

Нейрохимия, 2021, T. 38, № 1, стр. 14-20
Альфа-синуклеин в крови у мышей на нейротоксической модели болезни Паркинсона
В. Е. Блохин 1, М. В. Угрюмов 1
1 Федеральное государственное бюджетное учреждение науки
Институт биологии развития им. Н.К. Кольцова РАН
Москва, Россия
Поступила в редакцию 13.04.2020
После доработки 25.06.2020
Принята к публикации 29.06.2020
Аннотация
Одним из приоритетов нейронауки является борьба с социально значимыми нейродегенеративными заболеваниями, к которым относится болезнь Паркинсона (БП). Учитывая ограниченные возможности изучения патогенеза этого заболевания, важное значение приобретает создание и детальное изучение соответствующих экспериментальных моделей. Поэтому целью данной работы явилось продолжение многолетних исследований клеточных и молекулярных механизмов патогенеза болезни Паркинсона и их системных проявлений при моделировании этого заболевания у мышей с помощью 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП). В данной работе с помощью Вестерн блота показано, что у мышей на МФТП-модели клинической стадии БП в плазме крови практически вдвое снижается отношение мономерного α-синуклеина к олигомерному и наблюдается тенденция к снижению отношения общего α-синуклеина к форсфорелированному по серину-129. По данным ряда исследований, такие изменения характерны для больных при БП. В отличие от плазмы, в лимфоцитах не было обнаружено изменения соотношения указанных белков. Из совпадения наших данных по содержанию α-синуклеина и его изоформ в крови у паркинсонических мышей и данных литературы по содержанию α-синуклеина у больных при БП следует, что наша модель БП может быть использована для углубленной оценки периферических проявлений этого заболевания, а также для усовершенствования дифференциальной диагностики БП, анализа эффективности используемой терапии и скрининга новых лекарственных средств.
ВВЕДЕНИЕ
Одним из важнейших приоритетов современной нейронауки является борьба с социально значимыми нейродегенеративными заболеваниями – в первую очередь с болезнью Альцгеймера и болезнью Паркинсона (БП) [1]. При этом к наиболее актуальным задачам относится изучение клеточных и молекулярных механизмов патогенеза этих болезней и, в соответствии с методологией трансляционной медицины, разработка ранней и дифференциальной диагностики, а также усовершенствование существующей симптоматической терапии и превентивной нейропротекторной терапии [2]. Однако возможности для изучения механизмов патогенеза нейродегенеративных заболеваний у больных по ряду причин крайне ограничены. Так, клинические исследования ограничены тем, что нейродегенеративные заболевания диагностируют по характерным симптомам только через 20–30 лет после начала патологического процесса. Патологоанатомические исследования ограничены тем, что больные умирают уже при полной деградации ключевых регуляторных систем мозга и исчерпания компенсаторных резервов организма [3].
Ограничения в исследовании молекулярных механизмов патогенеза нейродегенеративных заболеваний у человека можно отчасти компенсировать путем их изучения на экспериментальных моделях. При этом необходимо учитывать, что модели любого заболевания способны адекватно воспроизводить только отдельные элементы патогенеза. Поэтому разработке моделей нейродегенеративных заболеваний и их сравнению с патогенезом у больных уделяется большое внимание [4]. Только после тщательного изучения экспериментальных моделей и получения доказательств их соответствия по определенным метаболическим путям патогенезу БП, они могут быть использованы для разработки инновационных методов диагностики и лечения.
