

Нейрохимия, 2021, T. 38, № 1, стр. 3-13
Влияние гимантана на изменения Са2+ и Nа+, вызванные активацией NMDA-каналов в культивируемых нейронах мозга крысы
Н. А. Воронина 1, О. Ю. Лисина 1, И. А. Красильникова 2, В. Г. Кучеряну 1, И. Г. Капица 3, Т. А. Воронина 3, А. М. Сурин 1, 2
1 ФГБНУ “НИИ общей патологии и патофизиологии”
Москва, Россия
2 ФГАУ “НМИЦ Здоровья детей” Минздрава России
Москва, Россия
3 ФГБНУ “НИИ фармакологии им. В.В. Закусова”
Москва, Россия
Поступила в редакцию 17.12.2020
После доработки 23.12.2020
Принята к публикации 24.12.2020
Аннотация
При болезни Паркинсона одним из ведущих механизмов нейродегенерации является эксайтотоксичность глутамата, основного возбуждающего нейромедиатора мозга, развивающаяся в результате избыточной стимуляции ионотропных глутаматных рецепторов NMDA-типа. Препараты, способные обратимо ингибировать NMDA рецепторы, рассматривают в качестве перспективных фармакологических агентов, предотвращающих гибель нейронов и замедляющих развитие нейродегенерации. В данной работе исследовано влияние N-(2-адамантил)-гексаметиленимина гидрохлорида (гимантана, Hmn) на изменения внутриклеточной концентрации Са2+ ([Ca2+]i), Nа+ ([Na+]i) и митохондриального потенциала (ΔΨm), индуцированные NMDA в первичных нейроглиальных культурах, приготовленных из коры головного мозга 1-2-дневных крыс Вистар. Измерения [Ca2+]i, [Na+]i и ΔΨm выполнены в индивидуальных нейронах методом флуоресцентной микроскопии с применением индикаторов Ca2+ и Na+, соответственно Fura-FF и SBFI, и потенциал-чувствительного зонда Rh123. Кратковременные добавки NMDA (2–3 мин, 10 мкМ) вызывали быстрые транзиторные подъемы [Ca2+]i, которые в ~30% нейронов были частично и обратимо ингибированы высокими концентрациями Mg2+ (10 мМ) и Hmn (100 мкМ). Длительное действие нейротоксической концентрации NMDA (16 мин, 500 мкМ) вызывало развитие отсроченной кальциевой дизрегуляции (ОКД), устойчивый подъем [Na+]i и сильное падение ΔΨm. Гимантан снижал рост [Ca2+]i, отдалял развитие ОКД, уменьшал деполяризацию митохондрий и способствовал восстановлению исходных значений [Ca2+]i, [Na+]i и ΔΨm после прекращения действия NMDA. В отличие от Hmn, высокоаффинный неконкурентный ингибитор NMDA-каналов МК-801 необратимо ингибировал изменения [Ca2+]i и ΔΨm. Очевидно, Hmn проявляет нейропротекторные свойства, благодаря снижению потоков Са2+ и Na+ сквозь NMDA-каналы и меньшему росту [Ca2+]i и [Na+]i. Обратимость ингибирования NMDA-каналов может благоприятствовать нормализации функционирования мозга после отмены гимантана.
ВВЕДЕНИЕ
При болезни Паркинсона (БП) нарушается система отрицательной обратной связи межнейрональных контактов. В результате этого возбужденные нейроны не получают или не воспринимают быстрого и сбалансированного ингибирующего сигнала от других нейронов. В структуре мозга, называемой “черной субстанцией” (substantia nigra pars compacta), которая в значительной степени контролирует моторно-двигательную сигнализацию, отрицательную обратную связь осуществляют дофаминэргические нейроны. Их постепенная гибель по мере старения организма является причиной недостаточной компенсации возбуждающей сигнализации [1, 2].
В центральной нервной системе основным возбуждающим нейромедиатором является глутаминовая кислота, которая при физиологических рН существует в форме цвиттериона, глутамата (Glu). Возбуждение нейронов возникает после связывания Glu с несколькими типами ионотропных рецепторов, в структуру которых входит канал для ионов Ca2+, Na+ и K+ [3, 4]. Для указанных катионов максимальной проводимостью и минимальной скоростью инактивации обладают рецепторы NMDA-типа. Избыточная стимуляция этих рецепторов глутаматом, так называемая глутаматная нейротоксичность или эксайтотоксичность, приводит к гибели нейронов [5–9]. В этой связи одним из направлений разработки лекарств против БП является поиск ингибиторов NMDA-рецепторов [10–15].
Препараты, которые конкурируют с глутаматом или глицином в сайтах их связывания на NMDA-рецепторе, блокируют нормальную функцию и, таким образом, до сих пор не прошли клинических испытаний из-за побочных эффектов (сонливость, галлюцинации и даже кома) [16]. Аллостерический ингибитор NMDA-рецепторов МК-801 (дизоцильпин), который часто используют в экспериментах на клеточных культурах или с животными, прочно связывается с внеканальной частью рецептора и почти необратимо тормозит межнейрональную сигнализацию в мозге в течение клинически неприемлемого времени. Поэтому актуальным остается поиск и создание новых фармакологических препаратов, которые эффективно и селективно блокируют NMDA-рецепторы, но при этом достаточно быстро выводятся из организма.
