигнального каскада. Этот результат показывает, что кошаперон CSP-α важен для удержания RGL клеток в состоянии покоя. Однако, является ли взаимодействие CSP-α с компонентами mTORC1-сигнального каскада прямым или опосредованным, еще предстоит выяснить.
Покой RGL клеток во взрослом гиппокампе контролируется через аутокринные механизмы. RGL клетки зубчатой извилины продуцируют фактор фагоцитоза Mfge8 (в англ. яз. лит-ре: Milk Fat Globule-EGF 8) и имеют на клеточной мембране неканонический рецептор к нему – интегрин α8β1. С использованием генетических инструментов было показано, что у взрослых мышей, у которых ген Mfge8 нокаутирован в клетках герминальной линии, нейрогенез снижен, а пул RGL клеток истощен [76]. Индукция же кондиционного нокаутирования Mfge8 в RGL клетках приводила к увеличению числа делящихся RGL клеток через 3 дня. В тоже время, анализ препаратов мозга животных спустя длительное время (30 дней) после индукции кондиционного нокаутирования Mfge8 показал истощение пула RGL клеток и снижение нейрогенеза. При этом увеличилась продукция астроцитов. Иными словами, нокаутирование приводило к избыточной активации покоящихся RGL клеток с последующей необратимой дифференцировкой в астроциты [49]. Аутокринный механизм действия Mfge8 на RGL клетки через интегрин β1 был подтвержден в культуре in vitro. Было установлено, что добавление рекомбинантного Mfge8 подавляло пролиферацию клеток-предшественников, выделенных из зубчатой извилины гиппокампа зрелого мозга, тогда как в присутствии антител, специфичных к интегину β1, но не α8, этот эффект пропадал.
Похожим образом работает система аутокринной регуляции, в которой протейн S (в англоязычной литературе: Protein S или PROS1), взаимодействуя с рецепторами семейства TAM (семейство протеин киназ Tyro3, Axl и Mer), удерживает RGL клетки в состоянии покоя. PROS1 известен тем, что активирует протеин C, который в свою очередь ингибирует процесс свертывания крови. Так было показано, что и PROS1, и TAM-рецепторы синтезируются в RGL клетках зубчатой извилины гиппокампа, а их нокаутирование изменяет гиппокампальный нейрогенез [113, 114]. Генетическая абляция PROS1 приводила к активации деления покоящихся RGL клеток, как оказалось, через подавление экспрессии генов, кодирующих внутриклеточные компоненты Notch-зависимого сигнального пути, Notch1-рецептор и транскрипционный фактор Hes5 [114]. Как и в случае Mfge8, нокаутирование гена PROS1 вызывало увеличение продукции астроцитов. Однако, в отличие от Mfge8, генетическая абляция PROS1 не приводила к истощению пула RGL клеток.
Состояния покоя и деления RGL клеток отличаются между собой биосинтезом специфических клеточных компонентов и функционированием определенных энергетических систем в клетках. Переход из одного состояния в другое всегда сопряжен с существенной перестройкой биосинтезов и энергетического метаболизма. В состоянии покоя RGL клетка должна обеспечивать функционирование всего многообразия путей внутри- и межклеточной сигнализации. При активации и вхождении в клеточный цикл RGL клетка запускает программу биосинтезов, необходимых для репликации геномной ДНК, удвоения жизненно важных органелл, увеличения поверхности плазмалеммы и создания аппарата клеточного деления. Сравнительно недавно появились публикации, в которых исследовался вопрос о роли метаболизма в регуляции жизненного цикла стволовых клеток. Так, с помощью фармакологического ингибирования и кондиционного нокаутирования было показано, что Fasn-зависимый липогенез (Fasn – синтаза жирных кислот, которая является одним из ключевых ферментов, катализирующих de novo синтез липидов в клетке) необходим для активации покоящихся RGL клеток и поддержания нормального уровня их пролиферации [115]. Далее было обнаружено, что для функционирования RGL клеток помимо глюкозы и лактата, традиционных источников энергии в клетках мозга, необходимо окисление жирных кислот (бета-окисление) [75]. При этом интенсивность окисления жирных кислот оказалась высокой в покоящихся RGL клетках и снижалась при их активации и переходе в пролиферативное состояние. Похожим образом с помощью фармакологического ингибирования и кондиционного нокаутирования фермента Cpt1a, ограничивающего скорость синтеза жирных кислот из молекулы ацил-CoA, было показано, что сдвиг в интенсивности окисления жирных кислот заставляет RGL выходить из состояния покоя. Тот факт, что покоящиеся RGL клетки имеют более разнообразные источники энергии по сравнению с делящимися RGL клетками, оказался совершенно новым и неожиданным. По-видимому, следствием многообразия источников энергии является и неожиданно высокий уровень активных форм кислорода (АФК) в покоящихся RGL клетках [116]. Содержание АФК, как правило, отражает уровень метаболической активности клеток. Поэтому в совокупности предыдущие работы показывают, что покоящиеся RGL клетки – это “метаболически активные” клетки. Интересно, что при переходе RGL клеток от покоящегося к пролиферативному состоянию происходит постепенное снижение уровня АФК по мере того, как RGL клетка входит в клеточный цикл и начинает делиться [116]. Вероятно, это связано с реорганизацией биосинтезов и энергетического метаболизма при активации RGL клеток. Однако, чтобы это выяснить, требуются дальнейшие исследования.
Такое многообразие механизмов автономной регуляции (рис. 2) покоя и деления RGL клеток зубчатой извилины гиппокампа, по-видимому, прежде всего необходимо для удержания RGL клеток в состоянии покоя. Это, с одной стороны, должно воспрепятствовать ускоренному истощению их пула и, следовательно, преждевременному прекращению генерации новых нервных клеток в зубчатой извилине. С другой стороны – уменьшение числа делений должно препятствовать накоплению потенциально опасных мутаций, возникающих при регулярной репликации ДНК, обеспечивая, таким образом, рождение нормальных нейронов на протяжении всей жизни и снижая риск трансформации клеток и развития опухоли. В отсутствие внешних стимулов баланс между покоем и делением RGL клеток, по-видимому, регулируется преимущественно механизмами автономной регуляции. Поэтому, идентификация новых внутренних регуляторов покоя и деления RGL клеток и исследование того, как взаимодействуют между собой уже известные внутренние регуляторы, имеют важное значение для объяснения динамики расходования пула RGL клеток на протяжении всей жизни. Кроме того, изучение механизмов автономной регуляции покоя и деления RGL клеток важно, чтобы вести поиск мишеней для терапии дефицита пластичности гиппокампа из-за снижения нейрогенеза в результате преждевременного истощения пула стволовых клеток при воздействии паталогического стимула.
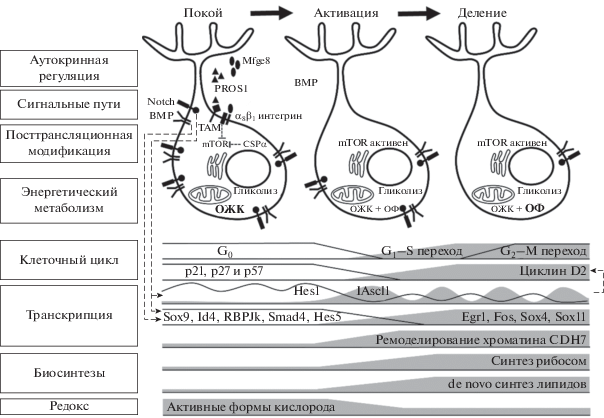
Рис. 2.