Целью данной работы явилось продолжение многолетних исследований клеточных и молекулярных механизмов патогенеза БП и их системных проявлений – физиологических, биохимических и молекулярно-биологических – у мышей при моделировании этого заболевания с помощью 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП). МФТП – предшественник нейротоксина, который в организме превращается в МФП+ – специфический токсин дофаминергических нейронов [5]. С помощью МФТП удается воспроизвести гибель дофаминергических нейронов нигростриатной системы мозга – ключевого звена регуляции моторной функции, а также ряд других центральных и периферических элементов патогенеза БП [6]. По данным литературы, на МФТП-моделях, наряду с дегенерацией нигростриатных дофаминергических нейронов, воспроизводится и второй ключевой механизм патогенеза БП – нарушение метаболизма альфа-синуклеина [7]. При α-синуклеинопатии происходит внутринейрональное накопление и агрегация α-синуклеина, который приобретает цитотоксические свойства, вызывая дегенерацию центральных и периферических нейронов. Считается, что секретируемый агрегированный токсичный α-синуклеин быстро распространяется по организму у больных прионоподобным способом – от нейрона к нейрону или через гуморальные среды (ликвор, кровь), вызывает системную нейродегенерацию [8]. В данной работе была поставлена задача впервые оценить содержание α-синуклеина в крови у мышей на модели ранней клинической стадии БП.
МАТЕРИАЛЫ И МЕТОДЫ
Животные. В данной работе использованы 20 самцов мышей линии C57Bl/6J в возрасте от 2 до 2.5 мес. и весом от 23 до 25 г. Животных содержали в виварии с 12 часовым циклом дня и ночи и свободным доступом (ad libitum) к пище и воде. Все манипуляции с животными были проведены в соответствии с национальными и международными требованиями и правилами, утвержденными комитетом по охране животных ФГБУН Института биологии развития им. Н.К. Кольцова РАН, Протокол № 27 от 04.07.2019 г.
Моделирование болезни Паркинсона и пробоподготовка для анализа. Мышам в контрольной группе (n = 10) 4 раза с интервалом в 2 ч подкожно вводили физиологический раствор. Для моделирования БП мышам (n = 10) четырежды с интервалом в 2 ч подкожно вводили МФТП в физрастворе (Sigma-Aldrich, США) в однократной дозе 10 мг/кг. Через 2 нед. после введения МФТП или физраствора мышей наркотизировали изофлураном (KentScientific, США), декапитировали, собирали кровь из шеи и вычленяли мозг.
Из мозга при 4°C под контролем бинокулярной лупы выделяли ростро-дорзальную область стриатум – центральное звено регуляции моторной функции, в соответствии с атласом мозга мышей [9]. Стриатум взвешивали, замораживали в жидком азоте и хранили при –70°С до измерения содержания дофамина. Кровь собирали в 1.5 мл пробирки, содержащие 20 мкл 5%-го раствора ЭДТА (Sigma-Aldrich, США). Общий объем раствора доводили до 1 мл фосфатносолевым буфером (ФСБ) и этот раствор аккуратно наслаивали в 15 мл пробирки содержащие 5 мл раствора фикола (р = 1.09). Полученную смесь центрифугировали в течение 15 мин при 4°C со скоростью 1500 об./мин. Из расслоившейся после центрифугирования крови отбирали плазму и лимфоциты. Фракцию лимфоцитов дополнительно центрифугировали 10 мин при 10 000 об./мин и промывали трижды раствором ФСБ, центрифугируя 5 мин после каждого промывания при скорости 10 000 об./мин. Полученные образцы плазмы и лимфоцитов замораживали в жидком азоте и хранили при –70°C до дальнейшего анализа.
Определение содержания дофамина в образцах стриатума. Содержание дофамина в образцах стриатума определяли с помощью высокоэффективной жидкостной хроматографии с электрохимической детекцией (ВЭЖХ-ЭД) по описанной ранее методике [10]. Перед измерением дофамина в образцы ткани добавляли 400 мкл раствора 0.1 М HClO4 и 250 пМ 2,3-дигидроксибензойной кислоты (ДГБА, внутренний стандарт) и их гомогенизировали при 4°С. Часть гомогената отбирали для измерения концентрации белка с помощью набора “BCA Protein Assay Kit” по протоколу производителя (The Thermo Scientific Pierce, США). Остаток гомогената ткани центрифугировали 20 мин со скоростью 18 000 g при 4°C. Супернатант отбирали для измерения дофамина в тканях стриатума.
