

Нейрохимия, 2021, T. 38, № 2, стр. 154-160
Нейропластические изменения в гиппокампе крыс после ишемического инсульта в неокортексе: гипоталамо-гипофизарно-надпочечниковая система и система нейротрофинов
М. В. Онуфриев 1, Ю. В. Моисеева 1, М. Н. Волобуева 1, А. А. Квичанский 1, Л. В. Третьякова 1, Н. В. Гуляева 1
1 Институт высшей нервной деятельности и нейрофизиологии РАН
Москва, Россия
Поступила в редакцию 05.01.2020
После доработки 20.01.2021
Принята к публикации 21.01.2021
Аннотация
Окклюзия средней мозговой артерии (ОСМА) явлется причиной большинства ишемических инсультов, в результате которой развивается фокальное поражение мозга, затрагивающее ни только различные области коры больших полушарий, но регионы мозга, которые располагаются за пределами области инфаркта, в том числе и гиппокамп. Активация гипоталамо-гипофизарно-надпочечниковой системы (ГГНС), приводящая к повышению циркулирующего уровня гюкокортикоидов, является одной из первых реакций организма на церебральный инсульт. Баланс между степенью активации ГГНС и нейропротекторным потенциалом системы нейротрофинов может предопределять интенсивность повреждения гиппокампа, как региона мозга, экспрессирующего высокий уровень кортикостероидных рецепторов. В настоящей работе исследована динамика изменения уровня кортикостерона, BDNF и NGF в крови и регионах гиппокампа крыс после транзиторной ОСМА. Повышение уровня кортикостерона в крови, происходило на ранних сроках после ОСМА и сопровождалось появлением стресс-гормона в дорсальном гиппокампе (ДГ) ишемического полушария. В отличие от неизменившегося уровня циркулирующего BDNF, экспрессия белка данного нейротрофина увеличилась на ранних сроках реперфузии в ДГ и вентральном гиппокампе (ВГ), тогда как уровень NGF повысился в крови и в дальнейшем только в ДГ. Таким образом, ишемический инсульт в неокортексе индуцировал активацию ГГНС, аккумуляцию кортикостерона и повышение экспрессии белка BDNF в большей степени, чем NGF, в регионах ипсилатерального гиппокампа на ранних сроках после ОСМА.
ВВЕДЕНИЕ
Инсульт – тяжелая неврологическая патология, которая характеризуется высокой смертностью, развитием постинсультного неврологического дефицита и когнитивных нарушений. Повреждение после инсульта не ограничивается только областью инфаркта, но также распространяется в неишемические регионы мозга, вызывая их вторичное повреждение. Так, после фокального повреждения неокортекса и/или стриатума вторичные изменения наблюдаются в таламусе, черной субстанции, гиппокампе и спинном мозге [1–3].
Активация гипотоламо-гипофизарно-надпочечниковой системы (ГГНС) является одним из первых физиологических ответов на церебральную ишемию, происходит в первые часы после ишемии и приводит к длительному повышению в крови концентрации глюкокортикоидов [4–6]. Показано, что как высокий, так и низкий уровень кортизола в крови усиливает риск смерти в течение 28 дней от наступления инсульта [7]. Предполагают, что дисбалланс между центральными кортикостероидными рецепторами лежит в основе дисрегуляции ГГНС и предрасположенности к стресс-индуцированным психиатрическим заболеваниям, в том числе и депрессии [8]. Модуляция активности ГГНС отчасти осуществляется гиппокампом [9], в том числе и за счет минерало- и глюкокортикоидных рецепторов (МР и ГР), высокий уровень экспрессии которых наблюдается в поляx СА1, СА2, СА3 и зубчатой извилине [10]. Известно, что гиппокамп неоднородная структура и в септо-темпоральном направлении выделяют дорсальную, промежуточную и вентральную части. Более того, дорсальный (ДГ) и вентральный гиппокамп (ВГ) выполняют различные функции, причем первый преимущественно связан с когнитивными функциями, а второй с реакциями на стресс и эмоциями [11, 12].