В настоящее время для снижения симптоматики БП применяют только один ингибитор NMDA-рецепторов, мемантин, который является аминопроизводным адамантана. Способность блокировать NMDA-каналы для неорганических катионов подробно изучена методами электрофизиологии для производных адамантана [17, 18]. Однако внутриклеточные концентрации этих катионов, прежде всего Са2+ и Na+, определяются не только скоростью их поступления из окружающей клетки среды в цитозоль по NMDA и другим каналам, но и скоростью удаления этих катионов из цитозоля соответствующими ионными насосами и обменниками, локализованными как на плазматической, так и на внутриклеточных мембранах. Помимо этого, остается открытым вопрос о динамике восстановления нормальной концентрации ионов Ca2+ и Na+ после удаления Glu и ингибитора NMDA-каналов.
Ранее на экспериментальном паркинсонизме мышей, вызванном системным введением пронейротоксина МФТП показано, что аналог мемантина гимантан (Hmn) при предварительном применении уменьшал паркинсоническую симптоматику (гипокинезию и мышечную ригидность животных [19]), снижал уровень продуктов перекисного окисления липидов в мозге мышей с МФТП-индуцированным паркинсоническим синдромом [20].
В данной работе сопоставлено влияние гимантана (N-(2-адамантил)-гексаметиленимина гидрохлорида, Hmn) с влиянием МК-801 и Mg2+ на изменения внутриклеточной концентрации Са2+ ([Ca2+]i), Nа+ ([Na+]i) и митохондриального потенциала ΔΨm, индуцированные NMDA в культивируемых нейронах. Показано, что Hmn снижает индуцированный NMDA рост [Ca2+]i и уменьшает деполяризацию митохондрий в нейронах первичной культуры из коры головного мозга крысы в меньшей степени и более обратимо, чем МК-801, и с различной эффективностью в разных клетках. Hmn меньше влияет на [Na+]i, чем на [Ca2+]i, что может благоприятствовать нормализации функционирования мозга после отмены гимантана.
МАТЕРИАЛЫ И МЕТОДЫ
Первичные нейрональные культуры приготавливали из коры головного мозга 1-дневных крыс породы Вистар, как описано ранее [21]. Кратко, животное анестезировали, декапитировали и извлекали кору головного мозга. Суспензию клеток получали, обрабатывая ткань коры папаином, диссоциировали пипетированием и отмывали от разрушенных клеток двукратным осаждением в центрифуге. Полученную суспензию клеток в нейробазальной среде с добавлением 2% Supplement B-27 и 0.5 мМ L-глутамина наносили на покровные стекла, прикрепленные к лункам 35 мм пластиковых чашек Петри (“MatTek”, США). Стекла предварительно покрывали полиэтиленимином (1 мг/мл, 30 мин); плотность посадки 2.5 × × 105 клеток/лунку. Клетки росли при 37°С в атмосфере 5% СО2/95% воздуха при 100% влажности.
Клеточные культуры использовали спустя 12–15 дней после посадки (12–15 дней в культуре, ДВК). Клетки нагружали низкоаффинными Ca2+-индикаторами Fura-FF или X-rhod-FF и Na+ индикатором SBFI в форме ацетоксиметиловых эфиров (соответственно Fura-FF/AМ, 4–6 µM, X-rhod-FF/AM, 1 µM и SBFI/AM, 6–8 µM) при 36°С в течение 50–60 мин. Изменения величины митохондриального потенциала ΔΨm выполнены с помощью потенциал-чувствительного флуоресцентного зонда родамина-123 (Rh123; клетки нагружали 15 мин, 2,5 µМ, 36°С).
Измерения проводили в буферных растворах состава (мМ): 134 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, 5 glucose; pH = 7.4 при температуре 27–29°С. В тех случаях, когда NMDA-каналы ингибировали, повышая концентрацию Mg2+, его концентрацию в буфере увеличивали до 10 мМ; остальные компоненты сохраняли в той же концентрации. В экспериментах с применением длительных воздействий высоких концентраций NMDA (500 µМ, 16 мин), удаление агониста проводили номинально бескальциевым буфером, в котором 2 мМ CaCl2 замещали на 0.1 мМ Ca2+-хелатора ЭГТА (этиленгликоль-бис(β-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота). Калибровку максимальных сигналов Ca2+-индикаторов осуществляли в конце каждого эксперимента, добавляя 2 µM Ca2+-ионофора иономицина в присутствии 5 мМ CaCl2.
Наступление отсроченной кальциевой дизрегуляции (лаг-период ОКД) определяли, как время второго максимум на графике первой производной сигнала X-rhod-FF (график зависимости ΔF/ΔT от Т, где F – интенсивность флуоресценции индикатора; Т – время от момента добавки NMDA; Δ – шаг дифференцирования, 10с).
Флуоресцентные измерения выполнены с помощью системы получения и анализа изображений на базе инвертированного микроскопа Olympus IX-71 (Япония), включающей 175 Вт лампу, систему освещения с фильтровыми колесами каналов возбуждения и эмиссии Sutter Lambda 10-2 (США), перемещение которых синхронизовано с регистрацией изображений охлаждаемой CCD-камерой CoolSnap HQ-2 (Photometrics, США). Управление системой, сохранение изображений и последующий их анализ осуществляли с помощью программ MetaFluor и MetaFluor Analyst (Molecular Devices, USA).
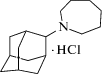
Гимантан (рис. 1) (N-(2-адамантил)-гексаметиленимина гидрохлорид) синтезирован в ФГБНУ “НИИ фармакологии имени В.В. Закусова” из адамантан-2-она и гексаметиленимина в условиях реакции Лейкарта и представляет собой белое кристаллическое вещество, хорошо растворимое в воде [15, 22, 23].
Флуоресцентные индикаторы Ca2+, Na+ и Rh123 приобретены у Molecular Probes (Thermo Fisher, USA). Остальные реагенты Sigma (USA).