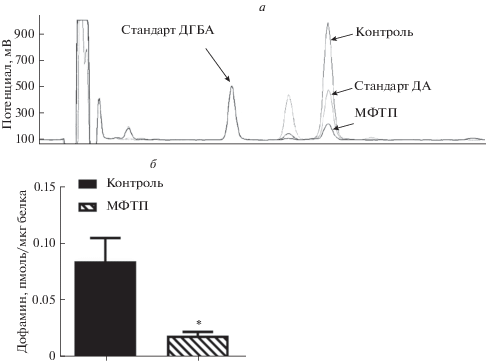
Определение дофамина проводили на обращенно-фазовой колонке (100 × 4 мм ReproSil-Pur C18, 3 мкм) (Dr. Maisch, Германия). Подвижной фазой служил 0.1 М цитратно-фосфатный буфер, содержащий 0.25 мМ октансульфоната натрия, 0.1 М ЭДТА и 6% ацетонитрила (pH 2.55). Потенциал веществ, выходящих из колонки, определяли с помощью электрохимического детектора DECADE II (Antec Leyden, Нидерланды). Пики дофамина и внутреннего стандарта (ДГБА) идентифицировали по времени выхода относительно раствора внешних стандартов (ДГБА и дофамина). Содержание дофамина рассчитывали как отношение площадей пиков в образце к стандартам (рис. 1а). Площадь пиков измеряли с помощью программного обеспечения LabSolutions (Shimadzu, Япония). Содержание дофамина нормировали на концентрацию белка.
Рис. 1.
Определение уровня дофамина (ДА) в стриатуме у мышей через 2 нед. после четырехкратного подкожного введения физраствора (контроль) или четырехкратного введения 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП) в разовой дозе 10 мг/кг (модель болезни Паркинсона). а – Хроматограмма выхода пиков стандартов 2,3-дигидроксибензойной кислоты (ДГБА) и дофамина (ДА), а также пик ДА у мышей в контроле и после введения МФТП. б – Концентрация ДА (пмоль/мкг белка) в стриатуме у мышей после введения физраствора или МФТП. *Р < 0.05.
Определение соотношения мономерного и олигомерного α-синуклеина в крови
В плазме и лимфоцитах с помощью Вестерн блота определяли содержание мономерного и олигомерного α-синуклеина, а также общего и фосфорелированного по серину-129 α-синуклеина. В конечном итоге с помощью анализа изображений и программного обеспечения рассчитывали отношения между различными изоформами α-синуклеина – мономерный/олигомерный, общий/фосфорелированный. Перед измерением α-синуклеина образцы плазмы и лимфоцитов размораживали. Далее плазму разводили в 5 раз RIPA-буфером, отбирали 10 мкл полученного раствора для определения концентрации белка в плазме при помощи ВСА-метода, согласно протоколу производителя. К лимфоцитам добавляли 100 мкл RIPA-буфера и гомогенизировали ультразвуком. После этого отбирали 10 мкл полученного гомогената для определения концентрации белка в лимфоцитах при помощи ВСА-метода согласно протоколу производителя.