В различных исследованиях показано, что нейротрофины принимают участие в структурном и функциональном восстановлении нервной ткани после ишемического повреждения [13, 14]. Хорошо известно, что связывание зрелых форм нейротрофинов, в частности, BDNF и NGF, с соответствующими высокоаффинными тирозин-киназными рецепторами (mNGF-TrkА; mBDNF-TrkВ) необходимо для нейропротекции, дифференцировки клеток и образования синапсов [15, 16]. Однако проформы BDNF и NGF, взаимодействуя с низкоаффинными рецепторами р75NTR, участвуют в регуляции роста нейритов, гибели клеток во время развития ЦНС и при патологических состояниях [17]. Известно, что активация ГГНС и повышение уровня глюкокортикоидов при стрессе приводит к снижению экспрессии нейротрофинов в гиппокампе [18, 19], но этот эффект зависит от интенсивности и продолжительности стресса [20]. С другой стороны, однократная инъекция синтетического глюкокортикоида дексаметазона ни только не влияет на содержание и секрецию BDNF, NGF, NT3 в коре и гиппокампе, а также в кортикальных и гиппокампальных культурах клеток, но и вызывает активацию Trk рецепторов, усиливая степень их фосфорилирования [21].
В связи с вышеизложенным цель данного исследования заключалась в выявлении связи между активацией ГГНС, которую характеризует уровень стресс-гормона кортикостерона, и содержанием нейротрофинов в регионах гиппокампа в динамике на ранних и отдаленных сроках после инсульта.
МЕТОДЫ ИССЛЕДОВАНИЯ
Самцы крыс линии Вистар были получены из Филиала “Столбовая” Федерального государственного бюджетного учреждения науки “Научного центрабиомедицинских технологий Федерального медико-биологического агентства” (Московская обл., РФ) и размещены в клетках вивария.
Модель ишемического инсульта. Ишемический инсульт создавали посредством окклюзии средней мозговой артерии [22]. Наркотизацию крыс линии Вистар весом 200–300 г осуществляли с помощью ингаляционного наркоза (изофлуран). Через левую внешнюю сонную артерию вводили филамент, продвигая его через внутреннюю сонную артерию до пересечения со средней мозговой артерией. Длительность окклюзию составляла 60 мин, при этом температуру тела крысы поддерживали в интервале 37 ± 0.5°С. По окончании окклюзии филамент извлекали и восстанавливали кровоток по ипсилатеральной общей сонной артерии, которую освобождали от лигатуры после извлечения филамента. У ложнооперированных крыс проделывали все этапы операции кроме введения филамента. Число крыс в каждой группе составляло 6–8 животных на каждую временнýю точку.
Оценка неврологического дефицита. 1. 5-Балльная шкала. Тест основан на 5-балльной поведенческой шкале [23] и позволяет оценить функциональное состояние контралатеральной передней лапы крыс, наличие поворотов и циркуляции в контралатеральную сторону, а также подвижность животных. 2. Тест на степень высовывания языка. Данный показатель определяли по способности крысы вылизывать арахисовое масло из стеклянного цилиндра с последующим измерением расстояния от начала цилиндра до уровня оставшегося масла [24].
Подготовка биологического материала. На 1, 3, 7 и 14 сутки после ОСМА крысы были выведены из эксперимента. После декапитации животных был получен следующий биологический материал: кровь (сыворотка), дорсальная и вентральная части гиппокампа ипсилатерального полушария. Образцы гиппокампа гомогенизировали и после центрифугирования была получена растворимая фракция белков (супернатант), которую аликвотировали и хранили при –80°С до проведения биохимических исследований.
Определение кортикостерона. Для определения уровня кортикостерона в сыворотке крови и супернатантах гиппокампа использовали наборы для иммуноферментного анализа (DRG, Германия), с помощью которых детектировали как свободный, так и связанный с транспортными белками кортикостерон методом конкурентного ИФА.
Определение нейротрофинов. В крови и супернатантах гипокампа общий уровень зрелых и проформ BDNF и NGF определяли с использованием наборов для ИФА (R&D Systems, USA) в соответствии с инструкцией производителя.