Автономные механизмы регуляции покоя и деления RGL клеток зубчатой извилины гиппокампа и графическое резюме изменений их функционирования при активации покоящихся RGL клеток. Штриховой линией показаны активирующие (▶) и подавляющие ($ \top $) взаимодействия между различными внутренними регуляторами покоя и деления RGL клеток. Рисунок адаптирован из [82, 83, 103, 106, 116]. Обозначения: ОЖК - β-окисление жирных кислот, ОФ - окислительное фосфорилирирование.
РОЛЬ СИГНАЛОВ “СТВОЛОВОЙ” НИШИ В РЕГУЛЯЦИИ ЖИЗНЕННОГО ЦИКЛА RGL КЛЕТОК
Гиппокамп и входящая в его состав зубчатая извилина являются компонентами лимбической системы мозга. Эта система получает входы всех сенсорных модальностей организма, воспринимает сигналы от внутренних органов, участвует в формировании целостного поведения организма и регулирует множество физиологических функций. Иными словами, гиппокамп и зубчатая извилина напрямую или опосредованно связанны со всеми системами организма и воспринимают разнообразные физиологические стимулы. Нейрогенез, как уже говорилось выше, восприимчив к различным физиологическим и патологическим стимулам. За последнее десятилетие появился целый ряд исследований, которые были направлены на выяснение механизмов передачи таких стимулов нейрогенному каскаду. Особое место в этом направлении исследований занимает идентификация тех молекулярных и физиологических механизмов, которые непосредственно вовлечены в регуляцию жизненного цикла (в управление покоем и делением) именно RGL клеток. Это важно для того, чтобы понимать активация или, наоборот, подавление каких сигнальных путей может приводить к патологическому преждевременному истощению пула стволовых клеток, а каких – к сохранению пула стволовых клеток и длительному поддержанию нейрогенеза.
Как показывают исследования, баланс между покоем и делением RGL клеток поддерживается нейротрофическими и ростовыми факторами, факторами клеточной адгезии, регуляторами процессов эмбрионального развития, нейромедиаторами и электрической активностью локальных нервных сетей и афферентных проекций в зубчатую извилину (рис. 3 и 4).
Рис. 3.
Внеклеточные механизмы регуляции покоя и деления RGL клеток и основные источники сигнальных молекул. Обозначения: П. RGL – покоящаяся RGL клетка (стволовая клетка), Д. RGL – делящаяся RGL клетка (стволовая клетка), НБ – нейробласт, ГН– гранулярный нейрон, А – зрелый астроцит, МГ – микроглия, МС – молекулярный слой, ГС – гранулярный слой, СГЗ – субгранулярная зона, Хил – хилус. Размер букв в названии ключевых внеклеточных сигнальных путей отражает их активность в состоянии покоя или деления RGL клеток.
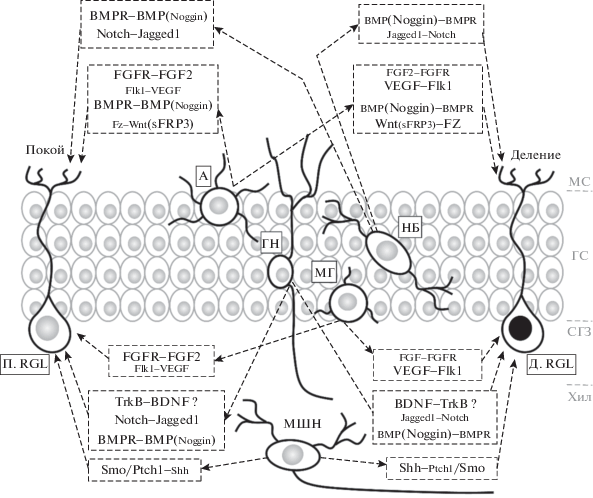
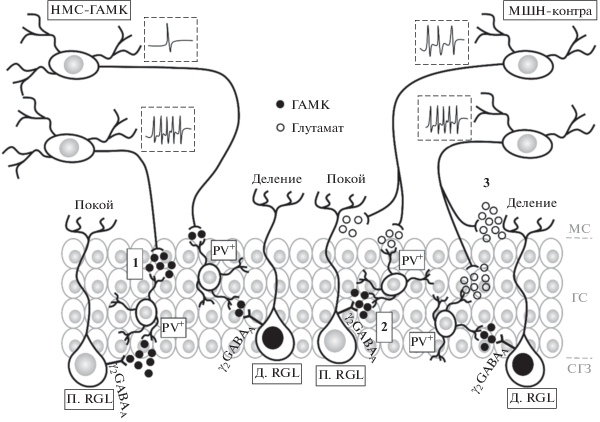
Рис. 4.
Графическое резюме роли электрической активности некоторых афферентных проекций в зубчатую извилину. 1 – ГАМК, высвобождаемый нейронами медиального септума, деполяризует тормозные парвальбумин-позитивные интернейроны зубчатой извилины. 2 – при среднем уровне электрической активности комиссуральных проекций мшистых нейронов хилуса покой RGL клеток обеспечивается опосредовано через парвальбумин-позитивные интернейроны зубчатой извилины. 3 – при высоком уровне электрической активности комиссуральных проекций мшистых нейронов хилуса активация покоящихся RGL клеток обеспечивается напрямую за счет высвобождения глутамата. Обозначения: П. RGL – покоящаяся RGL клетка (стволовая клетка), Д. RGL – делящаяся RGL клетка (стволовая клетка), PV+ – тормозные парвальбумин-позитивные интернейроны, НМС-ГАМК – ГАМКергические нейроны медиального септума, МШН-контра – мшистые нейроны хилуса контралатерального полушария, g2GABAA – ГАМКA-рецепторы, содержащих g2 субъединицу, МС – молекулярный слой, ГС – гранулярный слой, СГЗ – субгранулярная зона.
Нейротрофины представляют собой группу факторов роста, которые являются регуляторами развития и функционирования нервной системы [117–120]. Они, взаимодействуя со своими рецепторами, управляют выживанием новорожденных нервных клеток, специализацией нервных структур и дифференцировкой клеток в эмбриональном развитии, ростом и навигацией аксонов и дендритов, формированием нервных связей, регенерацией периферических нервов и т.д. Среди нейротрофинов млекопитающих, наиболее интенсивно изучалось действие белка BDNF (в англ. яз. лит-ре: brain derived neurotrophic factor) на гиппокампальный нейрогенез. Основным источником BDNF в зубчатой извилине гиппокампа являются гранулярные нейроны [121]. Однако, результаты многочисленных исследований оказались противоречивыми в отношении влияния фактора BDNF и активации его рецептора TrkB (в англ. яз. лит-ре: tropomyosin receptor kinase B) на пролиферацию и генерацию новых нейронов в зубчатой извилине [122–126]. Так, было показано, что при внутригиппокампальной инъекции BDNF стимулирует нейрогенез [125], а нокаут гена BDNF с помощью РНК-интерференции снижает нейрогенез в зубчатой извилине и приводит к формированию депрессивного поведения у животных [126]. Похожие эффекты на пролиферацию в зубчатой извилине, нейрогенез и поведение имеет кондиционный нокаут гена, кодирующего рецептор к BDNF TrkB (в англоязычной литературе: tropomyosin receptor kinase B), в GFAP-положительных клетках-предшественниках [124]. В то же время гетерозиготные мыши BDNF+/– и трансгенные мыши с избыточной экспрессией гена укороченной формы TrkB рецептора (trkB.T1) показывают увеличенную пролиферацию в субгранулярной зоне [127]. Более того, фармакологическая блокада TrkB рецептора приводила активации деления покоящихся RGL клеток [123]. Эти противоречия прежде всего связаны с различиями в способах демонстрации функциональной роли BDNF и его рецептора TrkB в нейрогенезе, а выяснение этой роли требует дополнительных исследований. Интересно, что мемантин значительно увеличивает пролиферацию RGL клеток в гиппокампе [61] и при этом усиливает экспрессию генов, кодирующих BDNF и его рецептор TrkB во взрослом мозгу [128]. Эти наблюдения могут указывать роль BDNF и TrkB в активации деления покоящихся RGL клеток.
