

Нейрохимия, 2020, T. 37, № 4, стр. 358-367
Получение и характеристика глиальных клеток из индуцированных плюрипотентных стволовых клеток человека
Е. В. Новосадова 1, Е. Л. Арсеньева 1, С. А. Антонов 1, Е. А. Казанцева 1, Л. В. Новосадова 1, О. Д. Курко 1, С. Н. Иллариошкин 2, В. З. Тарантул 1, И. А. Гривенников 1
1 ФГБУ Институт молекулярной генетики Национального исследовательского центра
“Курчатовский институт”
Москва, Россия
2 ФГБНУ Научный центр неврологии
Москва, Россия
Поступила в редакцию 07.05.2020
После доработки 11.06.2020
Принята к публикации 26.06.2020
Аннотация
Технология получения индуцированных плюрипотентных стволовых клеток (ИПСК) человека и возможность их направленной дифференцировки в специализированные клетки всех тканей организма открыла уникальные возможности для изучения молекулярно-генетических основ патогенеза нейродегенеративных заболеваний in vitro и эффективного скрининга соединений с нейропротекторной активностью. Целью настоящей работы явилось получение из ИПСК здорового донора и пациента с семейной формой болезни Паркинсона (мутация G2019S в гене LRRK2) культур глиальных клеток и их характеристика. На первом этапе работы было проведено сравнение трех описанных ранее протоколов дифференцировки глиальных клеток из нейральных предшественников ИПСК человека и был выбран метод, наиболее приемлемый в наших условиях с точки зрения качества и времени получения соответствующих культур. Полученные таким методом культуры глиальных клеток были охарактеризованы по уровням экспрессии ряда генов нейроглиальной дифференцировки. Также в полученных культурах была проанализирована экспрессия генов некоторых нейротрофических факторов (GDNF, BDNF, NGF, NT3). Было показано, что кондиционированная среда, полученная при культивировании глиальных клеток от пациента с болезнью Паркинсона, оказывала негативное влияние на рост нейритов дофаминергических нейронов в дифференцированных культурах здорового донора, уменьшая их длину примерно в 2 раза.
ВВЕДЕНИЕ
Технология получения индуцированных плюрипотентных стволовых клеток (ИПСК) человека [1] и возможность их направленной дифференцировки в специализированные типы нейронов открыла уникальные возможности для изучения молекулярно-генетических основ патогенеза нейродегенеративных заболеваний in vitro и эффективного скрининга соединений с нейропротекторной активностью [1–3].
Однако, как известно, в нервной системе животных, включая человека, наряду с нейронами присутствуют и глиальные клетки, которые играют важную роль в поддержании и обеспечении их нормальной работы [4, 5]. Так, кроме трофической функции, глиальные клетки принимают участие в обмене нейротрансмиттеров, формировании и созревании синапсов, регуляции рН, водного и ионного гомеостаза и формировании гематоэнцефалического барьера [5]. В последние время накапливаются данные, что большинство, если не все нейродегенеративные заболевания, прямо или опосредованно, могут быть связаны с нарушениями функций глиальных клеток [6–8].
Так, например, при развитии бокового амиотрофического склероза астроциты могут продуцировать токсические факторы и не оказывать должную поддержку соответствующим мотонейронам [9]. При семейной форме болезни Паркинсона (БП) было показано, что астроциты, полученные из соответствующих ИПСК пациента, способны продуцировать белковые факторы, которые оказывали негативное воздействие на жизнеспособность нейронов in vitro [10]. В связи с появлением новых знаний о роли глиальных клеток в функционировании головного мозга в норме и при различных патологиях, много исследований сосредоточено на осуществлении направленной дифференцировки ИПСК человека в глиальном направлении и получении соответствующих культур клеток с последующей их характеристикой и дальнейшим исследованием [11–14]. Действительно, в настоящее время описаны методы получения культур глиальных клеток человека из ИПСК как здоровых доноров, так и пациентов с различными патологиями ЦНС [14–18]. Однако, как эпигенетическое разнообразие ИПСК от разных доноров, так и отличающиеся методы культивирования и дифференцировки клеток в отдельных лабораториях могут в итоге приводить к получению культур, которые разнятся по своим свойствам [19, 20]. Поэтому получение новых культур глиальных клеток человека, основанное на технологии репрограммирования, и их дальнейшая характеристика по-прежнему остаются актуальной задачей.
Ранее в нашем коллективе были получены линии ИПСК от здоровых доноров и больных с диагносцированной БП, среди которых были пациенты как со спорадической, так и с семейными формами этого заболевания [21, 22]. В дальнейшем эти ИПСК были дифференцированы в нейрональном направлении и получены культуры клеток, обогащенные дофаминергическими нейронами [23]. Однако, как упоминалось выше, для более адекватной оценки молекулярных механизмов развития нейродегенеративных изменений in vitro необходимо также исследовать роль клеток нейроглии в этом процессе. Поэтому целью настоящей работы явилось получение из ИПСК здорового донора и пациента с семейной формой БП (мутация G2019S в гене LRRK2) соответствующих культур глиальных клеток и их характеристика.