Статистическая обработка результатов. Статистическую обработку результатов проводили в программе Statistica 10. После определения нормальности распределения данных динамику уровней кортикостерона и нейротрофинов оценивали с помощью дисперсионного анализа (ANOVA) с последующим Fisher post-hoc тестом. Использовали влияние факторов “группа” и “время”. Данные представлены в виде M ± SEM. Различия считали достоверными при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
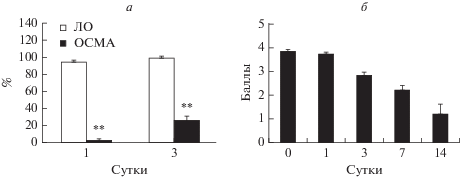
В результате ОСМА развивался неврологический дефицит у крыс и статистически значимое снижение степени вытягивания языка наблюдалось на 1 и 3 сутки, что составляло от уровня до операции 2.86 ± 1.33% на 1 сут и 26.70 ± 4.59% на 3 сут, причем в группе ЛО статистически значимого уменьшения этого показателя не наблюдалось (рис. 1а). По 5-балльной шкале в группе ОСМА максимальный неврологический дефицит детектировали в день операции, на 1 и 3 сут после окклюзии и затем он постепенно уменьшался к 14 сут (рис. 1б). У крыс в группе ЛО неврологический дефицит отсутствовал.
Рис. 1.
Динамика изменения неврологического дефицита в тесте с арахисовым маслом (а) и по 5-балльной шкале (б). ** p < 0.01 – отличия от группы ЛО.
После ОСМА активация ГГНС по уровню кортикостерона происходила на 1 сут и содержание гормона в крови статистически значимо возросло в 1.6 раза (табл. 1). Исследуемые факторы не оказывали статистически значимого влияния на этот показатель. Уровень BDNF в крови крыс после ОСМА существенно не отличался от уровня нейротрофина в группе ЛО на всех сроках исследования. В отличие от BDNF динамика изменения циркулирующего уровня NGF после ОСМА статистически значимо превышала данный показатель в группе ЛО на ранних сроках исследования. Так, на 1 и 3 сут после ОСМА уровень NGF статистически значимо возрос в 1.57 раза и в 1.58 раза соответственно, но в на 7 и 14 сут уже не отличался от контрольных значений. На динамику изменения уровня NGF в крови оказывали влияние фактор “время” (F(3,58) = 2.93; p = = 0.042), фактор “группа” на уровне тенденции к достоверности (F(1,58) = 2.98; p = 0.089) и взаимодействие факторов “время” и “группа” (F(3,58) = = 3.44; p = 0.023).
Таблица 1.
Динамика изменения уровня кортикостерона (КС), BDNF и NGF в крови крыс после ОСМА
| Показатель | Группа | 1 сутки | 3 сутки | 7 сутки | 14 сутки |
|---|---|---|---|---|---|
| КС, нМ | ЛО | 1042 ± 205 | 1159 ± 178 | 1027 ± 201 | 1191 ± 79 |
| ОСМА | 1668 ± 266* | 1178 ± 158 | 1151 ± 195 | 943 ± 151 | |
| BDNF, пг/мл | ЛО | 724 ± 94 | 574 ± 19 | 756 ± 95 | 553 ± 59 |
| ОСМА | 613 ± 69 | 616 ± 81 | 741 ± 127 | 566 ± 55 | |
| NGF, пг/мл | ЛО | 1.54 ± 0.30 | 2.03 ± 0.24 | 2.50 ± 0.21 | 3.12 ± 0.43 |
| ОСМА | 2.42 ± 0.25* | 3.21 ± 0.43* | 2.47 ± 0.26 | 2.58 ± 0.20 |
Возросший уровень кортикостерона в крови на 1 сут сопровождался его аккумуляцией в ДГ ишемического полушария, где его содержание достоверно увеличилось в 1.44 раза (табл. 2). На уровень кортикостерона в ДГ ипсилатерального полушария с тенденцией к достоверности оказывали влияние факторы “время” (F(3,51) = 2.29; p = 0.088) и “группа” (F(1,51) = 2.98; p = 0.090). Так же как и кортикостерон, на 1 сут после ОСМА повысился уровень BDNF в ДГ ишемического полушария, хотя и на уровне тенденции к достоверности (р < 0.1).