Ростовые факторы FGF2 (основной фактор роста фибробластов, в англ. яз. лит-ре: basic fibroblast growth factor) и VEGF (фактор роста эндотелия, в англ. яз. лит-ре: vascular endothelial growth factor) оказывают стимулирующий эффект на пролиферацию в зубчатой извилине и продукцию новых нейронов в целом, что, в частности, было продемонстрировано путем их прямого введения в гиппокамп [129–131]. Основными источниками этих ростовых факторов в зубчатой извилине зрелого мозга являются астроциты и сами RGL клетки [132, 133], а по некоторым последним данным и клетки микроглии [134]. Их рецепторы присутствуют на клеточной мембране RGL клеток [86, 132]. Поэтому помимо паракринного действия можно предположить аутокринную регуляцию жизненного цикла RGL клеток факторами FGF2 и VEGF. Кондиционное нокаутирование сразу трех генов Fgfr1, Fgfr2 и Fgfr3, которые кодируют рецепторы ростового фактора FGF2, в RGL клетках приводило к снижению их общего числа по сравнению с контролем [86]. В тоже время, искусственное увеличение экспрессии Fgfr3 не влияло на общее число RGL клеток. Полученные результаты показывают, что сигналинг с участием FGF2 необходим для поддержания пула RGL клеток, а его недостаток может приводить к преждевременному истощению пула покоящихся RGL клеток. Роль фактора VEGF в жизни RGL клеток была показана на модели электроконвульсивной терапии (ЭКТ) [65]. Было установлено, что ЭКТ усиливает продукцию фактора VEGF в зубчатой извилине гиппокампа, а применение блокатора Flk1-рецептора (рецептора к VEGF) существенно снижало эффект ЭКТ на пролиферацию RGL. Кроме того, внутригиппокампальное введение рекомбинантного VEGF двукратно увеличивало число делящихся RGL клеток. Эти наблюдения показывают, что фактор VEGF напрямую способен стимулировать выход RGL клеток из состояния покоя. ЭКТ также вызывает повышение экспрессии генов, кодирующих BDNF и TrkB [121]. Поэтому возможно, что в активации деления покоящихся RGL клеток под действием ЭКТ [65] также задействован и механизм, связанный с BDNF и TrkB.
Wnt является эволюционно древним консервативным сигнальным путем, который регулирует многие процессы в организме в период эмбрионального развития [135, 136]. Взаимодействие лигандов Wnt c трансмембранными рецепторами семейства Frizzled (Fz), а также с тирозинкиназными рецепторами Ryk и Ror2, активирует канонический β-катенин-зависимый внутриклеточный каскад или неканонические каскады, вызывающие повышение внутриклеточного кальция или активацию JNK-зависимого пути [137]. Wnt-сигнальный путь регулирует нейрогенез в зубчатой извилине гиппокампа [87, 137, 138]. Лиганды Wnt, в частности Wnt-3, продуцируются астроцитами зубчатой извилины [87]. Введение в зубчатую извилину гиппокампа лентивирусных конструкций, которые кодируют либо блокатор Wnt-сигнального пути мутантный Wnt1, либо его агонист Wnt-3, показало, что активация Wnt-сигнального пути повышает продукцию новых нейронов, тогда как блокирование наоборот подавляет ее. При поиске генов, вовлеченных в активацию деления RGL клеток при ЭКТ, было обнаружено существенное снижение экспрессии гена, кодирующего белок sFRP3. Этот белок секретируется в большом количестве в межклеточное пространство нервными клетками зубчатой извилины, а при их гиперактивации происходит снижение его секреции. В межклеточном пространстве sFRP3 связывает лиганды Wnt, блокируя таким образом их взаимодействие с Fz-рецепторами. Генетическая абляция sFRP3 приводила к активации деления покоящихся RGL клеток [139]. Хотя экспериментально это не было показано, авторы исследования полагают, что sFRP3 важен для предотвращения преждевременного истощения пула покоящихся стволовых клеток. Аналогичный эффект был получен при абляции гена Dkk1 в RGL клетках зубчатой извилины. Этот ген кодирует белок dickkopf1, который связывается с ко-рецептором LRP6 рецептора Fz, блокируя таким образом Wnt-сигнальный путь. Генетическая абляция Dkk1 приводила увеличению числа делящихся RGL клеток [140]. Таким образом, блокирование Wnt-сигнального пути имеет важное значение для поддержания пула покоящихся RGL клеток и предотвращения его преждевременного истощения.
Выход RGL клеток из состояния покоя и их пролиферация управляются морфогеном Sonic Hedgehog (Shh). В зубчатой извилине гиппокампа источником белка Shh являются мшистые клетки хилуса (в англ. яз. лит-ре: hilar mossy cells) [141]. Активация Shh-зависимого сигнального пути осуществляется через взаимодействие белка Shh с рецепторным комплексом, состоящим из белков Patched1 (Ptch1) and Smoothened (Smo). Когда лиганд Shh связывается с Ptch1, ингибирующее действие, оказываемое Ptch1 на Smo, ослабляется, что приводит к активации внутриклеточного сигнального каскада. Оказалось, что конститутивное подавление экспрессии гена Ptch1 приводило помимо многочисленных структурных нарушений гиппокампа к ускоренному истощению пула покоящихся RGL клеток через активацию их пролиферации [142].
Роль BMP-зависимого и Notch-зависимого сигнальных путей в регуляции покоя и деления RGL клеток уже обсуждалась нами в предыдущем разделе. Здесь необходимо упомянуть об источниках лигандов и блокаторов этих сигнальных путей в зубчатой извилине и о том, как изменение в уровне лигандов и блокаторов влияет на покой и деление RGL клеток. Источником лигандов BMP в гиппокампе являются клетки эндотелия и микроглии [143]. В зубчатой извилине гиппокампа канонический лиганд Notch-зависимого сигнального пути белок Jagged1 был обнаружен в RGL клетках, даблкортин-позитивных нейробластах и гранулярных нейронах [144, 145]. Избыточная экспрессия гена, кодирующего антагонист BMP-лигандов Noggin, в гранулярных нейронах зубчатой извилины вызывает выход RGL клеток из состояния покоя [146], а кондиционный нокаут гена, кодирующего лиганд Notch-зависимого сигнального пути белок Jagged1, в потомках RGL клеток приводит к временному увеличению продукции новых нейронов с последующим истощением пула RGL клеток [144].
Одним из важных компонентов ниши стволовых клеток зубчатой извилины является внеклеточный матрикс. Способность гепаран-сульфата и хондроитин-сульфата связывать и удерживать факторы роста, лиганды сигнальных путей, играет важную роль в жизни гиппокампальных стволовых клеток и регуляции нейрогенеза [147, 148]. Интегрины являются трансмембранными рецепторами, которые состоят из α и β субъединиц и опосредуют контактные межклеточные взаимодействия и взаимодействия между клетками и внеклеточным матриксом. Выше мы уже обсуждали роль α8β1 интегрина в аутокринной регуляции покоя и деления RGL клеток [76]. Однако, еще в одном исследовании было показано, что кондиционное нокаутирование β1 интегрина в RGL клетках, во-первых, дезорганизует цитоархитектуру зубчатой извилины, а, во-вторых, с течением времени истощает пул RGL клеток, вызывая при этом избыточную продукцию зрелых астроцитов [149].