МЕТОДЫ ИССЛЕДОВАНИЯ
Культивирование ИПСК, получение нейральных предшественников и дифференцированных нейрональных культур. Получение ИПСК от здоровых доноров и пациентов с различными формами БП, осуществляли с помощью соответствующих векторов для лентивирусной экспрессии LeGO-hOCT4, LeGO-hSOX2, LeGO-hc-Myc и LeGO-hKLF4 как описано ранее [21, 22]. Полученные таким образом ИПСК культивировали в среде mTeSR (STEM CELL Technologies) на подложке матригеля. Дальнейшую дифференцировку ИПСК до стадии нейральных предшественников (НП) осуществляли по методу, описанному ранее [22].
Для получения дифференцированных культур, обогащенных дофаминергическими (ДА) нейронами, НП культивировали в среде следующего состава: (DMEM/F12 (Gibco, США), 2% заменитель сыворотки (Gibco, США), добавка B27 (×50) – 1% (Gibco, США), 2 мМ L-глутамин (ICN Biomedicals, США), 1% смесь аминокислот (ПанЭко, РФ), 50 ед./мл пенициллин–стрептомицин (ПанЭко, РФ), 20 нг/мл BDNF (PeproTech, США), 20 нг/мл GDNF (PeproTech, США), 200 мкМ аскорбиновая кислота (ПанЭко, РФ) и 4 мкМ форсколин (Stemgent, США) в течение не менее 14 дней [23].
Получение глиальных клеток из нейральных предшественников. Для получения глиальных клеток из НП использовали три приведенных ниже протокола дифференцировки, описанных ранее [17, 18].
Протокол № 1. Протокол № 1 основан на рекомендациях фирмы производителя: STEMCELL Technologies, Канада ( https://www.stemcell.com).
НП, полученные из ИПСК, рассевали на чашки Петри (диаметр 35 мм), покрытые Matrigel (BD Biosciences, США), из расчета 250 000–400 000 клеток на см2 и культивировали в нейрональной среде (НС): DMEM/F12, 2% заменитель сыворотки (Gibco, США), 1 мМ заменимые аминокислоты (ПанЭко, РФ), 2 мМ L-глутамин, 1% N2 (Life Technologies, США), пенициллин–стрептомицин (50 ед./мл; 50 мкг/мл) в СO2-инкубаторе при 37°С в течение 24 ч. На следующий день нейрональную среду заменяли на коммерческую среду STEMdiff Astrocyte Differentiation Kit (STEMCELL Technologies, Канада) и культивировали клетки в течение 20–21 дня. Согласно рекомендациям производителя, на протяжении первых 6–7 дней среду меняли каждый день, а последующие дни смену среды осуществляли через день. Пересев осуществляли раз в 6–7 дней с помощью 0.05% раствора трипсина (ICN Biomedicals, США). Для дальнейшей дифференцировки клеток в глиальном направлении на 20–21-й день культивирования происходила замена среды на коммерческую среду STEMdiff Astrocyte Maturation Kit (STEMCELL Technologies, Канада), в которой клетки культивировали в течение 14–15 дней. Среду меняли через день, пересев осуществляли раз в 6–7 дней с помощью 0.05% раствора трипсина (ICN Biomedicals, США). На 19–20 и 28–30 дни дифференцировки часть клеток тестировали на экспрессию маркеров глиальных клеток S100b и GFAP.
Протокол № 2. Протокол № 2 основан на методических рекомендациях, описанных в статье Lundin A. et al. [17].
НП, полученные из ИПСК, рассевали на чашки Петри (диаметр 35 мм), покрытые Matrigel (BD Biosciences, США), из расчета 250 000–400 000 клеток на см2 и культивировали в нейрональной среде НС в СO2-инкубаторе при 37°С в течение 24 ч. На следующий день нейрональную среду заменяли на среду для культивирования глиальных клеток (DMEM/F12 (Gibco, США), 1% N2 добавки (Gibco, США), 1 мМ глутамин (ICN Biomedicals, США), β-FGF (Gibco, США), 10 нг/мл Heregulin β1 (ThermoFisher, США), 200 нг/мл IGF 1 (ProSpec, США), Activin A (Gibco, США)) и культивировали клетки в течение 28 дней. По мере нарастания до полного монослоя клетки пересевали 1 : 3 с помощью 0.05% раствора трипсина (ICN Biomedicals, США). На 19–20-й и 28–30-й дни дифференцировки часть клеток тестировали на экспрессию маркеров глиальных клеток S100b и GFAP.
Протокол № 3. Протокол № 3 основан на методических рекомендациях, описанных в статье Mormone E. et al. [18].
НП, полученные из ИПСК, рассевали на чашки Петри (диаметр 35 мм), покрытые Matrigel (BD Biosciences, США), из расчета 250 000–400 000 клеток на см2 и культивировали в нейрональной среде НС в СO2-инкубаторе при 37°С в течение 24 ч. На следующий день нейрональную среду заменяли на среду для культивирования глиальных клеток (DMEM/F12 (HyСlone, США), FBS (HyClone, США), 1% N2 добавки (Gibco, США), 1 мМ глутамин (ICN Biomedicals, США), 1% смеси аминокислот (“ПанЭко”, Россия), 50 ед./мл пенициллин–стрептомицина (“ПанЭко”, Россия), 10 нг/мл β-FGF (Gibco, США), 20 нг/мл CNTF (MACS, США)), культивирование в которой продолжалось на протяжении 3 дней в СO2-инкубаторе при 37°С.