Статистический анализ данных, построение графиков, вычисление площадей под кривыми выполняли, используя программы GraphPad Prism 8 и Microsoft Excel 2010. Результаты статистической обработки представлены как среднее ± SEM (стандартная ошибка среднего). Количество клеток было от 40 до 60, если не отмечено особо. Статистически отличия между сравниваемыми группами данных считали достоверными, если p < 0.05 (two-tailed t-test).
РЕЗУЛЬТАТЫ
Изменения внутриклеточной концентрации Са2+ ([Ca2+]i) вызванные одним NMDA и в присутствии ингибиторов NMDA-рецепторов. Ранее электрофизиологические измерения на свежеизолированных нейронах гиппокампа крысы показали, что высокая концентрация гимантана (Hmn, 100 мкМ) ингибирует >90% ионного тока через NMDA-рецепторы, причем Hmn, как и Mg2+, располагается внутри канала [17, 18]. Поэтому мы проверили, 1) будут ли 100 мкМ Hmn столь же эффективно ингибировать рост [Ca2+]i в первичной культуре нейронов коры головного мозга крысы и 2) будет ли эффективность ингибирования гимантаном и ионами магния одинаковой в одних и тех же клетках. Измерения с помощью низкоаффинного Са2+-индикатора Fura-FF, показали, что ~75% клеток (29 из 39 в поле наблюдения) отреагировали на кратковременное добавление NMDA (10 µM, 2 мин) быстрым подъемом [Ca2+]i, причем амплитуда роста была индивидуальной для каждой клетки (рис. 2а). Остальные клетки, не изменившие [Ca2+]i в ответ на NMDA, очевидно, не содержали NMDA-рецепторов и относились к глие либо к нейронам, лишенным этих ионотропных рецепторов. Эти клетки исключены из рассмотрения при анализе данных на рис. 2г и 2в.
Рис. 2.
Изменения внутриклеточной концентрации свободного кальция ([Ca2+]i), вызванные одним NMDA и в присутствии ингибиторов канала – Mg2+ и гимантана (Hmn). а–г – изменения [Ca2+]i представлены как отношения сигналов флуоресцентного низкоаффинного Ca2+-индикатора Fura-FF, измеренных при возбуждении на 340 и 380 нм (F340/F380) и регистрации при 525 нм. Графики нормированы по оси ординат для каждого нейрона согласно уравнению: (R-Rmin)/(Rmax-Rmin), где R – текущее значение F340/F380, Rmin и Rmax – соответственно минимальное наблюдаемое в начале эксперимента и максимальное в присутствии Ca2+ ионофора иономицина (2 мкМ +5 мМ внеклеточного Ca2+) в конце эксперимента. Концентрации агентов во время первой (1st) и четвертой (4th) добавок: 10 µM NMDA, 10 µM Gly, 0 Mg2+, 2 mM Ca2+; во время второй (2nd) и третьей (3rd) добавок концентрации Mg2+ и Hmn составляли соответственно 10 и 0.1 мM; концентрации остальных компонентов сохраняли такими же, как во время первой и четвертой добавок.
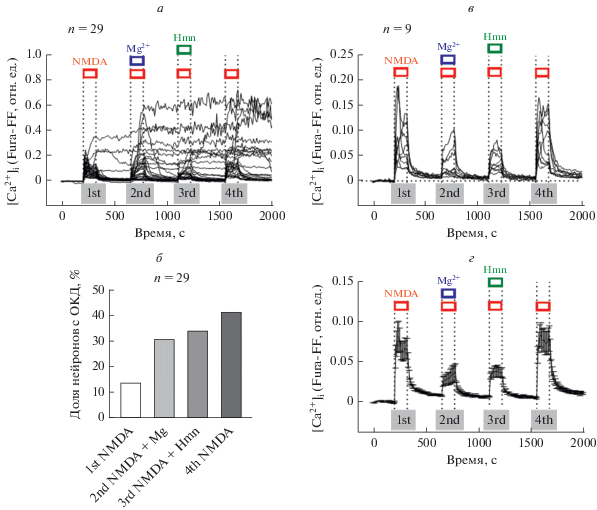
Примерно 14% клеток, которые отреагировали на первую аппликацию NMDA, не смогли восстановить исходное значение [Ca2+]i после отмывания агониста буфером (рис. 2а, б). В них развилась отсроченная кальциевая дизрегуляция (ОКД) [7]. Последующее чередование добавления и отмывания NMDA последовательно увеличивало долю клеток, в которых развилась ОКД (рис. 2а, б), несмотря на присутствие Hmn и Mg2+. По-видимому, в этих нейронах в период между добавками не успевала восстановиться [Na+]i и, соответственно, потенциал плазматической мембраны [24]. Подобный кумулятивный эффект наблюдали при повторных кратковременных добавках глутамата к культивируемым гиппокампальным нейронам [25].
Увеличение концентрации Mg2+ в буфере до 10 мМ уменьшило скачок [Ca2+]i, вызванный повторной добавкой NMDA, но не во всех клетках и в разной степени (рис. 2а). Доля клеток, в которых развилась ОКД, увеличилась с ~14 до ~31% (рис. 2б). В этих нейронах [Ca2+]i оставалась высокой на протяжении остальной части эксперимента независимо от наличия NMDA.
Поскольку Hmn входит, как отмечалось выше, только в предварительно открытый агонистом канал, то добавление этого ингибитора осуществляли одновременно с добавлением NMDA. Гимантан (100 мкМ) снизил амплитуду [Ca2+]i ответов на NMDA, однако, как и в случае с предшествующей добавкой 10 мМ Mg2+, ингибирование было отмечено только в части нейронов (рис. 2а, б). Доля нейронов, в которых [Ca2+]i осталась на высоком уровне, несмотря на удаление NMDA и Hmn, увеличилась до ~35% (рис. 2б).