Вопрос о том, какие еще механизмы, которые обеспечивают контактные взаимодействия RGL клеток зубчатой извилины гиппокампа с клеточными компонентами “стволовой” ниши, участвуют в регуляции покоя и деления RGL клеток, остается открытым. В то же время, в литературе имеются данные о значимости таких белков клеточной адгезии, как VCAM1 (в англ. яз. лит-ре: vascular cell adhesion molecule 1) и N-кадгерин, в поддержании покоя стволовых клеток субвентрикулярной зоны боковых желудочков мозга [150, 151]. Ранее было показано, что гены этих белков клеточной адгезии экспрессируются астроцитами нейрогенной ниши в раннем постнатальном и зрелом гиппокампе [152]. Поэтому, можно предположить, что также, как и в субвентрикулярной зоне, эти белки клеточной адгезии будут важны для поддержания состояния покоя RGL клеток зубчатой извилины.
Жизнь RGL клеток зубчатой извилины гиппокампа протекает в окружении множества возбуждающих и тормозных нейронов. Зубчатая извилина получает множество афферентных входов из различных отделов мозга: коры, септума, ядер шва, маммилярных тел, и др. Поэтому, как полагают многие исследователи, электрическая активность афферентных проекций в зубчатую извилину и локальных нервных сетей должна оказывать влияние на нейрогенез. Действительно, в последнее время появилось достаточно много работ, которые показывают, что электрическая активность афферентных проекций в зубчатую извилину и локальных нервных сетей регулирует различные этапы нейрогенеза [80, 81, 153–157] (рис. 4). Считается, что этот механизм важен для адаптации процесса генерации новых нервных клеток в зубчатой извилине к изменениям в окружающей среде.
Если говорить о физиологических механизмах регуляции покоя и деления RGL клеток, то одним ключевых посредников между электрической активностью и RGL клетками является гамма амино-масляная кислота (ГАМК). Ее основным источником являются тормозные интернейроны, расположенные в гранулярном слое и в субгранулярной зоне зубчатой извилины [80]. Чтобы показать роль этих нейронов в регуляции покоя и деления была использована гетерологичная экспрессия светочувствительных каналов channelrhodopsin-2 (ChR2) для индукции деполяризации и halorhodopsin (eNpHR3.0) для индукции гиперполяризации в тормозных нейронах зубчатой извилины. Исследование влияния активации и подавления тормозных нейронов на деление и покой стволовых клеток с помощью оптогенетических инструментов показали следующие результаты. Во-первых, RGL клетки способны отвечать на ГАМК путем тонического изменения мембранного потенциала, и этот эффект опосредуется присутствующими на RGL клетках GABAA-рецепторов, содержащих γ2 субъединицу. Во-вторых, среди трех типов тормозных интернейронов (парвальбумин+, соматостатин+ и VIP+ (в англ. яз. лит-ре: vasoactive intestinal polypeptide) интернейроны) активация или торможение только парвальбумин+ интернейронов влияет на покой и деление RGL клеток. В-третьих, активация парвальбумин+ интернейронов удерживает RGL клетки в состоянии покоя, тогда как их торможение активирует деление покоящихся RGL клеток.
Локальные тормозные интернейроны получают синаптические входы от ГАМКергических нервных клеток медиального септума. Было показано, что в отличие от своей обычной тормозящей функции ГАМК, высвобождающийся из окончаний проекций из медиального септума, деполяризует мембрану парвальбумин+ интернейронов зубчатой извилины [154]. Функциональный анализ активации и торможения ГАМКергических проекций из медиального септума в зубчатую извилину гиппокампа с помощью методов опто- и хемогенетики показал, что электрическая активность этих проекций ведет к снижению пролиферации RGL клеток, предотвращая истощение их пула. В то же время “молчание” этих проекций активировало деление покоящихся RGL клетки, истощая их пул [154].
Комиссуральные проекции глутаматергических мшистых клеток хилуса, как оказалось, имеют прямой контакт с апикальными отростками RGL клеток [81]. Более того, они имеет синаптические контакты с локальными тормозными интернейронами зубчатой извилины. Таким образом, они могут на прямую и опосредовано влиять на жизненный цикл RGL клеток. Оказалось, что разная степень активации этих проекций по-разному влияет на покой и деление RGL клеток [81]. Различная степень активации достигалась с помощью метода хемогенетики путем доставки различных концентраций клозапин-N-оксида к контралатеральным проекциям мшистых клеток, в которых продуцировался модифицированный возбуждающий ацетилхолиновый рецептор hM3Dq. Если активировать проекции низкой дозой клозапин-N-оксида, то это приводит к доминированию пути, опосредованного через тормозные нейроны и высвобождение ГАМК, что приводит к удержанию RGL клеток в состоянии покоя. В случае введения высокой дозы клозапина-N-оксида начинает доминировать прямой путь активации RGL клеток с высвобождением глутамата. Это приводит к повышенной пролиферации RGL клеток и преждевременном истощению их пула.
Таким образом, к настоящему моменту удалось определить достаточно много внеклеточных путей регуляции покоя и деления RGL клеток (рис. 3 и 4), а для некоторых из них еще и установить роль в передаче конкретных стимулов RGL клеткам. В то же время сегодня отсутствуют данные о роли нейроиммунных взаимодействий и гормонов стресса в регуляции жизненного цикла стволовых клеток, хотя их значение для гиппокампального нейрогенеза уже было неоднократно продемонстрировано. Интересно, что, несмотря на обилие внеклеточных сигнальных систем, регулирующих покой и деление RGL клеток, описано не так много реальных стимулов, которые бы влияли на жизненный цикл именно RGL клеток. К таким стимулам относятся электроконвульсивный шок, по-видимому, некоторые модели эпилепсии, а также введение препарата мемантин. Все эти стимулы, вызывают избыточную активацию делений RGL клеток, приводя в конечном счете к истощению их пула и снижению базального уровня нейрогенеза в оставшийся период жизни организма. При этом полностью отсутствуют данные о каких-либо стимулах, которые, наоборот, сохраняли бы пул RGL клеток, обеспечивая более высокий базальный уровень нейрогенеза при старении.
ЗАКЛЮЧЕНИЕ
Баланс между покоем и делением стволовых клеток зубчатой извилины гиппокампа регулируется разнообразными автономными и внешними механизмами. Поддержание покоя гиппокампальных стволовых клеток важно, с одной стороны, для защиты генетического материала от накопления потенциально опасных мутаций, которые могут возникнуть в результате регулярной репликации ДНК, а с другой – для обеспечения продукции новых нейронов на протяжении всей жизни организма. Многообразие автономных и внешних механизмов, по-видимому, необходимо для удержания стволовых клеток в состоянии покоя во избежание преждевременного истощения их пула. В то же время способность стволовых клеток отвечать на изменение электрической активности локальных нервных сетей и афферентных проекций в гиппокамп имеет важное значение для адаптации процесса рождения новых нервных клеток к актуальным обстоятельствам внешней среды. Углубление знаний в этой области и создание подходов для контролируемого управления покоем и делением гиппокампальных стволовых клеток поможет в будущем разработать терапию для коррекции дефицита пластичности гиппокампа при разнообразных патологических состояниях и старении мозга.
Список литературы
Gould E., Reeves A.J., Fallah M., Tanapat P., Gross C.G., Fuchs E. // Proc. Natl. Acad. Sci. 1999. V. 96. № 9. P. 5263–5267.