Следующие 3 дня клетки растили в среде для культивирования глиальных клеток (DMEM/F12 (Hyclone, США), FBS (HyClone, США), 1% N2 добавки (Gibco, США), 1 мМ глутамин (ICN Biomedicals, США), 1% смеси аминокислот (“ПанЭко”, Россия), 50 ед./мл пенициллин–стрептомицина (“ПанЭко”, Россия), 10 нг/мл β-FGF (Gibco, США), 20 нг/мл EGF (Gibco, США), 20 нг/мл CNTF (MACS, США)).
Дальнейшее культивирование глиальных клеток проводили в среде следующего состава: DMEM/F12 (Gibco, США), FBS (HyClone, США), 1% N2 добавки (Gibco, США), 1 мМ глутамин (ICN Biomedicals, США), 1% смеси аминокислот (“ПанЭко”, Россия), 50 ед./мл пенициллин–стрептомицина (“ПанЭко”, Россия), 20 нг/мл CNTF (MACS, США). Последующий пересев осуществляли по мере достижения монослоя с помощью 0.05% раствора трипсина (HyClone, США). На 19–20 и 28–30 дни дифференцировки часть клеток тестировали на экспрессию маркеров глиальных клеток S100b и GFAP.
Иммунофлуоресцентная детекция. Клетки однократно промывали раствором PBS (HyClone, США) и фиксировали в 4% параформальдегиде (ПФА) (Sigma-Aldrich, США) 20–30 мин при комнатной температуре. Потом клетки промывали три раза раствором PBS (HyClone, США), а затем инкубировали в растворе PBS содержащем 0.1% Triton Х-100 (Sigma-Aldrich, США), 5% FBS (HyClone, США) в течение 15 мин для пермеабилизации и уменьшения неспецифической сорбции антител. Далее фиксированные клетки инкубировали в растворе PBS, содержащем 0.1% Triton X-100 (Sigma-Aldrich, США), 5% FBS (HyClone, США) и первичные антитела, в течение ночи при +4°С. После трехкратных отмывок по 10 мин раствором PBS–0.1% Tween 20 (Sigma-Aldrich, США) клетки инкубировали в растворе PBS, содержащем 0.1% Triton Х-100 (Sigma-Aldrich, США), 5% FBS (HyClone, США) и вторичные антитела, меченные флуоресцентным красителем, в течение 1.5 ч при комнатной температуре. После трехкратных отмывок по 10 мин раствором PBS (HyClone, США), содержащем 0.1% Tween 20 (Sigma-Aldrich, США), к клеткам добавляли раствор DAPI (Sigma-Aldrich, США) для окраски ядер в концентрации 0.1 мкг/мл, и инкубировали 1 мин при комнатной температуре. Избыток красителя удаляли 3-кратной промывкой раствором PBS–0.1% Tween 20. Визуализацию результатов осуществляли на микроскопе Axio Imager Z1 (Zeiss, Германия) с помощью программы AxioVision Rel.4.8. Для характеристики различных стадий глиальной дифференцировки ИПСК использовали следующие антитела:
мышиные моноклональные к GFAP, (G3893) (Sigma, США), 1 : 400;
кроличьи поликлональные к S100b, (IR50461-2) (Dako, США), 1 : 200.
В качестве вторичных антител в работе использовались следующие антитела (“Invitrogen”, США):
козьи к иммуноглобулинам мыши, конъюгированные с Alexa Fluor 488, (А11001), 1 : 2000;
козьи к иммуноглобулинам кролика, конъюгированные с Alexa Fluor 546, (А11010), 1 : 2000.
Выделение тотальной РНК из клеточных культур и ПЦР в реальном времени. Тотальную РНК из клеточных культур выделяли с помощью коммерческого набора “Trizol RNA purification kit” (Invitrogen, USA) согласно рекомендации производителя. Примеси ДНК в полученных пробах удаляли с помощью набора “DNA-free™ DNA Removal Kit” (ThermoFischer, USA). Для синтеза кДНК использовали 2 мкг тотальной РНК и обратную транскриптазу MMLV (“Евроген”, Россия). Полученную кДНК амплифицировали с помощью CFX96 Touch™Real-Time PCR Detection System (Bio-Rad, USA), используя реакционную смесь qPCRmix-HS SYBR (“Евроген”, Россия) и праймеры, приведенные в табл. 1.
Таблица 1.