Таблица 2.
Динамика изменения уровня кортикостерона (КС), BDNF и NGF в дорсальном (ДГ) и вентральном (ВГ) гиппокампе ишемического полушария
| Показатель | РГ | Группа | 1 сутки | 3 сутки | 7 сутки | 14 сутки |
|---|---|---|---|---|---|---|
| КС, пмоль/г ткани | ДГ | ЛО | 296 ± 56 | 287 ± 28 | 310 ± 29 | 279 ± 32 |
| ОСМА | 428 ± 59* | 349 ± 20 | 288 ± 20 | 269 ± 26 | ||
| ВГ | ЛО | 346 ± 93 | 152 ± 13 | 268 ± 44 | 249 ± 24 | |
| ОСМА | 296 ± 49 | 206 ± 27 | 272 ± 60 | 231 ± 22 | ||
| BDNF, нг/г ткани | ДГ | ЛО | 18.6 ± 1.0 | 18.4 ± 1.2 | 18.3 ± 1.3 | 19.0 ± 1.6 |
| ОСМА | 22.9 ± 2.5# | 24.0 ± 1.8* | 15.8 ± 1.1 | 19.0 ± 1.2 | ||
| ВГ | ЛО | 20.3 ± 1.1 | 17.5 ± 1.3 | 19.5 ± 1.6 | 20.9 ± 1.0 | |
| ОСМА | 26.8 ± 2.9* | 21.0 ± 2.1 | 20.8 ± 1.1 | 18.3 ± 1.3 | ||
| NGF, пг/г ткани | ДГ | ЛО | 527 ± 22 | 509 ± 22 | 519 ± 17 | 515 ± 29 |
| ОСМА | 515 ± 30 | 602 ± 26* | 533 ± 19 | 584 ± 25 | ||
| ВГ | ЛО | 731 ± 43 | 753 ± 40 | 732 ± 19 | 803 ± 23 | |
| ОСМА | 804 ± 70 | 795 ± 55 | 719 ± 34 | 824 ± 22 |
Он был в 1.23 раза выше, чем в группе ЛО. Высокий уровень BDNF в ДГ сохранялся и на 3 сутки, статистически значимо в 1.30 раза, превышая таковой в группе ЛО. На уровень BDNF в ДГ оказывал статистически значимое влияние фактор “время” (F(3,56) = 3.10; p = 0.034) и взаимодействие факторов “время” и “группа” (F(3,56) = 3.0; p = 0.038), а также с тенденцией к достоверности фактор “группа” (F(1,56) = 3.0; p = 0.089). В ВГ ишемического полушария уровень BDNF статистически значимо возрос в 1.32 раза также на 1 сут после ОСМА, тогда как на 3, 7 и 14 сут содержание нейротрофина практически не отличалось от такового в группе ЛО. На уровень BDNF в ВГ ипсилатерального полушария оказывали статистически значимое влияние фактор “время” (F(3,57) = = 2.90; p = 0.043) и взаимодействие факторов “время” и “группа” (F(3,57) = 2.88; p = 0.044), а также с тенденцией к достоверности фактор “группа” (F(1,57) = 3.75; p = 0.058). В отличие от BDNF уровень NGF в меньшей степени изменился в ДГ ишемического полушария, в котором уровень нейротрофина только на 3 сут достоверно увеличился в 1.18 раза, а на его содержание влиял с тенденцией к достоверности фактор “группа” (F(1,58) = 3.59; p = = 0.063). В ВГ ипсилатерального полушария ОСМА существенно не повлияла на уровень NGF и никаких различий между группами в исследованном постинсультном периоде не наблюдалось.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Активация ГГНС начинается в первые часы после ОСМА и может быть достаточно протяженной с сохранением повышенного уровня глюкокортикоидов кортизола/кортикостерона в крови даже в течение нескольких суток [25, 26]. Как известно, глюкокортикоиды обладают противовоспалительным и иммуносупрессивным действием и широко используются в терапии для блокирования воспалительных процессов. Тем не менее, накапливаются экспериментальные свидетельства в пользу того, что глюкокортикоиды в соответствующих условиях могут обладать как анти-, так и провоспалительными эффектами в ЦНС [27, 28]. Активация ГГНС по нашим данным происходила кратковременно и на ранних сроках после ОСМА, т. к. возросший уровень кортикостерона в крови наблюдали только на 1 сут после фокального инсульта, что сопровождалось также аккумуляцией стресс-гормона в ДГ, но не в ВГ, в это же время.