Для проверки того, что влияние 10 мМ Mg2+ и 100 мкМ Hmn на изменения [Ca2+]i связано с их ингибирующим действием, была сделана четвертая добавка NMDA в буфере того же состава, что и первая добавка (рис. 2). Доля нейронов, в которых возникла ОКД, возросла до ~41% (рис. 2б).
Учитывая сложный характер [Ca2+]i ответов клеток на действие как одного NMDA, так и в присутствии Mg2+ или Hmn, мы рассмотрели динамику ответов на стимулы только в тех нейронах, в которых первая (1st) и последняя (4th) добавки NMDA вызывали примерно одинаковые по амплитуде и полностью обратимые изменения [Ca2+]i (рис. 2в, г). Профиль кривых [Ca2+]i не был одинаковым во время каждой из четырех добавок даже в тех клетках, в которых были одинаковы амплитуды подъемов [Ca2+]i. Поэтому количественным показателем была выбрана суммарная [Ca2+]i за время действия соответствующих агентов, т.е. площадь под кривой изменений [Ca2+]i (area under curve, AUC).
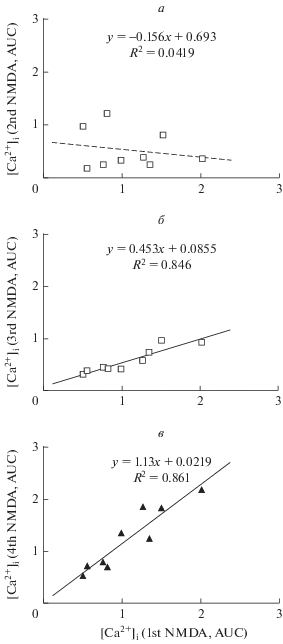
График соотношения площади под кривой [Ca2+]i во время первой (1st) и последней (4th) добавки NMDA был близок к линейной функции с наклоном 1.13 ± 0.17 (Mean ± SEM, n = 9) (рис. 3в). Чем больше была амплитуда подъема [Ca2+]i (и соответственно AUC) во время первой добавки NMDA, тем больше она была и при последнем четвертом воздействии агониста. Такое же соотношение наблюдалось и при действии NMDA в присутствии Hmn, но наклон был в 2.5 раза меньше (0.453 ± 0.073, n = 9), что отражает ингибирующее действие антагониста (рис. 3б). Неожиданным оказалось отсутствие корреляции между интегральным ответом [Ca2+]i на первую добавку NMDA и на вторую при его действии в присутствии 10 мМ Mg2+ (рис. 3а). Не выявлено также корреляции между действием NMDA в присутствии 10 мМ Mg2+ и 100 мкМ Hmn (график не показан). При этом средние амплитуды изменений [Ca2+]i (рис. 2г) и средние величины AUC (рис. 3а, б) при действии NMDA в присутствии 10 мМ Mg2+ и 100 мкМ Hmn практически одинаковые (в диапазоне 0.6–0.7).
Рис. 3.
Соотношение площадей под графиками изменений [Ca2+]i, вызванных одним NMDA и NMDA в присутствии Mg2+ или Hmn (см. рис. 2а, в, г). Площади под кривыми изменений [Ca2+]i (AUC, отн. ед.) определяли в течение первых 150 с после добавки агентов. По осям абсцисс представлены значения параметра AUC после самой первой добавки NMDA (соответствует 1st на рис. 2а, в, г). По осям ординат представлены значения AUC после добавки NMDA в присутствии 10 мМ Mg2+ (а) (2nd на рис. 2) и NMDA в присутствии 0.1 мМ Hmn (б) (3rd, на рис. 2). Последняя четвертая добавка NMDA (в) (4th, на рис. 2) сделана для проверки воспроизводимости изменений [Ca2+]i.
Гимантан частично и обратимо ингибирует изменения [Ca2+]i и ΔΨm, индуцированные высокой концентрацией NMDA. Нейропротекторные свойства ингибиторов глутаматных каналов NMDA-типа особенно важны при высоких, нейротоксических концентрациях глутамата. Поэтому мы сравнили изменения [Ca2+]i и ΔΨm, применяя высокие концентрации NMDA в присутствии Hmn. Для сравнения был использован аллостерический ингибитор NMDA-рецепторов МК-801, место связывания которого, в отличие от Hmn, находится вне канала [17, 18].
Действие 500 мкМ NMDA вызывало быстрый, но относительно небольшой подъем [Ca2+]i, который после короткого лаг-периода переходил во вторую фазу подъема [Ca2+]i, названную отсроченной кальциевой дизрегуляцией (ОКД) (рис. 4а) [6, 7]. Синхронно с ОКД происходила вторая фаза падение ΔΨm, которая проявлялась в быстром и значительном увеличении флуоресценции Rh123 (рис. 4б, см. также рис. 4д, е) [26]. Отмывание NMDA приводило к медленному снижению [Ca2+]i, начало которого, как и наступление ОКД, было индивидуальным в каждой клетке. Снижение сигнала потенциал-чувствительного митохондриального зонда Rh123 в период отмывания NMDA (рис. 4б) обусловлено частично восстановлением потенциала (ростом ΔΨm), а частично вытеканием зонда из клеток сквозь деполяризованную плазматическую мембрану [26].
Рис. 4.