Kuhn H.G., Dickinson-Anson H., Gage F.H. // J. Neurosci. 1996. V. 16. № 6. P. 2027–2033.
Siwak-Tapp C.T., Head E., Muggenburg B.A., Milgram N.W., Cotman C.W. // Neurobiol. Learn. Mem. 2007. V. 88. № 2. P. 249–259.
Van Praag H., Schinder A.F., Christle B.R., Toni N., Palmer T.D., Gage F.H. // Nature. 2002. V. 415. № 6875. P. 1030–1034.
Boldrini M., Fulmore C.A., Tartt A.N., Simeon L.R., Pavlova I., Poposka V., Rosoklija G.B., Stankov A., Arango V., Dwork A.J., Hen R., Mann J.J. // Cell. 2018. V. 22. № 4. P. 589–599.e5.
Eriksson P.S., Perfilieva E., Björk-Eriksson T., Alborn A.M., Nordborg C., Peterson D.A., Gage F.H. // Nat. Med. 1998. V. 4. № 11. P. 1313–1317.
Knoth R., Singec I., Ditter M., Pantazis G., Capetian P., Meyer R.P., Horvat V., Volk B., Kempermann G. // PLoS One. 2010. V. 5. № 1. P. e8809.
Moreno-Jiménez E.P., Flor-García M., Terreros-Roncal J., Rábano A., Cafini F., Pallas-Bazarra N., Ávila J., Llorens-Martín M. // Nat. Med. 2019. V. 25. № 4. P. 554–560.
Spalding K.L., Bergmann O., Alkass K., Bernard S., Salehpour M., Huttner H.B., Boström E., Westerlund I., Vial C., Buchholz B.A., Possnert G., Mash D.C., Druid H., Frisén J. // Cell. 2013. V. 153. № 6. P. 1219.
Tobin M.K., Musaraca K., Disouky A., Shetti A., Bheri A., Honer W.G., Kim N., Dawe R.J., Bennett D.A., Arfanakis K., Lazarov O. // Cell Stem Cell. 2019. V. 24. № 6. P. 974–982.
Altman J. // Science. 1962. V. 135. № 3509. P. 1127–1128.
Altman J. // Anat. Rec. 1963. V. 145. № 4. P. 573–591.
Altman J., Das G.D. // J. Comp. Neurol. 1965. V. 124. № 3. P. 319–335.
Anacker C., Hen R. // Nature Reviews Neuroscience. 2017. V. 18. № 6. P. 335–346.
Gonçalves J.T., Schafer S.T., Gage F.H. // Cell. 2016. V. 167. № 4. P. 897–914.
Schoenfeld T.J., Cameron H.A. // Neuropsychopharmacology. 2015. V. 40. № 1. P. 113–128.
Snyder J.S., Soumier A., Brewer M., Pickel J., Cameron H.A. // Nature. 2011. V. 476. № 7361. P. 458–462.
Saxe M.D., Battaglia F., Wang J.W., Malleret G., David D.J., Monckton J.E., Garcia A.D.R., Sofroniew M.V., Kandel E.R., Santarelli L., Hen R., Drew M.R. // Proc. Natl. Acad. Sci. 2006. V. 103. № 46. P. 17501–17506.
Tunc-Ozcan E., Peng C.Y., Zhu Y., Dunlop S.R., Contractor A., Kessler J.A. // Nat. Commun. 2019. V. 10. № 1. P. 1–9.
Nakashiba T., Cushman J.D., Pelkey K.A., Renaudineau S., Buhl D.L., McHugh T.J., Barrera V.R., Chittajallu R., Iwamoto K.S., McBain C.J., Fanselow M.S., Tonegawa S. // Cell. 2012. V. 149. № 1. P. 188–201.
Sahay A., Scobie K.N., Hill A.S., O’Carroll C.M., Kheirbek M.A., Burghardt N.S., Fenton A.A., Dranovsky A., Hen R. // Nature. 2011. V. 472. № 7344. P. 466–470.
Anderson M.L., Sisti H.M., Curlik D.M., Shors T.J. // Eur. J. Neurosci. 2011. V. 33. № 1. P. 175–181.
Encinas J.M., Hamani C., Lozano A.M., Enikolopov G. // J. Comp. Neurol. 2011. V. 519. № 1. P. 6–20.
Gould E., Beylin A., Tanapat P., Reeves A., Shors T.J. // Nat. Neurosci. 1999. V. 2. № 3. P. 260–265.
Malberg J.E., Eisch A.J., Nestler E.J., Duman R.S. // J. Neurosci. 2000. V. 20. № 24. P. 9104–9110.
Santarelli L., Saxe M., Gross C., Surget A., Battaglia F., Dulawa S., Weisstaub N., Lee J., Duman R., Arancio O., Belzung C., Hen R. // Science. 2003. V. 301. № 5634. P. 805–809.
Van Praag H., Kempermann G., Gage F.H. // Nat. Neurosci. 1999. V. 2. № 3. P. 266–270.
Veena J., Srikumar B.N., Mahati K., Bhagya V., Raju T.R., Shankaranarayana Rao B.S. // J. Neurosci. Res. 2009. V. 87. № 4. P. 831–843.
Gould E., McEwen B.S., Tanapat P., Galea L.A.M., Fuchs E. // J. Neurosci. 1997. V. 17. № 7. P. 2492–2498.
Ngwenya L.B., Danzer S.C. // Front. Neurosci. 2019. V. 13. № 1014. P. 1–8.
Pereira Dias G., Hollywood R., Bevilaqua M.C. do N., da Luz A.C.D. da S., Hindges R., Nardi A.E., Thuret S. // Neuro. Oncol. 2014. V. 16. № 4. P. 476–492.
Raber J., Rola R., LeFevour A., Morhardt D., Curley J., Mizumatsu S., VandenBerg S.R., Fike J.R. // Radiat. Res. 2004. V. 162. № 1. P. 39–47.
Reif A., Fritzen S., Finger M., Strobel A., Lauer M., Schmitt A., Lesch K.P. // Mol. Psychiatry. 2006. V. 11. № 5. P. 514–522.
Sahay A., Hen R. // Growth Factors and Psychiatric Disorders. 2008. V. 289. P. 152–160.
Marxreiter F., Regensburger M., Winkler J. // CMLS. 2013. V. 70. № 3. P. 459–473.
Woitke F., Ceanga M., Rudolph M., Niv F., Witte O.W., Redecker C., Kunze A., Keiner S. // PLoS One. 2017. V. 12. № 9. P. e0183463.
Drew M., Hen R. // CNS Neurol. Disord. 2008. V. 6. № 3. P. 205–218.
Miller B.R., Hen R. // Curr. Opin. Neurobiol. 2015. V. 30. P. 51–58.
Erickson C.A., Barnes C.A. // Experimental Gerontology. 2003. V. 38. № 1–2. P. 61–69.
Kronenberg G., Bick-Sander A., Bunk E., Wolf C., Ehninger D., Kempermann G. // Neurobiol. Aging. 2006. V. 27. № 10. P. 1505–1513.
Dayu Sun, Junhua Chen, Xiaohang Bao, Yulong Cai, Jinghui Zhao, Jing Huang, Wei Huang, Xiaotang Fan, Haiwei Xu // Mol. Neurobiol. 2015. V. 52. № 1. P. 464–477.
Jonas P., Lisman J. // Front. Neural Circuits. 2014. V. 8. P. 107
Imayoshi I., Sakamoto M., Ohtsuka T., Takao K., Miyakawa T., Yamaguchi M., Mori K., Ikeda T., Itohara S., Kageyama R. // Nat. Neurosci. 2008. V. 11. № 10. P. 1153–1161.