В отличие от кортикостерона, никаких изменений уровня BDNF в крови не удалось выявить. Отсутствие реактивности циркулирующего уровня BDNF на инсульт соответствует данным других авторов, полученных в экспериментальных и клинических условиях. Разная степень тяжести мультифокального ишемического инсульта, моделированная введением в кровоток крыс микросфер, не повлияла на уровень циркулирующего BDNF, который также не коррелировал с повышенным уровнем нейротрофина в мозге через 2 ч, 4 ч и 8 дней после эмболизации [29]. В течение четырех дней после ишемического инсульта уровень BDNF не изменялся в плазме пациентов [30]. Тем не менее, низкий уровень BDNF в крови в острой фазе ишемического инсульта был связан с медленным функциональным восстановлением пациентов [31].
Уровень NGF в системном кровотоке оказался более реактивным после ишемического инсульта, чем BDNF. Разнонапрваленные изменения уровня NGF в крови наблюдали на экспериментальных моделях и у пациентов после ишемического инсульта. Через 24 ч после 2 ч ОСМА детектировали снижение уровня NGF в крови крыс [32]. У пациентов с “лучшим” функциональным восстановлением через 24 ч после ишемического инсульта уровень NGF в крови был выше, чем в контрольной группе и группе с “худшим” восстановлением [33]. Согласно полученным нами результатам повышение уровня NGF в крови на 1 и 3 сут после 1 ч ОСМА, вероятно, свидетельствует о нормальном функциональном восстановлении животных.
Постинсультное повышение продукции нейротрофинов в нейронах и микроглии периинфарктной зоны в неокортексе рассматривают как компенсаторный механизм, направленный на предотвращение избыточной гибели нейронов [34]. Любые воздействия, приводящие к повышению эндогенного уровня нейротрофинов, будут играть важную роль в предотвращении последствий инсульта. Так, ежедневные упражнения на беговой дорожке в течение трех недель до ОСМА привели к уменьшению неврологического дефицита у крыс и снижению объема инфаркта в фронтопариетальной коре и дорсолатеральном стриатуме и этот эффект коррелировал с повышением уровня экспрессии BDNF и NGF в нейронах коры и глиальных клетках стриатума после курса тренировки [35].
Экспрессия генов нейротрофинов и их рецепторов зависит от интенсивности фокальной ишемии. Повышение уровня экспрессии мРНК BDNF, NGF, TrkA и TrkB регистрировали в первые часы после транзиторной (2 ч) ОСМА в различных областях коры больших полушарий и полях гиппокампа, а к 24 ч уровень экспрессии исследованных генов не отличался от контрольных значений. При менее интенсивном варианте ОСМА (15 мин) возросший уровень экспрессии мРНК BDNF был выявлен через 2 ч реперфузии в цингулярной и фронтальной коре, но не в гиппокампе [36]. В результате фокальной ишемии, индуцированной инъекцией микросфер и вызывающей повреждение гиппокампа, высокий уровень белка BDNF в этом регионе сохранялся через неделю после воздействия [37]. После четырехчасовой ОСМА с использованием in situ гибридизации повышенная экспрессия мРНК BDNF и TrkB зарегистрирована в зубчатой фасции и СА1–СА4 полях гиппокампа билатерально [38].