Структура праймеров, использованных для проведения ПЦР в реальном времени
| Название гена | Температура отжига °С | Последовательность |
|---|---|---|
| 18 s | 60 | 5'-CGGCTACCACATCCAAGGAA-3' 5'-GCTGGAATTACCGCGGCT-3' |
| GDNF | 60 | 5'-ACCTGGAGTTAATGTCCAACC-3' 5'-GGCATATTTGAGTCACTGCT-3' |
| BDNF | 60 | 5'-TTTGGTTGCATGAAGGCTGC-3' 5'-GCCGAACTTTCTGGTCCTCA-3' |
| b-III-tub | 60 | 5'-CTCAGGGGCCTTTGGACATC-3' 5'-CAGGCAGTCGCAGTTTTCAC-3' |
| NGF | 60 | 5'-CACACTGAGGTGCATAGCGT-3' 5'-TGATGACCGCTTGCTCCTGT-3' |
| NT3 | 65 | 5'-AACTGCTGCGACAACAGAGA-3' 5'-CCAGCCCACGAGTTTATTGT-3' |
| S100b | 60 | 5'-CTGGAGAAGGCCATGGTTGC-3' 5'-CTCCAGGAAGTGAGAGAGCT-3' |
| GFAP | 60 | 5'-TCCTGGAACAGCAAAACAAG-3' 5'-CAGCCTCAGGTTGGTTTCAT-3' |
| EATT1 | 57 | 5'-CGAAGCCATCATGAGACTGGTA-3' 5'-TCCCAGCAATCAGGAAGAGAA-3' |
| GLUT1 | 57 | 5'-GCTGTGCTTATGGGCTTCTC-3' 5'-CACATACATGGGCACAAAGC-3' |
| MBP | 57 | 5'-CTGAGCAGATTTAGCTGGGG-3' 5'-TAGGTAACAGGGGCAAGTGG-3' |
| AQP4 | 57 | 5'-GGCCGTAATCTGACTCCCAG-3' 5'-TGTGGGTCTGTCACTCATGC-3' |
| Vim | 60 | 5'-ATTCCACTTTGCGTTCAAGG-3' 5'-CTTCAGAGAGAGGAAGCCGA-3' |
| α-Syn | 56 | 5'-AGTGACAAATGTTGGAGGAG-3' 5'-GCTTCAGGTTCGTAGTCTTG-3' |
| NRF2 | 56 | 5'-GGTTTCTTCGGCTACGTTT-3' 5'-TAACTCAGGAATGGATAATAGCTC-3' |
| ALDH1L1 | 55.5 | 5'-TCACAGAAGTCTAACCTGCC-3' 5'-AGTGACGGGTGATAGATGAT-3' |
| PLP | 60 | 5'-GGCGCAGTCAGGCAGATC-3' 5'-CCCTTGCCGCAGATGGT-3' |
| GALC | 59 | 5'-AGTTTACTCAACCTGGCTGG-3' 5'-AACAAGAATTGGCTCTGAACC-3' |
| CNP | 60 | 5'-CACCATGCACCTCTCCCAGC-3' 5'-ATGGAGCCGATCCGGTCCAG-3' |
| MAG | 55 | 5'-TCTGGATTATGATTTCAGCC-3' 5'-GCTCTGAGAAGGTGTACTGG-3' |
| PMP22 | 58 | 5'-ACCCAGTGCATCCAAC-3' 5'-ATCCATAGCACCATTTCAAAG-3' |
GDNF – нейротрофический фактор глии; BDNF – нейротрофический фактор мозга; NGF – фактор роста нервов; NT3 – нейротрофин-3; GFAP – кислый фибриллярный белок глии; EATT1 – транспортер глутамата; GLUT1 – транспортер глюкозы; MBP – основной белок миелина; AQP4 – аквапорин 4; Vim – виментин; α-Syn – альфа-синуклеин; NRF2 – транскрипционный фактор NRF2; ALDH1L1 – алкогольдегидрогеназа; PLP – протеолипид миелина; GALC – галактоцереброзидаза; CNP – фосфодиэстераза циклических 2'–З'-нуклеотидов; MAG – гликопротеин ассоциированный с миелином; PMP22 – периферический белок миелина 22.
Условия ПЦР: денатурация при 95°С (3 мин), далее 40 циклов (95°С, 15 с; 60°С, 20 с; 72°С, 45 с). В качестве референсного гена использовали 18S рРНК. Оценку изменений в экспрессии генов проводили с помощью 2∆∆Ct метода.
Статистическая обработка результатов. Статистическую обработку результатов проводили на основе стандартного пакета статистических программ “Statistica 7.0” для WinXP с использованием непараметрических методов сравнения двух независимых между собой групп (критерий Манна–Уитни). Статистически значимыми считали результаты при р < 0.05.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Сравнительная эффективность разных протоколов получения линий глиальных клеток. Как было уже отмечено, ранее нами были получены несколько линий ИПСК от здоровых доноров и пациентов с диагностированной болезнью БП. Из них, через стадию НП, были получены культуры дифференцированных нейронов [21–23]. В настоящей работе НП были использованы для получения глиальных клеток. На первом этапе работы было проведено сравнение трех описанных ранее протоколов дифференцировки глиальных клеток из ИПСК человека, для выбора более адекватного с точки зрения финансовых затрат и времени получения этих клеток [17, 18].