Ninkovic J., Mori T., Götz M. // J. Neurosci. 2007. V. 27. № 40. P. 10906–10911.
Seri B., García-Verdugo J.M., McEwen B.S., Alvarez-Buylla A. // J. Neurosci. 2001. V. 21. № 18. P. 7153–7160.
Kempermann G., Jessberger S., Steiner B., Kronenberg G. // Trends Neurosci. 2004. V. 27. № 8. P. 447–452.
Lugert S., Basak O., Knuckles P., Haussler U., Fabel K., Götz M., Haas C.A., Kempermann G., Taylor V., Giachino C. // Cell Stem Cell. 2010. V. 6. № 5. P. 445–456.
Bonaguidi M.A., Wheeler M.A., Shapiro J.S., Stadel R.P., Sun G.J., Ming G.L., Song H. // Cell. 2011. V. 145. № 7. P. 1142–1155.
Encinas J.M., Michurina T. V., Peunova N., Park J.H., Tordo J., Peterson D.A., Fishell G., Koulakov A., Enikolopov G. // Cell Stem Cell. 2011. V. 8. № 5. P. 566–579.
Mignone J.L., Kukekov V., Chiang A.S., Steindler D., Enikolopov G. // J. Comp. Neurol. 2004. V. 469. № 3. P. 311–324.
Sierra A., Martín-Suárez S., Valcárcel-Martín R., Pascual-Brazo J., Aelvoet S.A., Abiega O., Deudero J.J., Brewster A.L., Bernales I., Anderson A.E., Baekelandt V., Maletić-Savatić M., Encinas J.M. // Cell Stem Cell. 2015. V. 16. № 5. P. 488–503.
Gebara E., Bonaguidi M.A., Beckervordersandforth R., Sultan S., Udry F., Gijs P.J., Lie D.C., Ming G.L., Song H., Toni N. // Stem Cells. 2016. V. 34. № 4. P. 997–1010.
Moss J., Gebara E., Bushong E.A., Sánchez-Pascual I., O’Laoi R., El M’Gharia I., Kocher-Braissant J., Ellisman M.H., Toni N. // Proc. Natl. Acad. Sci. 2016. V. 113. № 18. P. E2536–E2545.
Shtaya A., Sadek A.R., Zaben M., Seifert G., Pringle A., Steinhäuser C., Gray W.P. // Glia. 2018. V. 66. № 11. P. 2397–2413.
Nicola Z., Fabel K., Kempermann G. // Front. Neuroanat. 2015. V. 9. № 53. P. 1–13
Suh H., Consiglio A., Ray J., Sawai T., D’Amour K.A., Gage F.H.H. // Cell Stem Cell. 2007. V. 1. № 5. P. 515–528.
Lazutkin A., Podgorny O., Enikolopov G. // Behav. Brain Res. 2019. V. 374. P. 112-118.
Brandt M.D., Jessberger S., Steiner B., Kronenberg G., Reuter K., Bick-Sander A., Von Der Behrens W., Kempermann G. // Mol. Cell. Neurosci. 2003. V. 24. № 3. P. 603–613.
Hattiangady B., Shetty A.K. // Neurobiol. Aging. 2008. V. 29. № 1. P. 129–147.
Mineyeva O.A., Enikolopov G., Koulakov A.A. // Sci. Rep. 2018. V. 8. № 1. P. 1–12.
Namba T., Maekawa M., Yuasa S., Kohsaka S., Uchino S. // Glia. 2009. V. 57. № 10. P. 1082–1090.
Urbán N., Van Den Berg D.L.C., Forget A., Andersen J., Demmers J.A.A., Hunt C., Ayrault O., Guillemot F. // Science. 2016. V. 353. № 6296. P. 292–295.
Pilz G.A., Bottes S., Betizeau M., Jörg D.J., Carta S., Simons B.D., Helmchen F., Jessberger S. // Science. 2018. V. 359. № 6376. P. 658–662.
Sierra A., Encinas J.M., Deudero J.J.P., Chancey J.H., Enikolopov G., Overstreet-Wadiche L.S., Tsirka S.E., Maletic-Savatic M. // Cell Stem Cell. 2010. V. 7. № 4. P. 483–495.
Segi-Nishida E., Warner-Schmidt J.L., Duman R.S. // Proc. Natl. Acad. Sci. 2008. V. 105. № 32. P. 11352–11357.
Cheung T.H., Rando T.A. // Nat. Rev. Mol. Cell Biol. 2013. V. 14. № 6. P. 329–340.
Dhawan J., Rando T.A. // Trends Cell Biol. 2005. V. 15. № 12. P. 666–673.
Lay K., Kume T., Fuchs E. // Proc. Natl. Acad. Sci. 2016. V. 113. № 11. P. 1506–1515.
Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E., Lió P., MacDonald H.R., Trumpp A. // Cell. 2008. V. 135. № 6. P. 1118–1129.
Clevers H. // Cell. 2013. V. 154. № 2. P. 274.
Li N., Clevers H. // Science. 2010. V. 327. № 5965. P. 542–545.
Andersen J., Urbán N., Achimastou A., Ito A., Simic M., Ullom K., Martynoga B., Lebel M., Göritz C., Frisén J., Nakafuku M., Guillemot F. // Neuron. 2014. V. 83. № 5. P. 1085–1097.
Furutachi S., Matsumoto A., Nakayama K.I., Gotoh Y. // EMBO J. 2013. V. 32. № 7. P. 970–981.
Jones K.M., Saric N., Russell J.P., Andoniadou C.L., Scambler P.J., Basson M.A. // Stem Cells. 2015. V. 33. № 1. P. 196–210.
Knobloch M., Pilz G.A., Ghesquière B., Kovacs W.J., Wegleiter T., Moore D.L., Hruzova M., Zamboni N., Carmeliet P., Jessberger S. // Cell Rep. 2017. V. 20. № 9. P. 2144–2155.
Zhou Y., Bond A.M., Shade J.E., Zhu Y., Davis C.-H.O., Wang X., Su Y., Yoon K.-J., Phan A.T., Chen W.J., Oh J.H., Marsh-Armstrong N., Atabai K., Ming G.-L., Song H. // Cell Stem Cell. 2018. V. 23. № 3. P. 444–452.
Urbach A., Witte O.W. // Front. Cell Dev. Biol. 2019. V. 7. № 55. P. 1–22.
Cope E.C., Gould E. // Cell Stem Cell. 2019. V. 24. № 5. P. 690–705.
Mira H., Andreu Z., Suh H., Chichung Lie D., Jessberger S., Consiglio A., Emeterio J.S., Hortigüela R., Marqués-Torrejón M.Á., Nakashima K., Colak D., Götz M., Fariñas I., Gage F.H. // Cell Stem Cell. 2010. V. 7. № 1. P. 78–89.
Song J., Zhong C., Bonaguidi M.A., Sun G.J., Hsu D., Gu Y., Meletis K., Huang Z.J., Ge S., Enikolopov G., Deisseroth K., Luscher B., Christian K.M., Ming G.L., Song H. // Nature. 2012. V. 489. № 7414. P. 150–154.
Yeh C.Y., Asrican B., Moss J., Quintanilla L.J., He T., Mao X., Cassé F., Gebara E., Bao H., Lu W., Toni N., Song J. // Neuron. 2018. V. 99. № 3. P. 493–510.