В нашем исследовании в результате умеренной транзиторной ОСМА уровень белка BDNF возрос в ДГ и ВГ на 1 и 3 сут, а NGF только в ДГ ишемического полушария на 3 сут. Увеличение уровня белка BDNF в ДГ на 1 сут совпадало с появлением кортикостерона в этом регионе гиппокампа, что несколько противоречит известным реципрокным изменениям уровня нейротрофина и стресс-гормона. Тем не менее, есть данные о положительном влиянии кортикостерона на нейротрофиновый сигналинг. Если однократная инъекция высокой дозы кортикостерона способствовала снижению уровня экспрессии мРНК BDNF в зубчатой фасции и в СА1 и СА3 полях гиппокампа, то полсе низкой дозы кортикостерона регистрировали повышение уровня экспрессии мРНК TrkB в СА3 и зубчатой фасции [39]. Также в пользу положительной связи между кортикостероидным и нейротрофиновым сигналингом свидетельствует и тот факт, что у мышей, нокаутных по гену глюкокортикоидных рецепторов, нарушен BDNF/TrkB сигналинг в мозге [40].
ЗАКЛЮЧЕНИЕ
Таким образом, в результате умеренной ОСМА активность ГГНС по уровню кортикостерона в крови повысилась на 1 сутки и сопровождалась аккумуляцией стресс-гормона только в ДГ ипсилатерального полушарии, но не в ВГ. На ранних сроках после ишемического инсульта произошла активация нейротрофиновой системы в виде увеличения циркулирующего уровня NGF и повышения экспрессии белка BDNF и NGF в обеих исследованных регионах гиппокампа ипсилатерального полушария, причем возрастание уровня BDNF в ДГ и ВГ произошло раньше, чем NGF, однако в дальнейшем уровень обоих нейротрофинов сохранялся высоким в ДГ, обеспечивая нейропластический и нейропротекторный потенциал данного региона гиппокампа, реактивность которого на активацию ГГНС первична по сравнению с ВГ.
Список литературы
Nakane M., Tamura A., Sasaki Y., Teraoka A. // Neuroradiology. 2002. V. 44. P. 915–920.
Butler T., Kassed C., Sanberg P., Willing A., Pennypacker K. // Brain Res. 2002. V. 929. P. 252–260.
Block F., Dihné M., Loos M. // Prog. Neurobiol. 2005. V. 75. P. 342–365.
Olsson T. // J. Intern. Med. 1990. V. 228. P. 177–181.
Fassbender K., Schmidt R., Mössner R., Daffertshofer M., Hennerici M. // Stroke. 1994. V. 25. P. 1105–1108.
Johansson A., Olsson T., Carlberg B., Karlsson K., Fagerlund M. // J. Neurol. Sci. 1997. V. 147. P. 43–47.
Marklund N., Peltonen M., Nilsson T.K., Olsson T. // J. Intern. Med. 2004. V. 256. P. 15–21.
Sapolsky R. // Arch. Gen. Psychiatry. 2000. V. 57. P. 925–935.
Jacobson L., Sapolsky R. // Endocr. Rev. 1991. V. 12. P. 118–134.
Sarabdjitsingh R., Meijer O., Schaaf M., de Kloet E. // Brain Res. 2009. V. 1249. P. 43–53.
Segal M., Richter-Levin G., Maggio N. // Hippocampus. 2010. V. 20. P. 1332–1338.
Гуляева Н.В. // Рос. физиол. журн. им. И.М. Сеченова. 2013. Т. 99. С. 3–16.
Holtzman D., Sheldon R., Jaffe W., Cheng Y., Ferriero D. // Ann. Neurol. 1996. V. 39. P. 114–122.
Berretta A., Tzeng Y.C., Clarkson A. // Expert. Rev. Neurother. 2014. V. 14. P. 1335–1344.
Friedman W., Greene L. // Exp. Cell Res. 1999. V. 253. P. 131–42.
Reichardt L. // Philos. Trans. Biol. Sci. 2006. V. 361. P. 1545–1564.