Изменения [Ca2+]i и митохондриального потенциала (ΔΨm), вызванные добавлением одного NMDA и в присутствии обратимого (гимантана, Hmn) и необратимого (МК-801) ингибиторов NMDA-каналов. Серыми линиями показаны графики [Ca2+]i и ΔΨm индивидуальных клеток, черными линиями – средние значения (± ошибка среднего). а – изменения [Ca2+]i в контрольном эксперименте при действии одного NMDA (500 мкМ), в – в присутствии Hmn (100 мкМ) и ж – в присутствии МК-801 (10 мкМ). Изменения [Ca2+]i измерены, как сказано в подписи к рис. 2. б, г, з – изменения величины митохондриального потенциала ΔΨm выполнены с помощью потенциал-чувствительного флуоресцентного зонда родамина-123 (Rh123). Сигналы Rh123 (F) нормированы относительно исходной величины в покоящихся нейронах (Fo). д, е – изменения [Ca2+]i и ΔΨm совмещены для двух представительных нейронов, один из которых имел ОКД (д), а другой устоял против развития ОКД (е). Клеточная культура кортикальных нейронов в возрасте 14 дней. Концентрации (мкМ): NMDA – 500, Gly – 10, Hmn – 100, MK-801 – 10. NMDA добавляли в безмагниевом буфере.
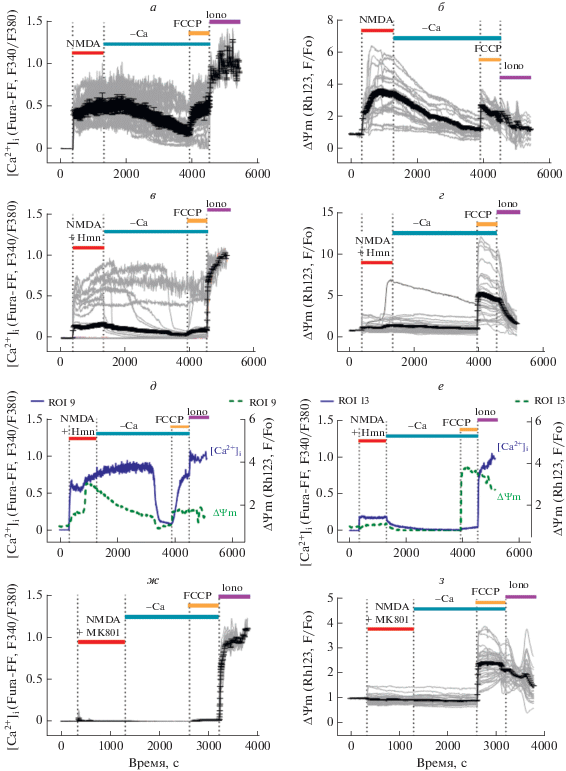
Для оценки количества Са2+, которое митохондрии накопили во время действия NMDA и смогли удержать после отмывания агониста бескальциевым буфером, в конце эксперимента добавляли протонофор FCCP (1 мкМ). Протонофор вызывал полную деполяризацию митохондрий, ликвидируя электрофоретическую силу, удерживавшую Са2+ в матриксе митохондрий.
Видно, что те клетки, которые в присутствии Hmn имели небольшой подъем [Ca2+]i, смогли накопить и удержать относительно небольшое количество Са2+ по сравнению с теми нейронами, в которых, несмотря на присутствие ингибитора, развилась ОКД (рис. 4а, в).
Две представительные клетки, в одной из которых произошла ОКД с последующим восстановлением низкой [Ca2+]i после отмывания NMDA, а во второй ОКД не успела возникнуть за время действия агониста, показаны, соответственно на рис. 4д и 4е. Нейрон, в котором развилась ОКД, смог удержать небольшое количество Rh123 в митохондриях (рис. 4д), тогда как нейрон, не имевший ОКД (рис. 4е), высвободил небольшое количество Са2+ из митохондрий, но зато относительно много Rh123, поскольку митохондрии сохранили высокий ΔΨm во время действия NMDA.
Гимантан частично ингибирует изменения [Ca2+]i и внутриклеточной концентрации Nа+ ([Na+]i), вызванные NMDA. NMDA каналы хорошо проницаемы для ионов натрия. Мы сопоставили изменения [Ca2+]i и [Na+]i, вызванные одной NMDA и в присутствии Hmn. Ингибитор уменьшил долю нейронов, в которых произошла ОКД за время действия NMDA (рис. 5а, в, ж). Этот феномен проявился также в увеличении лаг-периода ОКД в присутствии Hmn (рис. 5ж).
Рис. 5.
Изменения внутриклеточной концентрации [Ca2+]i и [Na+]i, вызванные добавлением NMDA и гимантана Hmn. Серыми линиями показаны графики [Ca2+]i и [Na+]i индивидуальных клеток, черными линиями – средние значения (± ошибка среднего). а, в – измерения [Ca2+]i выполнены с помощью флуоресцентного Ca2+-индикатора X‑rhod-FF. б, г – изменения [Na+]i представлены как отношения сигналов флуоресцентного Na+-индикатора SBFI, измеренных при возбуждении на 340 и 380 нм (F340/F380) и регистрации при 525 нм; значения F340/F380 нормированы относительно исходной величины в покоящихся нейронах. ж – зависимость доли клеток, имевших ОКД от лаг-периода ОКД (определение лаг-периода см. раздел “Материалы и методы”). д, е – изменения [Ca2+]i и [Na+]i совмещены для двух представительных нейронов, в одном из которых (д) началось развитие ОКД. з – площади под графиками изменения [Na+]i во время действия одного NMDA или NMDA + Hmn, а также в период после удаления NMDA (-Ca) или удаления (-Ca + Hmn). Клетки нагружали Ca2+ и Na+ индикаторами как описано в разделе “Материалы и методы”. Клеточная культура кортикальных нейронов в возрасте 12 дней. Концентрации добавляемых агентов как на рис. 4.