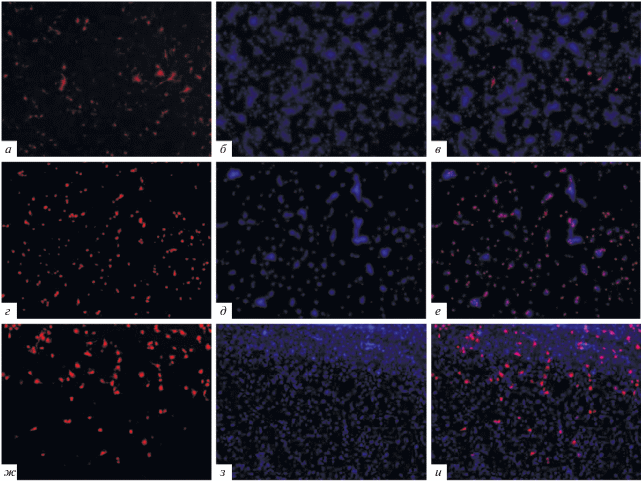
Сравнительную эффективность этих протоколов проводили, используя дифференцировку НП здорового донора и анализируя экспрессию глия-специфических антигенов: S100b и GFAP c помощью иммунофлуоресценции. У всех клеток, культивируемых согласно трем различным протоколам, на ранних этапах дифференцировки (19–20 дней дифференцировки in vitro) начинала проявляться положительная окраска на маркер астроцитов белок S100b (рис. 1а, г, ж).
Рис. 1.
Микрофотографии иммунофлуоресцентного окрашивания клеток на 19-й день дифференцировки согласно протоколу № 1 (а, б, в), протоколу № 2 (г, д, е), протоколу № 3 (ж, з, и), снятых в одном поле зрения, на маркер глиальных клеток S100b (увеличение ×100). а, г, ж – клетки, окрашенные антителами к S100b (красный); б, д, з – клетки, окрашенные ядерным красителем DAPI (синий); в, е, и – фотография наложения двух предыдущих (S100b, DAPI).
На следующем этапе исследования было проведено иммунофлуоресцентное окрашивание клеток на более поздних этапах дифференцировки (28 дней дифференцировки), культивированных согласно указанным выше протоколам (рис. 2). Как видно из представленных результатов, количество S100b-положительных клеток существенным образом увеличилось (рис. 2а, д, и) и появилась окраска на маркер глиальных клеток GFAP (рис. 2б, е, к), что свидетельствует о переходе глиальных клеток к зрелому фенотипу.
Рис. 2.
Микрофотографии иммуноцитохимического окрашивания клеток на 28-м дне культивирования согласно протоколу № 1 (а, б, в, г), протоколу № 2 (д, е, ж, з), протоколу № 3 (и, к, л, м), снятых в одном поле зрения, на маркеры глиальных клеток S100b и GFAP (увеличение ×100). а, д, и – клетки, окрашенные антителами к S100b; б, е, к – клетки, окрашенные антителами к GFAP; в, ж, л – ядра клеток, окрашенные ядерным красителем DAPI; г, з, м – фотография наложения трех предыдущих (S100b, GFAP, DAPI).
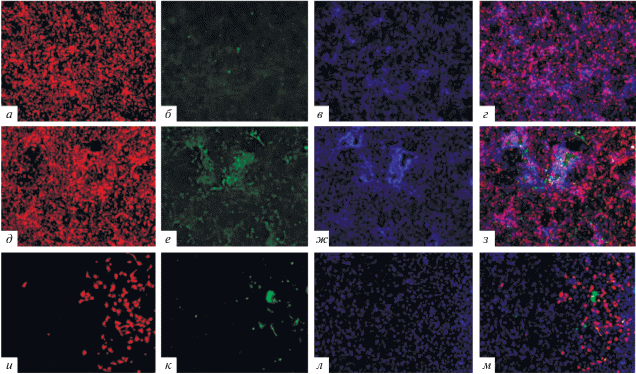
На основании анализа полученных данных для дальнейшей работы нами был выбран протокол № 2 как наиболее подходящий, вследствие того, что количество глиальных клеток, окрашенных на маркер GFAP и S100b именно в этом протоколе было максимальным.
В итоге нами были рассмотрены три протокола. Протокол 1, основанный на использовании коммерчески доступной среды, и Протокол 2 показали сопоставимые результаты, однако Протокол 2 оказался максимально отвечающим временны́м требованиям получения глиальных клеток зрелого фенотипа. Кроме того, по финансовым затратам Протокол 2 оказался более выгодным, поэтому он был выбран для дальнейшей работы.
Анализ отношения экспрессии ряда генов при индукции глиальной дифференцировки на 33-й день к 22-му дню дифференцировки in vitro. По данным литературы получаемые из ИПСК культуры глиальных клеток могут характеризоваться достаточно гетерогенным составом [10, 12–14, 16, 17]. Поэтому мы с помощью ПЦР в реальном времени оценили отношение экспрессии ряда генов, характерных для нейрональной и глиальной дифференцировки, при ее индукции на 33-ий день к 22-ому дню дифференцировки in vitro, в культурах клеток, полученных от здорового донора и от пациента с БП. Полученные результаты представлены в табл. 2.