Roux P., Barker P. // Prog. Neurobiol. 2002. V. 67. P. 203–233.
Hansson A., Sommer W., Rimondini R., Andbjer B., Strömberg I., Fuxe K. // J. Neurosci. 2003. V. 23. P. 6013–6022.
Smith M., Makino S., Kvetnansky R., Post R. // J. Neurosci. 1995. V. 15. P. 1768–1777.
Tapia-Arancibia L., Rage F., Givalois L., Arancibia S. // Front. Neuroendocrinol. 2004. V. 25. P. 77–107.
Jeanneteau F., Garabedian M., Chao M. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. P. 4862–4867.
Longa E., Weinstein P., Carlson S., Cummins R. // Stroke. 1989. V. 20. P. 84–91.
Hunter A., Hatcher J., Virley D., Nelson P., Irving E., Hadingham S., Parsons A. // Neuropharmacology. 2000. V. 39. P. 806–816.
Gulyaeva N., Thompson C., Shinohara N., Lazareva N, Onufriev M., Stepanichev M., Moiseeva Y. Fliss H., Hakim A. // J. Neurosci. Meth. 2003. V. 125. P. 183–193.
Fassbender K., Schmidt R., Mössner R., Daffertshofer M., Hennerici M. // Stroke. 1994. V. 25. P. 1105–1108.
Smith-Swintosky V., Pettigrew L., Sapolsky R., Phares C., Craddock S., Brooke S., Mattson M. // J. Cereb. Blood Flow Metab. 1996. V. 16. P. 585–598.
Duque Ede A., Munhoz C. // Front Endocrinol (Lausanne). 2016. V. 7. P. 78–80.
Frank M., Thompson B., Watkins L., Maier S. // Brain. Behav. Immun. 2012. V. 26. P. 337–345.
Béjot Y., Mossiat C., Giroud M., Prigent-Tessier A., Marie C. // PLoS One. 2011. V. 6. P. e29405.
Di Lazzaro V., Profice P., Pilato F., Dileone M., Florio L., Tonali P., Angelucci F. // Neurosci. Lett. 2007. V. 422. V. 128–130.
Stanne T., Åberg N., Nilsson S., Jood K., Blomstrand C, Andreasson U., Blennow K., Zetterberg H, Isgaard J., Svensson J., Jern C. // Stroke. 2016. V. 47. P. 1943–1945.
Piao J., Wu W., Yang Z., Li Y., Luo Q., Yu J. // Cell Physiol. Biochem. 2018. V. 46. P. 890–906.
Luan X., Qiu H., Hong X., Wu C., Zhao K., Chen H., Zhu Z., Li X., Shen H., He J. // Clin. Chim. Acta. 2019. V. 488. P. 20–24.
Tejeda G., Díaz-Guerra M. // Int. J. Mol. Sci. 2017. V. 18. P. E268.
Ding Y., Li J., Luan X., Ding Y., Lai Q., Rafols J., Phillis J., Clark J., Diaz F. // Neuroscience. 2004. V. 24. P. 583–591.
Kokaia Z., Zhao Q., Kokaia M., Elmér E., Metsis M., Smith M., Siesjö B., Lindvall O. // Exp. Neurol. 1995. V. 136. P. 73–88.
Miyake K., Yamamoto W., Tadokoro M., Takagi N., Sasakawa K., Nitta A., Furukawa S., Takeo S. // Brain Res. 2002. V. 935. P. 24–31.
Arai S., Kinouchi H., Akabane A., Owada Y., Kamii H., Kawase M., Yoshimoto T. // Neurosci. Lett. 1996. V. 211. P. 57–60.
Schaaf M., Hoetelmans R., de Kloet E., Vreugdenhil E. // J. Neurosci. Res. 1997. V. 48. P. 334–341.
Li Y., Huang L., Ma Q., Concepcion K., Song M., Zhang P., Fu Y., Xiao D., Zhang L. // Int. J. Mol. Sci. 2018. V. 19. P. E2428.
Дополнительные материалы отсутствуют.