Berg D.A., Su Y., Jimenez-Cyrus D., Patel A., Huang N., Morizet D., Lee S., Shah R., Ringeling F.R., Jain R., Epstein J.A., Wu Q.F., Canzar S., Ming G.L., Song H., Bond A.M. // Cell. 2019. V. 177. № 3. P. 654–668.
Shin J., Berg D.A., Zhu Y., Shin J.Y., Song J., Bonaguidi M.A., Enikolopov G., Nauen D.W., Christian K.M., Ming G.L., Song H. // Cell Stem Cell. 2015. V. 17. № 3. P. 360–372.
Walker T.L., Overall R.W., Vogler S., Sykes A.M., Ruhwald S., Lasse D., Ichwan M., Fabel K., Kempermann G. // Stem Cell Reports. 2016. V. 6. № 4. P. 552–565.
Ehm O., Göritz C., Covic M., Schäffner I., Schwarz T.J., Karaca E., Kempkes B., Kremmer E., Pfrieger F.W., Espinosa L., Bigas A., Giachino C., Taylor V., Frisén J., Lie D.C. // J. Neurosci. 2010. V. 30. № 41. P. 13794–13807.
Kang W., Hébert J.M. // J. Neurosci. 2015. V. 35. № 28. P. 10217–10223.
Lie D.C., Colamarino S.A., Song H.J., Désiré L., Mira H., Consiglio A., Lein E.S., Jessberger S., Lansford H., Dearie A.R., Gage F.H. // Nature. 2005. V. 437. № 7063. P. 1370–1375.
Mukherjee S., Brulet R., Zhang L., Hsieh J. // Nat. Commun. 2016. V. 7. № 1. P. 1–14.
Llorens-Bobadilla E., Zhao S., Baser A., Saiz-Castro G., Zwadlo K., Martin-Villalba A. // Cell Stem Cell. 2015. V. 17. № 3. P. 329–340.
Matsushime H., Roussel M.F., Ashmun R.A., Sherr C.J. // Cell. 1991. V. 65. № 4. P. 701–713.
Matsushime H., Quelle D.E., Shurtleff S.A., Shibuya M., Sherr C.J., Kato J.Y. // Mol. Cell. Biol. 1994. V. 14. № 3. P. 2066–2076.
Matsushime H., Ewen M.E., Strom D.K., Kato J.Y., Hanks S.K., Roussel M.F., Sherr C.J. // Cell. 1992. V. 71. № 2. P. 323–334.
Meyerson M., Harlow E. // Mol. Cell. Biol. 1994. V. 14. № 3. P. 2077–2086.
Sherr C.J., Roberts J.M. // Genes Dev. 2004. V. 18. № 22. P. 2699–2711.
Kowalczyk A., Filipkowski R.K., Rylski M., Wilczynski G.M., Konopacki F.A., Jaworski J., Ciemerych M.A., Sicinski P., Kaczmarek L. // J. Cell Biol. 2004. V. 167. № 2. P. 209–213.
Ansorg A., Witte O.W., Urbach A. // BMC Neurosci. 2012. V. 13. № 1. P. 46.
Artegiani B., Lindemann D., Calegari F. // J. Exp. Med. 2011. V. 208. № 5. P. 937–948.
Berdugo-Vega G., Arias-Gil G., López-Fernández A., Artegiani B., Wasielewska J.M., Lee C.C., Lippert M.T., Kempermann G., Takagaki K., Calegari F. // Nat. Commun. 2020. V. 11. № 1. P. 1–12.
Le O., Palacio L., Bernier G., Batinic-Haberle I., Hickson G., Beauséjour C. // Stem Cell Reports. 2018. V. 10. № 6. P. 1721–1733.
Pechnick R.N., Zonis S., Wawrowsky K., Pourmorady J., Chesnokova V. // Proc. Natl. Acad. Sci. 2008. V. 105. № 4. P. 1358–1363.
Qiu J., Takagi Y., Harada J., Topalkara K., Wang Y., Sims J.R., Zheng G., Huang P., Ling Y., Scadden D.T., Moskowitz M.A., Cheng T. // Stem Cells. 2009. V. 27. № 4. P. 920–927.
Andreu Z., Khan M.A., González-Gõmez P., Negueruela S., Hortigüela R., San Emeterio J., Ferrõn S.R., Martínez G., Vidal A., Fariñas I., Lie D.C., Mira H. // Stem Cells. 2015. V. 33. № 1. P. 219–229.
Morales A. V., Mira H. // Front. Cell Dev. Biol. 2019. V. 7. № 96. P. 1–10.
Jessberger S., Toni N., Clemenson G.D., Ray J., Gage F.H. // Nat. Neurosci. 2008. V. 11. № 8. P. 888–893.
Imayoshi I., Sakamoto M., Yamaguchi M., Mori K., Kageyama R. // J. Neurosci. 2010. V. 30. № 9. P. 3489–3498.
Sueda R., Imayoshi I., Harima Y., Kageyama R. // Genes Dev. 2019. V. 33. № 9–10. P. 511–523.
Froyen G., Corbett M., Vandewalle J., Jarvela I., Lawrence O., Meldrum C., Bauters M., Govaerts K., Vandeleur L., Van Esch H., Chelly J., Sanlaville D., van Bokhoven H., Ropers H.H., Laumonnier F., Ranieri E., Schwartz C.E., Abidi F., Tarpey P.S. et al. // Am. J. Hum. Genet. 2008. V. 82. № 2. P. 432–443.
Voronova A., Fischer A., Ryan T., Al Madhoun A., Skerjanc I.S. // PLoS One. 2011. V. 6. № 4. P. e19174.
Morikawa Y., Zehir A., Maska E., Deng C., Schneider M.D., Mishina Y., Cserjesi P. // Development. 2009. V. 136. № 21. P. 3575–3584.
Zhang R., Boareto M., Engler A., Louvi A., Giachino C., Iber D., Taylor V. // Cell Rep. 2019. V. 28. № 6. P. 1485–1498.
Ables J.L., DeCarolis N.A., Johnson M.A., Rivera P.D., Gao Z., Cooper D.C., Radtke F., Hsieh J., Eisch A.J. // J. Neurosci. 2010. V. 30. № 31. P. 10484–10492.
Nieto-González J.L., Gómez-Sánchez L., Mavillard F., Linares-Clemente P., Rivero M.C., Valenzuela-Villatoro M., Muñoz-Bravo J.L., Pardal R., Fernández-Chacón R. // Proc. Natl. Acad. Sci. 2019. V. 116. № 16. P. 8000–8009.
Ji R., Meng L., Li Q., Lu Q. // Metabolic Brain Disease. 2015. V. 30. № 3. P. 633–644.
Zelentsova K., Talmi Z., Abboud-Jarrous G., Sapir T., Capucha T., Nassar M., Burstyn-Cohen T. // Stem Cells. 2017. V. 35. № 3. P. 679–693.
Knobloch M., Braun S.M.G., Zurkirchen L., Von Schoultz C., Zamboni N., Araúzo-Bravo M.J., Kovacs W.J., Karalay Ö., Suter U., MacHado R.A.C., Roccio M., Lutolf M.P., Semenkovich C.F., Jessberger S. // Nature. 2013. V. 493. № 7431. P. 226–230.
Adusumilli V.S., Walker T.L., Overall R.W., Klatt G.M., Zeidan S.A., Fischer T.J., Zocher S., Sykes A.M., Reinhardt S., Dahl A., Kirova D.G., Mansfeld J., Rünker A.E., Kempermann G. // BioRxiv. 2019. P. 606186.
Huang E.J., Reichardt L.F. // Annu. Rev. Neurosci. 2001. V. 24. № 1. P. 677–736.