Таблица 2.
Отношение уровней транскрипции ряда генов на 33-й день к 22-му дню после начала индукции глиальной дифференцировки in vitro
| Гены | Отношение уровней транскрипции генов на 33-й день к 22-му дню дифференцировки in vitro | |
|---|---|---|
| здоровый донор | пациент с БП (мутация в гене LRRK2) |
|
| β-IIItub | 0.86 ± 0.05 | 7.76 ± 0.50*** |
| S100b | 1.11 ± 0.05 | 4.77 ± 3.5 |
| GFAP | 52.3 ± 20.3* | 4.05 ± 1.2* |
| EATT1 | 0.21 ± 0.02** | 6.91 ± 1.1** |
| GLUT1 | 0.95 ± 0.02 | 1.79 ± 0.71 |
| AQP4 | 0.16 ± 0.01* | 2.70 ± 0.21** |
| VIM | 0.81 ± 0.18* | 3.95 ± 0.10** |
| α-Syn | 17.65 ± 4.3* | 1.11 ± 0.02* |
| NRF2 | 1.25 ± 0.04* | 9.72 ± 0.51* |
| ALDH1L1 | 25.02 ± 1.76*** | 7.35 ± 2.28* |
| MBP PLP |
0.72 ± 0.13 0.76 ± 0.03 |
0.10 ± 0.03 1.41 ± 0.04 |
| CNP | 0.06 ± 0.02* | 0.16 ± 0.03* |
| PMP22 | 0.31 ± 0.02*** | 0.24 ± 0.03** |
| MAG | 178.9 ± 7.8*** | 9.22 ± 0.43** |
Из результатов, представленных в табл. 2, следует отметить отсутствие изменений или уменьшение в соотношении уровней транскрипции генов, специфичных для олигодендроцитов и клеток Шванна (MBP, PLP, CNP, PMP22), как в клеточных культурах здорового донора, так и от пациента с БП. Это может свидетельствовать об отсутствии увеличения доли этих клеток в глиальных культурах при более длительных временах дифференцировки (33 дня). Исключение представлял только ген MAG. Следует также отметить увеличение экспрессии гена GFAP на 33-й день дифференцировки как в клеточных культурах здорового донора, так и пациента с БП. Для гена S100b увеличение уровня экспрессии было отмечено только для глиальных клеток от пациента с БП. Также наблюдалось существенное увеличение экспрессии гена алкогольдегидрогеназы (ALDH1L1) в обоих типах культур на 33-й день дифференцировки.
Известно, что глиальные клетки, осуществляя трофические функции, способны синтезировать различные нейротрофические факторы [24–27].
Мы проанализировали экспрессию ряда этих факторов в полученных нами линиях клеток с помощью ПЦР в реальном времени. Результаты этих экспериментов, суммированные в табл. 3, представлены в виде соотношения уровней транскрипции этих генов по мере дифференцировки глиальных клеток (отношение уровня экспрессии гена на 33-й день к уровню его экспрессии на 22-й день).
Таблица 3.
Отношение уровней транскрипции генов нейротрофических факторов на 33-й день к 22-му дню после начала индукции глиальной дифференцировки in vitro
| Гены | Отношение уровней транскрипции генов нейротрофических факторов на 33-й день к 22-му дню дифференцировки in vitro | |
|---|---|---|
| здоровый донор | пациент с БП (мутация в гене LRRK2) |
|
| GDNF | 15.6 ± 5.4** | 1.9 ± 0.1** |
| NGF | 1.2 ± 0.1 | 2.8 ± 0.4* |
| NT-3 | 0.46 ± 0.04 | 2.1 ± 0.8 |
| BDNF | 0.40 ± 0.02** | 1.01 ± 0.04 |
Из приведенных в табл. 3 результатов видно, что только GDNF существенно увеличивал свою экспрессию на 33-й день дифференцировки, причем это увеличение было наиболее заметно в клетках здорового донора по сравнению с клетками, полученными от пациента с БП. Другие проанализированные нейротрофические факторы изменяли свою экспрессию незначительно. Безусловно данные ПЦР в дальнейшем предполагается подтвердить и данными по оценке уровней их белковых продуктов.
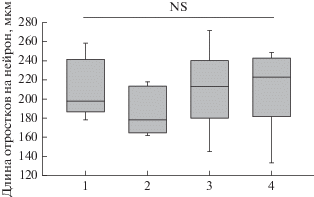
Оценка влияния кондиционированной среды от глиальных клеток здорового донора и пациента с БП, на рост нейритов в дифференцированных культурах, обогащенных дофаминергическими нейронами. На следующем этапе работы мы оценили влияние кондиционированной среды (КС) от глиальных клеток здорового донора и пациента с БП, на рост нейритов в дифференцированных культурах, обогащенных ДА-нейронами, полученных от здорового донора. Как видно из рис. 3, КС от глиальных клеток здорового донора, добавленная в разных количествах, не оказывала негативного влияния на рост нейритов ДА-нейронов в этих культурах при их культивировании в течение 7 дней.
Рис. 3.