Для фореза брали 25 мкг белка на 1 пробу и смешивали с SLB-буфером в соотношении 1 : 2 (v/v). Разделение белков производили в акриламид/бисакриламидном геле, состоящем из 4%-го концентрирующего геля и 14%-го разделяющего геля. Длительность фореза составляла 90 мин, из которых 20 мин – при 80 мВ, а 70 мин – при 180 мВ. Далее осуществляли мокрый перенос белков на нитроцеллюлозную мембрану в течение 75 мин при 290 мА. Перенос белков подтверждали окраской Ponceau. Мембраны отмывали в TNT буфере 3 раза по 5 мин, а затем блокировали 5%-м раствором сухого обезжиренного молока 1 ч. Далее мембраны инкубировали с первыми антителами к α-синуклеину (кролик, 1 : 250, SanaCruz, США), либо фосфорилированному α-синуклеину по серину-129 (кролик, 1 : 750, Invitrogen, USA) в течение 24 ч, отмывали TNT буфером 3 раза по 10 мин. После этого мембраны инкубировали 2 ч со вторыми антителами иммунореактивными к гамма-глобулинам кролика, связанными с пероксидазой хрена (Sigma, США). Затем мембраны отмывали TNT буфером 3 раза по 5 мин и проявляли при помощи раствора ECL (Bio-Rad) согласно протоколу производителя. В ходе реакции пероксидазы хрена с субстратом выделялась хемилюминесценция, которую детектировали при помощи прибора ChemiDoc Touch (Bio-Rad, США). Полученные изображения мембран анализировали с помощью программного обеспечения ImageLab для получения значений интенсивности свечения (Volume Intensity) (Bio-Rad, США).
Статистическая обработка. Полученные данные были статистически обработаны при помощи программы GraphPad Prism 6.0 (GraphPad Software, США). Для статистического сравнения выбранных групп использовали t-тест Стьюдента. Данные представлены как среднее ± стандартная ошибка среднего (SEM). Значение p < 0.05 считалось статистически значимым.
Отношение мономерного α-синуклеина к олигомерному α-синуклеину рассчитывали как:
ХЛмоно – показатель хемилюминесценции мономерного α-синуклеина,
ХЛолиго – показатель хемилюминесценции олигомерного α-синуклеина.
Отношение общего α-синуклеина к фосфорилированному α-синуклеину рассчитывали как:
ХЛмоно – показатель хемилюминесценции мономерного α-синуклеина,
ХЛолиго – показатель хемилюминесценции олигомерного α-синуклеина,
ХЛфосфо – показатель хемилюминесценции фосфорилированного α-синуклеина.
РЕЗУЛЬТАТЫ
Уровень дофамина в стриатуме. С помощью ВЭЖХ показано, что концентрация дофамина в стриатуме у мышей через 2 нед. после четырехкратного введения МФТП в однократной дозе 10 мг/кг снизилось на 76.5% по отношению к контролю после введения по той же схеме физраствора (рис. 1).
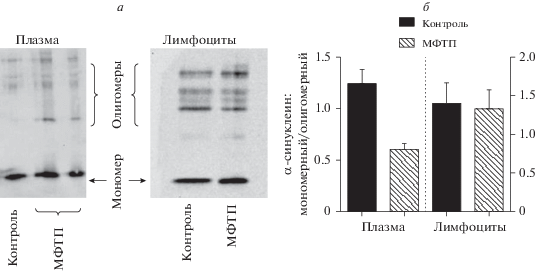
Соотношение содержания мономерного и олигомерного α-синуклеина в крови. Показано, что в плазме у мышей в контроле отношение мономерного альфа-синуклеина к олигомерному альфа-синуклеину в плазме равно 1.238, а в лимфоцитах – 1.404. У мышей через 2 нед. после введения МФТП отношение содержания мономерного α-синуклеина к олигомерному альфа-синуклеину в плазме равно 0.609, а в лимфоцитах – 1.339 (рис. 2).
Рис. 2.
Выявление с помощью Вестерн блота мономерного и олигомерного α-синуклеина в плазме и в лимфоцитах у мышей через 2 нед. после четырехкратного подкожного введения физраствора (контроль) или четырехкратного введения 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП) в разовой дозе 10 мг/кг (модель болезни Паркинсона). а – Мембраны после выявления α-синуклеина. В плазме и в лимфоцитах наблюдается разделение олигомерных форм α-синуклеина по массам, у мышей получавших инъекции МФТП содержание олигомеров увеличивается. В лимфоцитах также наблюдается разделение олигомерных форм α-синуклеина, однако изменений по содержанию α-синуклеина не наблюдается. б – Отношение содержания мономерного α-синуклеина к содержанию олигомерного α‑синуклеина в плазме и в лимфоцитах у мышей после введения физраствора или МФТП. *Р < 0.05.