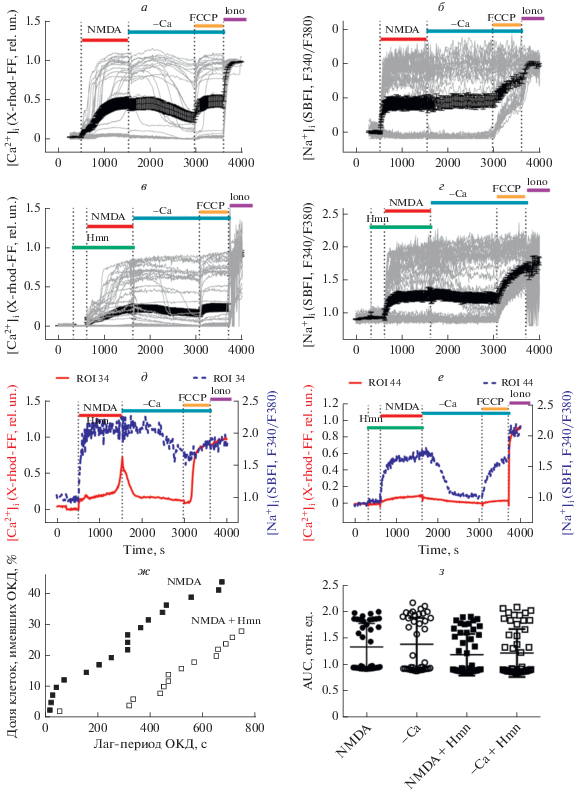
Увеличение [Na+]i в ответ на NMDA происходило без лаг-периода и [Na+]i оставалась на уровне высокого плато до конца действия агониста. Отмывание NMDA не привело к снижению [Na+]i даже в тех нейронах, в которых снизилась [Ca2+]i (рис. 5б). Похожее соотношение изменений [Ca2+]i и [Na+]i соответствует описанному ранее для культуры гиппокампальных [27] и кортикальных нейронов [24].
Присутствие Hmn увеличило долю нейронов, в которых не произошло роста [Na+]i (рис. 5г). Более того, появились нейроны, в которых удаление NMDA вызывало снижение [Na+]i с уровня высокого плато до почти базального. Однако эти отличия имели характер тенденции и достоверно не отличались (рис. 5з).
ОБСУЖДЕНИЕ
При болезни Паркинсона, как и при ряде других нейродегенеративных заболеваний, одним из ведущих механизмов гибели нейронов является эксайтотоксичность основного возбуждающего нейромедиатора мозга глутамата [2, 13, 14]. Эксайтотоксичность развивается в результате избыточной стимуляции ионотропных глутаматных рецепторов NMDA-типа. Препараты, способные обратимо ингибировать NMDA рецепторы, рассматривают в качестве перспективных фармакологических агентов, предотвращающих гибель нейронов и замедляющих развитие глутаматной эксайтотоксичности разной этиологии, включая нейродегенерацию. В данной работе методом флуоресцентной микроскопии измерены изменения [Ca2+]i, [Na+]i и ΔΨm в первичной нейро-глиальной культуре коры головного мозга и проведен анализ нейропротекторных возможностей Hmn в условиях, соответствующих эксайтотоксическому действию Glu.
Способность Hmn обратимо ингибировать NMDA-рецепторы была исследована ранее на свежеизолированных нейронах гиппокампа крысы методами электрофизиологии [17, 18]. Авторы обнаружили, что степень блокады увеличивалась с повышением концентрации Hmn, тогда как деблокирование не зависело от концентрации и усиливалось при гиперполяризации клеточной мембраны. Примечательно, что Hmn оставался “запертым” внутри канала “активационными воротами” и деблокирование канала происходило лишь при новом воздействии агониста на клетку и открывании “ворот”.
Данные, полученные нами с помощью кальциевого имиджинга индивидуальных нейронов, не во всем согласуются с результатами электрофизиологических экспериментов. Во-первых, Hmn в концентрации 100 мкМ, при которой на 90% ингибировал ионные токи через канал, блокировал индуцированный NMDA рост [Ca2+]i не во всех нейронах, а только в тех, в которых перед повторным добавлением NMDA [Ca2+]i смогла вернуться к базальному уровню (рис. 2в). В тех нейронах, в которых [Ca2+]i не вернулась к уровню в покоящихся нейронах перед повторной стимуляцией NMDA-рецепторов, Hmn не влиял на [Ca2+]i (третья добавка NMDA+Hmn на рис. 2а). Неспособность ингибиторов снижать [Ca2+]i в нейронах, если в них уже возникла ОКД, наблюдали ранее на культурах гиппокампа крысы [25] и гранулярных нейронов мозжечка [28]. Объясняли этот феномен тем, что во время высокого [Ca2+]i плато, которое наступает после завершения развития ОКД, скорости поступления Са2+ в цитоплазмы и удаления из нее становятся очень низкими и поэтому никакие ингибиторы NMDA-каналов не способны оказать заметного эффекта на уровень [Ca2+]i [7].