Влияние КС от глиальных клеток здорового донора, на рост нейритов у ДА-нейронов в дифференцированных культурах при их культивировании в течение 7 дней. Состав КС: DMEM/F12, 2% заменитель сыворотки, добавка B27 (×50) – 1%, 2 мМ L-глутамин, 1% смесь аминокислот, 50 ед./мл пенициллин–стрептомицинa, 20 нг/мл BDNF, 20 нг/мл GDNF, 200 мкМ аскорбиновая кислота, 4 мкМ форсколин. 1 – Без добавления КС. 2 – 20% КС. 3 – 50% КС. 4 – 80% КС. Культивирование с КС в течение 7 дней. После окончания культивирования клетки окрашивали антителами к тирозингидроксилазе. Окрашенные клетки анализировали c помощью программы Image J (Neurphology J). n = 6.
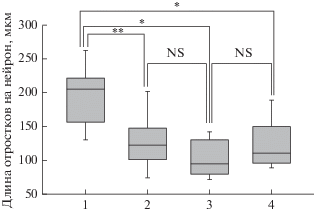
В то же время добавление КС от глиальных клеток, полученных от пациента с БП (мутация в гене LRRK), приводило к существенному торможению роста нейритов у ДА-нейронов в нейрональных культурах. Данные, представленные на рис. 4, показывают практически 2-кратное уменьшение длины отростков у ДА-нейронов при добавлении от 20 до 80% КС.
Рис. 4.
Влияние КС от глиальных клеток, полученных от пациента с БП (мутация в гене LRRK), на рост нейритов у ДА-нейронов в дифференцированных культурах при их культивировании в течение 7 дней. 1 – Без добавления КС. 2 – 20% КС. 3 – 50% КС. 4 – 80% КС. После окончания культивирования клетки окрашивали антителами к тирозингидроксилазе. Окрашенные клетки анализировали c помощью программы Image J (Neurphology J). n = 6. *р < 0.05, **р < 0.005. NS – статистически незначимые отличия.
Недавно Domenico с коллегами [10] продемонстрировали сходные результаты угнетающего действия кондиционированной среды от глиальных клеток, также полученных от пациента с БП (мутация в гене LRRK2), на рост нейритов у ДА-нейронов в нейрональных культурах. Более того, авторам удалось показать, что такое негативное воздействие связано с присутствием в этой среде альфа-синуклеина, который продуцировался “больными” глиальными клетками. Представляется важным в дальнейшем проверить продуцируют ли альфа-синуклеин глиальные клетки, полученные от пациентов с БП с другими мутациями и пациентов со спорадической формой этого заболевания.
ЗАКЛЮЧЕНИЕ
Таким образом, в ходе проведенной работы были получены линии глиальных клеток от здорового донора и пациента с семейной формой БП и подобраны условия для их эффективной дифференцировки. Полученные культуры были охарактеризованы по экспрессии ряда нейроглиальных маркеров, а также ряда нейротрофических факторов. Было показано, что КС, полученная при культивировании глиальных клеток от пациента с БП, оказывала негативное влияние на рост нейритов ДА-нейронов в дифференцированных культурах здорового донора, уменьшая их длину примерно в 2 раза. В дальнейшем предполагается получение дополнительных независимых линий глиальных клеток как от здоровых доноров, так и от пациентов с семейными и спорадическими формами БП для более надежного выявления и сравнения возможных патологических изменений в “больных” клетках по сравнению со “здоровыми”.
Список литературы
Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. // Cell. 2007. V. 131. P. 861–872.
Новосадова Е.В., Гривенников И.А. // Успехи биологической химии. 2014. Т. 54. С. 3–38.
Garcia-Leon J.A., Vitorica J., Gutierrez A. // Future Med. Chem. 2019. https://doi.org/10.4155/fmc-2018-0520
Von Bartheld C.S., Bahney J., Herculano-Houzel S. // J. Comp. Neurol. 2016. V. 524. P. 3865–3895.
Verkhratsky A., Nedergaard M. // Physiol. Rev. 2018. V. 98. P. 239–389.
Molofsky A.V., Krencik R., Ullian E.M., Tsai H.H., Deneen B., Richardson W.D., Barres B.A., Rowitch D.H. // Genes Dev. 2012. V. 26. P. 891–907.
Verkhratsky A., Sofroniew M.V., Messing A., Delanerolle N.C., Rempe D., Rodriguez J.J., Nedergaard M. // ASN Neuro. 2012. V. 4. pii: e00082. https://doi.org/10.1042/AN20120010
Palpagama T.H., Waldvogel H.J., Faull R.L.M., Kwakowsky A. // Front. Mol. Neurosci. 2019. V. 12. P. 258. https://doi.org/10.3389/fnmol.2019.00258
Bi F., Huang C., Tong J., Qiu G., Huang B.,Wu Q., Li F., Xu Z., Bowser R., Xia X.G., Zhou H. // Proc. Natl. Acad. Sci. U.S.A. 2013. V. 110. P. 4069–4074.
di Domenico A., Carola G., Calatayud C., Pons-Espinal M., Mun˜oz J.P., Richaud-Patin Y., Fernandez-Carasa I., Gut M., Faella A., Parameswaran J., Soriano J., Ferrer I., Tolosa E., Zorzano A., Cuervo A.M., Raya A., Consiglio A. // Stem Cell Reports. 2019. V. 12. P. 213–229.