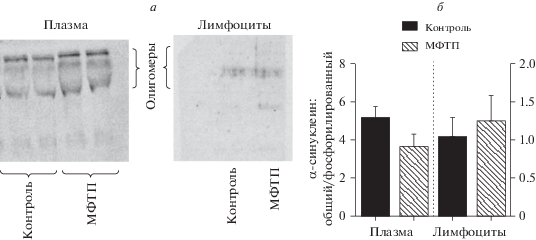
Соотношение содержания общего и фосфорелированного по серину-129 α-синуклеина. Показано, что в плазме у мышей в контроле отношение содержания общего α-синуклеина к фосфорелированному α-синуклеину в плазме равно 5.138, а в лимфоцитах – 10.437. У мышей через 2 нед. после введения МФТП отношение содержание общего α-синуклеина к фосфорелированному α-синуклеину в плазме равно 3.656, а в лимфоцитах – 12.541 (рис. 3).
Рис. 3.
Выявление с помощью Вестерн блота общего и фосфорилированного по серину-129 α-синуклеина в плазме и в лимфоцитах у мышей через две недели после четырехкратного подкожного введения физраствора (контроль) или четырехкратного введения 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП) в разовой дозе 10 мг/кг (модель болезни Паркинсона). а – Мембраны после выявления фосфорилированного по серину-129 α-синуклеина. В плазме наблюдается разделение по массам на 2 подгруппы фосфорилированного α-синуклеина, у мышей получавших инъекции МФТП его содержание увеличивается. В лимфоцитах изменений по содержанию фосфорилированного по серину-129 α-синуклеина не наблюдается. б – Отношение содержания общего α-синуклеина к содержанию α-синуклеина, фосфорелированного по серину-129, в плазме и в лимфоцитах у мышей после введения физраствора или МФТП. *Р < 0.05.
ОБСУЖДЕНИЕ
Многие годы в литературе господствовало представление о том, что α-синуклинопатию – один из ключевых патогенетических механизмов БП – можно воспроизвести только на трансгенных животных при усилении экспрессии гена α-синуклеина или при снижении экспрессии генов ферментов деградации протеосомной системы [11–13]. Однако моделирование БП на трансгенных животных имеет существенный недостаток – они, как правило, не воспроизводят пороговую деградацию нигростриатной дофаминергической системы при которой нарушается моторное поведение [14].
Оказалось, что постадийное развитие БП и, в частности, нарушение моторного поведения при пороговой деградации нигростриатной дофаминергической системы можно воспроизвести только на МФТП-моделях [4, 6, 15]. Так, в данной работе использована оригинальная острая модель клинической стадии БП, полученная на мышах с помощью МФТП [4]. Однако, учитывая то, что чувствительность мышей, полученных из разных питомников и в разное время, к МФТП может различаться [16], в данной работе была проведена коррекция режима его применения. В данном исследовании, так же как и в оригинальной работе по созданию модели [4], для воспроизведения ранней клинической стадии БП мышам четырехкратно с интервалом 2 ч подкожно вводили МФТП, однако в несколько меньшей разовой дозе – 10 мг/кг.
Оценка роли МФТП-моделей в изучении механизмов патогенеза БП значительно повысилась в последнее десятилетие. Так, в частности, было показано, что воздействие МФТП на мышей приводит к увеличению содержания в нигростриатной системе и в ряде других областей мозга α-синуклеина и его нейротоксичных изомеров – агрегированного и фосфорелированного по серину-129 α-синуклеина, что описано в ряде работ, но не во всех у больных при БП [7, 17–19]. Так, у ряда больных при БП отсутствует синуклеинопатия, и в этом случае нельзя ожидать изменений в содержании α-синуклеина и его патологических изоформ в гуморальных средах [20]. Более того, было показано, что на МФТП-моделях, как и у больных при БП, повышается проницаемость гемато-энцефалического барьера [17]. Это должно способствовать поступлению патологических белков из мозга в общую систему циркуляции и распространению их цитотоксического действия на периферии. Это не исключает широко распространенных представлений о том, что синуклеинопатия запускается на периферии токсическими внешними факторами микробиоты [21, 22].