Ceni C., Unsain N., Zeinieh M.P., Barker P.A. // Handb. Exp. Pharmacol. 2014. V. 220. P. 193–221.
Hallböök F. // Curr. Opin. Neurobiol. 1999. V. 9. № 5. P. 616–621.
Maisonpierre P.C., Belluscio L., Friedman B., Alderson R.F., Wiegand S.J., Furth M.E., Lindsay R.M., Yancopoulos G.D. // Neuron. 1990. V. 5. № 4. P. 501–509.
Nibuya M., Morinobu S., Duman R.S. // J. Neurosci. 1995. V. 15. № 11. P. 7539–7547.
Bergami M., Rimondini R., Santi S., Blum R., Götz M., Canossa M. // Proc. Natl. Acad. Sci. 2008. V. 105. № 40. P. 15570–15575.
Groves N., O’Keeffe I., Lee W., Toft A., Blackmore D., Bandhavkar S., Coulson E.J., Bartlett P.F., Jhaveri D.J. // Dev. Neurobiol. 2019. V. 79. № 9–10. P. 868–879.
Li Y., Luikart B.W., Birnbaum S., Chen J., Kwon C.H., Kernie S.G., Bassel-Duby R., Parada L.F. // Neuron. 2008. V. 59. № 3. P. 399–412.
Scharfman H., Goodman J., Macleod A., Phani S., Antonelli C., Croll S. // Exp. Neurol. 2005. V. 192. № 2. P. 348–356.
Taliaz D., Stall N., Dar D.E., Zangen A. // Mol. Psychiatry. 2010. V. 15. № 1. P. 80–92.
Sairanen M., Lucas G., Ernfors P., Castrén M., Castrén E. // J. Neurosci. 2005. V. 25. № 5. P. 1089–1094.
Marvanová M., Lakso M., Pirhonen J., Nawa H., Wong G., Castrén E. // Mol. Cell. Neurosci. 2001. V. 18. № 3. P. 247–258.
Woodbury M.E., Ikezu T. // J. Neuroimmune Pharmacology. 2014. V. 9. № 2. P. 92–101.
Jin K., Zhu Y., Sun Y., Mao X.O., Xie L., Greenberg D.A. // Proc. Natl. Acad. Sci. 2002. V. 99. № 18. P. 11946–11950.
Rai K.S., Hattiangady B., Shetty A.K. // Eur. J. Neurosci. 2007. V. 26. № 7. P. 1765–1779.
Bernal G.M., Peterson D.A. // Aging Cell. 2011. V. 10. № 3. P. 466–482.
Yang S.Z., Zhang L.M., Huang Y.L., Sun F.Y. // Anat. Rec. – Part A Discov. Mol. Cell. EV. 2003. V. 274. № 1. P. 851–856.
Diaz-Aparicio I., Paris I., Sierra-Torre V., Plaza-Zabala A., Rodríguez-Iglesias N., Márquez-Ropero M., Beccari S., Huguet P., Abiega O., Alberdi E., Matute C., Bernales I., Schulz A., Otrokocsi L., Sperlagh B., Happonen K.E., Lemke G., Maletic-Savatic M., Valero J. et al. // J. Neurosci. 2020. V. 40. № 7. P. 1453–1482.
Komiya Y., Habas R. // Organogenesis. 2008. V. 4. № 2. P. 68–75.
Gordon M.D., Nusse R. // J. Biol. Chem. 2006. V. 281. № 32. P. 22429–22433.
Zhang L., Yang X., Yang S., Zhang J. // Eur. J. Neurosci. 2011. V. 33. № 1. P. 1–8.
Varela-Nallar L., Inestrosa N.C. // Front. Cell. Neurosci. 2013. V. 7. № 100. P. 1–11.
Jang M.H., Bonaguidi M.A., Kitabatake Y., Sun J., Song J., Kang E., Jun H., Zhong C., Su Y., Guo J.U., Wang M.X., Sailor K.A., Kim J.Y., Gao Y., Christian K.M., Ming G.L., Song H. // Cell Stem Cell. 2013. V. 12. № 2. P. 215–223.
Seib D.R.M., Corsini N.S., Ellwanger K., Plaas C., Mateos A., Pitzer C., Niehrs C., Celikel T., Martin-Villalba A. // Cell Stem Cell. 2013. V. 12. № 2. P. 204–214.
Gonzalez-Reyes L.E., Chiang C.C., Zhang M., Johnson J., Arrillaga-Tamez M., Couturier N.H., Reddy N., Starikov L., Capadona J.R., Kottmann A.H., Durand D.M. // Sci. Rep. 2019. V. 9. № 1. P. 17402.
Antonelli F., Casciati A., Pazzaglia S. // Neural Regen. Res. 2019. V. 14. № 1. P. 59–61.
Yousef H., Morgenthaler A., Schlesinger C., Bugaj L., Conboy I.M., Schaffer D. V. // Stem Cells. 2015. V. 33. № 5. P. 1577–1588.
Lavado A., Oliver G. // Dev. Biol. 2014. V. 388. № 1. P. 11–21.
Stump G., Durrer A., Klein A.-L., Lütolf S., Suter U., Taylor V. // Mech. Dev. 2002. V. 114. № 1–2. P. 153–159.
Bonaguidi M.A., Peng C.Y., McGuire T., Falciglia G., Gobeske K.T., Czeisler C., Kessler J.A. // J. Neurosci. 2008. V. 28. № 37. P. 9194–9204.
Akita K., von Holst A., Furukawa Y., Mikami T., Sugahara K., Faissner A. // Stem Cells. 2008. V. 26. № 3. P. 798–809.
Sirko S., Von Holst A., Wizenmann A., Götz M., Faissner A. // Development. 2007. V. 134. № 15. P. 2727–2738.
Huang Z., Shimazu K., Woo N.H., Zang K., Müller U., Lu B., Reichardt L.F. // J. Neurosci. 2006. V. 26. № 43. P. 11208–11219.
Kokovay E., Wang Y., Kusek G., Wurster R., Lederman P., Lowry N., Shen Q., Temple S. // Cell Stem Cell. 2012. V. 11. № 2. P. 220–230.
Porlan E., Martí-Prado B., Morante-Redolat J.M., Consiglio A., Delgado A.C., Kypta R., López-Otín C., Kirstein M., Fariñas I. // Nat. Cell Biol. 2014. V. 16. № 7. P. 629–638.
Barkho B.Z., Song H., Aimone J.B., Smrt R.D., Kuwabara T., Nakashima K., Gage F.H., Zhao X. // Stem Cells Dev. 2006. V. 15. № 3. P. 407–421.
Catavero C., Bao H., Song J. // Cell and Tissue Research. 2018. V. 371. № 1. P. 33–46.
Bao H., Asrican B., Li W., Gu B., Wen Z., Lim S.A., Haniff I., Ramakrishnan C., Deisseroth K., Philpot B., Song J. // Cell Stem Cell. 2017. V. 21. № 5. P. 604–617. e5.
Klempin F., Beis D., Mosienko V., Kempermann G., Bader M., Alenina N. // J. Neurosci. 2013. V. 33. № 19. P. 8270–8275.
Dranovsky A., Picchini A.M., Moadel T., Sisti A.C., Yamada A., Kimura S., Leonardo E.D., Hen R. // Neuron. 2011. V. 70. № 5. P. 908–923.
Ge S., Goh E.L.K., Sailor K.A., Kitabatake Y., Ming G.L., Song H. // Nature. 2006. V. 439. № 7076. P. 589–593.
Дополнительные материалы отсутствуют.
Инструменты
Нейрохимия