Krencik R., Ullian E.M. // Frontiers in Cellular Neuroscience. 2013. V. 7. Article 25. P. 1–10. www.frontiersin.org.
Krencik R., Weick J.P., Liu Y., Zhang Z.J., Zhang S.C. // Nat. Biotechnol. 2011. V. 29. P. 528–534.
Santos R., Vadodaria K.C., Jaeger B.N., Mei A., Lefcochilos-Fogelquist S., Mendes A.P.D., Erikson G., Shokhirev M., Randolph-Moore L., Fredlender C., Dave S., Oefner R., Fitzpatrick C., Pena M., Barron J.J., Ku M., Denli A.M., Kerman B.E., Charnay P., Kelsoe J.R., Marchetto M.C., Gage F.H. // Stem Cell Reports. 2017. V. 8. P. 1757–1769.
Ye Zhang S.A., Sloan L.E., Clarke C., Caneda C.A., Plaza P.D., Blumenthal H., Vogel G.K., Steinberg M.S.B., Edwards G., Li J.A., Duncan S.H., Cheshier L.M., Shuer E.F., Chang G.A., Grant M.G., Gephart B.A.B. // Neuron. 2016. V. 89. P. 37–53.
Chen C., Jiang P., Xue H., Peterson S.E., Tran H.T., McCann A.E., Parast M.M., Li S., Pleasure D.E., Laurent L.C., Loring J.F., Liu Y., Deng W. // Nat. Commun. 2014. V. 5. P. 4430.
Shaltouki A., Peng J., Liu Q., Rao M.S., Zeng X. // Stem Cells. 2013. V. 31. P. 941–952.
Lundin A., Delsing L., Clausen M., Ricchiuto P., Sanchez J., Sabirsh A., Ding M., Synnergren J., Zetterberg H., Brolén G., Hicks R., Herland A., Falk A. // Stem Cell Reports. 2018. V. 10. P. 1030–1045.
Mormone E., D’Sousa S., Alexeeva V., Bederson M.M., Germano I.M. // Stem Cells and Development. 2014. V. 23. P. 2626–2636.
Kim K., Doi A., Wen B., Ng K., Zhao R., Cahan P., Kim J., Aryee M.J., Ji H., Ehrlich L.I., Yabuuchi A., Takeuchi A., Cunniff K.C., Hongguang H., McKinney-Freeman S., Naveiras O., Yoon T.J., Irizarry R.A., Jung N., Seita J., Hanna J., Murakami P., Jaenisch R., Weissleder R., Orkin S.H., Weissman I.L., Feinberg A.P., Daley G.Q. // Nature. 2010. V. 467. P. 285–290.
Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Müller F.J., Wang Y.C., Boscolo F.S., Fakunle E., Dumevska B., Lee S., Park H.S., Olee T., D’Lima D.D., Semechkin R., Parast M.M., Galat V., Laslett A.L., Schmidt U., Keirstead H.S., Loring J.F., Laurent L.C. // Cell Stem Cell. 2012. V. 10. P. 620–634.
Novosadova E.V., Nekrasov E.D., Chestkov I.V., Surdina A.V., Vasina E.M., Bogomazova A.N., Manuilova E.S., Arsenyeva E.L., Simonova V.V., Konovalova E.V., Fedotova E.Yu., Abramycheva N.Yu., Khaspekov L.G., Grivennikov I.A., Tarantul V.Z., Kiselev S.L., Illarioshkin S.N. // Sovremennye Tehnologii v Medicine. 2016. V. 8. P. 155–164.
Новосадова Е.В., Арсеньева Е.Л., Мануилова Е.С., Хаспеков Л.Г., Бобров М.Ю., Безуглов В.В., Иллариошкин С.Н., Гривенников И.А. // Биохимия. 2017. V. 82. P. 1732–1739.
Nenasheva V.V., Novosadova E.V., Makarova I.V., Lebedeva O.S., Grefenshtein M.A., Arsenyeva E.L., Antonov S.A., Grivennikov I.A., Tarantul V.Z. // Mol. Neurobiol. 2017. V. 54. P. 7204–7211.
Pöyhönen S., Er S., Domanskyi A., Airavaara M. // Front. Physiol. 2019. V. 10. P. 486. https://doi.org/10.3389/fphys.2019.00486
Bothwell M. F1000Res. 2016. V. 5. pii: F1000 Faculty Rev-1885. https://doi.org/10.12688/f1000research.8434.1
Ibáñez C.F., Andressoo J.O. // Neurobiol. Dis. 2017. V. 97(Pt B). P. 80–89. https://doi.org/10.1016/j.nbd.2016.01.021
Nasrolahi A., Mahmoudi J., Akbarzadeh A., Karimipour M., Sadigh-Eteghad S., Salehi R., Farhoudi M. // Rev. Neurosci. 2018. V. 29. P. 475–489.
Дополнительные материалы отсутствуют.