Однако до сих пор отсутствуют попытки проверить на нейротоксических моделях БП, отражается ли системная α-синуклеинопатия и повышение проницаемости гематоэнцефалического барьера на содержании α-синуклеина и его изомеров – агрегированного и фосфорелированного α-синуклеина – в крови у животных, что в ряде работ рассматривается как биомаркеры БП [23, 24].
В проведенном нами исследовании с помощью Вестерн блота показано, что у мышей на МФТП-модели клинической стадии БП в плазме крови практически вдвое снижается отношение мономерного α-синуклеина к олигомерному. Другими словами, у паркинсонических животных вдвое возрастает доля олигомерного токсичного α-синуклеина по сравнению с мономерным нетоксичным α-синуклеином, что хорошо воспроизводит изменение соотношения между содержанием мономерного и олигомерного α-синуклеина в плазме крови, описанные у ряда больных при спорадической БП [23]. В данной работе также показано, что у мышей на МФТП-модели БП в определенной степени воспроизводится увеличение доли фософорелированного α-синуклеина по сравнению с общим α-синуклеином в плазме, что также описано в литературе [23–25]. Однако при оценке отношения содержания общего α-синуклеина к фосфорелированному α-синуклеину была обнаружена только тенденция к снижению этого показателя (р = 0.1).
Учитывая то, что α-синуклеин принимает участие в аутофагии клеток, включая нейроны и лимфоциты [26], особое внимание в данной работе было обращено на экспрессию α-синуклеина и его изомеров – агрегированного и фосфорелированного α-синуклеина – в лимфоцитах у мышей после воздействия МФТП. Однако, в отличие от плазмы, в лимфоцитах не было обнаружено изменения соотношения мономерного и олигомерного α-синуклеина, а также соотношения общего α-синуклеина и фосфорелированного α-синуклеина.
Из сопоставления данных по содержанию α‑синуклеина и его изоформ в крови у мышей, полученных в этой работе, и у больных при БП, опубликованных ранее, следует, что, по крайней мере, по соотношению этих белков (α-синуклеин: мономерный/олигомерный, общий/фосфорилированный) в плазме крови наша модель хорошо воспроизводит патогенез БП, по крайней мере, у части больных и может быть использована для оценки периферических проявлений этого заболевания. Эта же модель также может быть использована для усовершенствования дифференциальной диагностики БП, оценки эффективности используемой терапии и скрининга новых лекарственных средств.
Таким образом, используемая нами модель клинической стадии БП у мышей воспроизводит изменения содержания α-синуклеина и его изоформ в плазме крови, характерные для ряда больных при БП, что может быть использовано для углубленной оценки периферических патогенетических процессов, а также для усовершенствования и создания новых технологий диагностики и лечения этого заболевания.
Список литературы
Trompetero A., Gordillo A., Del Pilar M.C., Cristina V.M., Bustos Cruz R.H. // Curr. Pharm. Des. 2018. V. 24. P. 22–45.
Maiti P., Manna J., Dunbar G.L. // Transl. Neurodegener. 2017. V. 6. P. 28–63.
Hartmann A. // Dialogues Clin. Neurosci. 2004. V. 6. P. 281–293.
Ugrumov M.V., Khaindrava V.G., Kozina E.A., Kucheryanu V.G., Bocharov E.V., Kryzhanovsky G.N., Kudrin V.S., Narkevich V.B., Klodt P.M., Rayevsky K.S., Pronina T.S. // Neuroscience. 2011. V. 181. P. 175–188.
Przedborski S., Vila M. // Adv. Neurol. 2001. V. 86. P. 177–186.
Meredith and Rademacher // J. Parkinsons Dis. 2011. V. 1. P. 19–33.