Второе отличие данных кальциевого имиджинга от электрофизиологических измерений состоит в том, что Hmn (100 мкМ) ингибировал рост [Ca2+]i на ~50% в тех клетках (~30% от общего числа n = 29), в которых индуцированный NMDA рост был обратим (рис. 2в). Ни в одном нейроне не отмечалось 90% ингибирования. Возможно, это отличие связано с типом клеток. В настоящей работе это нейроны из коры головного мозга 1-дневных крыс после 12 дней содержания в культуре, тогда как в работах [17, 18] использовали механически диссоциированные нейроны из свежевыделенного среза мозга крысы 2–3 месячного возраста. Эти различия могли проявиться в отличиях в субъединичном составе NMDA-рецепторов, других компонентов клетки, оказывающих влияние на функционирование рецепторов. Аффинность Hmn к NMDA рецепторам в полученных нами культурах могла быть заметно ниже, чем в остро изолированных нейронах из среза мозга и, соответственно, ингибирование менее эффективным. Другими словами, в части нейронов Hmn не входил в канал NMDA рецептора либо перекрывал его не полностью и поэтому в этих нейронах сохранялся, хоть и пониженный, транспорт Са2+ и [Ca2+]i не опускалась на 90% относительно амплитудной величины.
Поскольку Hmn подобно Mg2+ ингибирует ионный канал NMDA-рецептора потенциал-зависимым образом, мы ожидали подобия в изменениях [Ca2+]i при действии NMDA в присутствии ингибирующих концентраций Hmn и Mg2+. Оказалось, что Mg2+ (10 мМ) снижал площадь под кривой вызванных NMDA изменений [Ca2+]i тем сильнее, чем больше входило Са2+ в этот нейрон при действии одного NMDA (рис. 3а). Поэтому индуцированный NMDA подъем [Ca2+]i в присутствии высокой концентрации Mg2+ в буфере (10 мМ) был примерно одинаковым для клеток, имевших 3-кратное различие в соотношении скоростей поступления Са2+ в цитоплазму и удалении из нее (3-кратное различие площадей под кривыми изменений [Ca2+]i). В то же время, Hmn (100 мкМ) в одинаковой степени снижал рост [Ca2+]i в клетках с высоким и с низким подъемом [Ca2+]i. Поэтому динамическое равновесие между поступлением Са2+ в цитоплазму и его удалением из нее имело характер прямой пропорциональной зависимости (рис. 3б). Это наблюдение свидетельствует о том, что, несмотря на расположение внутри канала, Mg2+ и Hmn по-разному ограничивают поток Са2+ через NMDA-канал, что согласуется с моделью, предложенной в работе [17, 18] о том, что Mg2+ и Hmn располагаются на разной “глубине” внутри канала (на разном расстоянии от внутренней поверхности плазматической мембраны).
Высокие концентрации глутамата вызывают кальциевую дизрегуляцию (ОКД), которая происходит синхронно с сильной митохондриальной деполяризаций [6, 7]. На рис. 4д и 4е совмещены для наглядности изменения [Ca2+]i и ΔΨm двух нейронов, в одном из которых (рис. 4д) за время действия NMDA развилась ОКД и сильное падение ΔΨm (рост флуоресценции Rh123 во время вторичного подъема [Ca2+]i). Hmn не нарушил синхронность развития ОКД и падение ΔΨm, снижая долю нейронов, в которых развилась ОКД, и, соответственно, долю нейронов, в которых происходила сильная митохондриальная деполяризация (рис. 4а–4г). Неконкурентный ингибитор NMDA-рецепторов МК-801 (10 мкМ) вызывал практически полное блокирование индуцированных NMDA изменений [Ca2+]i и ΔΨm, причем во всех нейронах (рис. 4ж, з). Менее сильное ингибирование гимантаном роста [Ca2+]i и падения ΔΨm, связано, по-видимому, с тем, что МК-801 ингибирует NMDA-рецепторы не зависимо от величины потенциала плазмалеммы, тогда как действие Hmn зависит от этого потенциала [17, 18].
В зрелых нейрональных культурах часто наблюдается спонтанная активность, проявляющаяся в снижении потенциала плазматической мембраны и входе Са2+ по потенциал- и лиганд-зависимым каналам [29–31]. Возможно, эта спонтанная активность является одной из причин гетерогенности [Ca2+]i ответов нейронов на блокирующее действие Hmn.
Известно, что ионные каналы NMDA-рецепторов хорошо проницаемы не только для Са2+, но также для Na+ и K+. В культуре кортикальных нейронов Hmn (100 мкМ) вызывал лишь частичное ингибирование роста [Ca2+]i, удлиняя лаг-период развития ОКД (рис. 5ж). Hmn не оказывал заметного влияния на изменения [Na+]i при совместном добавлении с NMDA по сравнению с действием одного этого агониста, хотя прослеживалась тенденция к снижению общего количества Na+ прошедшего сквозь цитозоль (рис. 5з). Причиной неодинакового ингибирования изменений [Ca2+]i и [Na+]i, неконкурентными ингибиторами NMDA-каналов, включая Hmn, может быть, помимо отмеченной выше спонтанной электрической активности нейрональной культуры, то, что активация NMDA-рецепторов вызывает реверсию Na+/Ca2+-обмена [27].
В заключение отметим следующее. Быстрая обратимость ингибирования NMDA-каналов может благоприятствовать нормализации функционирования мозга после отмены применения Hmn. Более эффективное блокирование входа Са2+ по сравнению с Na+ будет сильнее ингибировать нейротоксическое воздействие Са2+, при этом в меньшей степени снижая поступление Na+, его деполяризующий эффект на плазматическую мембрану и, соответственно, передачу электрического сигнала между нейронами.
Список литературы
Amenta F., Zaccheo D., Collier W.L. // Mech. Ageing Dev. 1991. V. 61. № 3. P. 249–273.