Canerina-Amaro A., Pereda D., Diaz M., Rodriguez-Barreto D., Casañas-Sánchez V., Heffer M., Garcia-Esparcia P., Ferrer I., Puertas-Avendaño R., Marin R. // Front. Neurosci. 2019. V. 13. P. 382–403.
Vargas J.Y., Grudina C., Zurzolo C. // Ageing Res. Rev. 2019. V. 50. P. 89–101.
Franklin K.B.J., Paxinos G. // The Mouse Brain in Stereotaxic Coordinates, Compact 3rd Edition. London: Academic press, 2008.
Kozina E.A., Kim A.R., Kurina A.Y., Ugrumov M.V. // Neurobiol. Dis. 2017. V. 98. P. 108–121.
Chesselet M.F., Fleming S., Mortazavi F., Meurers B. // Parkinsonism Relat. Disord. 2008 V. 14. P. 1–12.
Bentea E., Verbruggen L., Massie A. // J. Parkinson’s Dis. 2017. V. 7. P. 31–63.
Henrich M.T., Geibl F.F., Lee B., Chiu W.H., Koprich J.B., Brotchie J.M., Timmermann L., Decher N., Matschke L.A., Oertel W.H. // Acta Neuropathol. Commun. 2018. V. 6. P. 39–58.
Dawson T.M., Ko H.S., Dawson V.L. // Neuron. 2010. V. 66. P. 646–661.
Muñoz-Manchado A.B., Villadiego J., Romo-Madero S., Suárez-Luna N., Bermejo-Navas A, Rodríguez-Gómez J.A., Garrido-Gil P., Labandeira-García J.L., Echevarría M., López-Barneo J., Toledo-Aral J.J. // J. Neurochem. 2016. V. 136. P. 373–387.
Jackson-Lewis V., Przedborski S. // Nat. Protoc. 2007. V. 2. P. 141–151.
Zhang Q.S., Heng Y., Mou Z., Huang J.Y., Yuan Y.H., Chen N.H. // Acta Pharmacol. Sin. 2017. V. 10. P. 1317–1328.
Matsuo K., Cheng A., Yabuki Y., Takahata I., Miyachi H., Fukunaga K. // Neuropharmacology. 2019. V. 150. P. 164–174.
Zhu J., Dou S., Wang C., Jiang Y., Wang C., Cheng B. // Brain Res. 2019. V. 1721. P. 1–12.
Johansen K.K., Torp S.H., Farrer M.J., Gustavsson E.K., Aasly J.O. // Case Rep. Neurol. Med. 2018. V. 2018. P. 2–5.
Clairembault T., Leclair-Visonneau L., Coron E., Bourreille A., Le Dily S., Vavasseur F., Heymann M.F., Neunlist M., Derkinderen P. // Acta Neuropathol. Commun. 2015. V. 10. P. 3–12.
Tremlett H., Bauer K.C., Appel-Cresswell S., Finlay B.B., Waubant E. // Ann. Neurol. 2017. V. 81. P. 369–382.
Chen X.Q., Niu J.P., Peng R.Q., Song Y.H., Xu N., Zhang Y.W. // Acta Neurol. Scand. 2019. V. 140. P. 268–273.
Foulds P.G., Mitchell J.D., Parker A., Turner R., Green G., Diggle P., Hasegawa M., Taylor M, Mann D., Allsop D. // FASEB J. 2011. V. 25. P. 4127–4137.
El-Agnaf O.M., Salem S.A., Paleologou K.E., Curran M.D., Gibson M.J., Court J.A., Schlossmacher M.G., Allsop D. // FASEB J. 2006 V. 20 P. 419–425.
Colasanti T., Vomero M., Alessandri C., Barbati C., Maselli A., Camperio C., Conti F., Tinari A., Carlo-Stella C., Tuosto L., Benincasa D., Valesini G., Malorni W., Pierdominici M., Ortona E. // Cell Death Dis. 2014. V. 5. P. 1–9.
Дополнительные материалы отсутствуют.