Колачева А.А., Козина Е.А., Хакимова Г.Р., Кучеряну В.Г., Кудрин В.С., Нигматуллина Р.Р., Базян А.С., Григорьян Г.А., Угрюмов М.В. Экспериментальное моделирование болезни Паркинсона в книге НЕЙРОДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ: от генома до целостного организма. // Под ред. Угрюмова М.В.. М.: Научный мир, 2014. Т. 1. С. 356–422.
Николлс Д.Г., Мартин А.Р., Валлас Б.Д., Фукс П.А. // От нейрона к мозгу. М.: Издательство Научной и учебной литературы УРСС, 2003. 671 с.
Sobolevsky A.I. // J. Physiol. 2015. V. 593. № 1. P. 29–38.
Dirnagl U., Iadecola C., Moskowitz M.A. // Trends Neurosci. 1999. V. 22. № 9. P. 391–397.
Nicholls D.G., Budd S.L. // Physiol. Rev. 2000. V. 80. № 1. P. 315–360.
Khodorov B. // Prog. Biophys. Mol. Biol. 2004. V. 86. № 2. P. 279–351.
Hardingham G.E. // Biochem. Soc. Trans. 2009. V. 37. № 6. P. 1147–1160.
Hardingham G.E., Bading H. // Nat. Rev. Neurosci. 2010. V. 11. № 10. P. 682–696.
Chen H.S.V., Pellegrini J.W., Aggarwal S.K., Lei S.Z., Warach S., Jensen F.E., Lipton S.A. // J. Neurosci. 1992. V. 12. № 11. P. 4427–4436.
Parsons C.G., Danysz W., Quack G. // Neuropharmacology. 1999. V. 38. № 6. P. 735–767.
Chen H.S.V., Lipton S.A. // J. Neurochem. 2006. V. 97. № 6. P. 1611–1626.
Stayte S., Vissel B. // Front. Neurosci. 2014. V. 8. article 113.
Freitas M.E., Fox S.H. // Neurodegener. Dis. Manag. 2016. V. 6. № 3. P. 249–268.
Капица И.Г., Иванова Е.А., Авдюнина Н.И., Воронина Т.А. // Хим.-фарм. журн. 2019. Т. 53. № 5. С. 3–7.
Olivares D., Deshpande V.K., Shi Y., Lahiri D.K., Greig N.H., Rogers J.T., Huang X. // Curr. Alzheimer Res. 2013. V. 9. № 6. P. 746–758.
Sobolevsky A.I., Yelshansky M.V. // J. Physiol. 2000. V. 526. № 3. P. 493–506.
Елшанская М.В., Соболевский А.И., Вальдман Е.А., Ходоров Б.И. // Эксперим. и клин. фармакол. 2001. Т. 64. № 1. С. 18–21.
Воронина Н.А., Кучеряну В.Г., Капица И.Г., Воронина Т.А. // Патогенез. 2019. Т. 17. № 4. С. 57–62.
Иванова Е.А., Капица И.Г., Золотов Н.Н., Вальдман Е.А., Непоклонов А.В., Колясникова К.Н., Воронина Т.А. // Фармакокинетика и Фармакодинамика. 2016. Т. 3. С. 9–12.
Krasil’nikova I., Surin A., Sorokina E., Fisenko A., Boyarkin D., Balyasin M., Demchenko A., Pomytkin I., Pinelis V. // Front. Neurosci. 2019. V. 13. article 1027.
Середенин С.Б., Воронина Т.А., Авдюнина Н.И., Морозов И.С., Быков Н.П., Неробкова Л.Н. Гидрохлорид N-2-адамантил)-гексаметиленимина, обладающий антикаталептической активностью.: патент № 1825499 Россия, 1991.
Nerobkova L.N., Val’dman E.A., Voronina T.A., Markina N.V., Sharkova L.M. // Eksp Klin Farmakol. 2000. V. 63. № 3. P. 3–6.
Шарипов Р.Р., Красильникова И.А., Пинелис В.Г., Горбачева Л.Р., Сурин А.М. // Биол. мембраны. 2018. Т. 35. № 5. С. 384–397.
Vergun O., Keelan J., Khodorov B.I., Duchen M.R. // J. Physiol. 1999. V. 519. № 2. P. 451–466.
Duchen M.R., Surin A., Jacobson J. // Methods Enzymol. 2003. V. 361. P. 353–389.
Brittain M.K., Brustovetsky T., Sheets P.L., Brittain J.M., Khanna R., Cummins T.R., Brustovetsky N. // Neurobiol. Dis. 2012. V. 46. № 1. P. 109–117.
Ходоров Б.И., Сторожевых Т.П., Сурин А.М., Сорокина Е.Г., Юравичус А.И., Бородин А.В., Винская Н.П., Хаспеков Л.Г., Пинелис В.Г. // Биол. мембраны. 2001. Т. 18. № 6. С. 421–432.
Генрихс Е.Е., Александрова О.П., Стельмашук Е.В., Новикова С.В., Воронков Д.Н., Исаев Н.К., Хаспеков Л.Г. // Анналы клинической и экспериментальной неврологии. 2019. Т. 13. № 4. С. 38–45.
Зинченко В.П., Туpовcкая М.В., Теплов И.Ю., Беpежнов А.В., Туpовcкий Е.А. // Биофизика. 2016. Т. 61. № 1. С. 102–111.
Aqrawe Z., Patel N., Vyas Y., Bansal M., Montgomery J., Travas-Sejdic J., Svirskis D. // PLoS One. 2020. V. 15. № 8.
Дополнительные материалы отсутствуют.
Инструменты
Нейрохимия