

Нейрохимия, 2021, T. 38, № 3, стр. 267-277
Влияние умеренной гипотермии на каталитические характеристики синаптической ацетилхолинэстеразы при субтотальной ишемии головного мозга у крыс
Н. К. Кличханов 1, А. М. Джафарова 1
1 ФГБОУ ВО “Дагестанский государственный университет”
Махачкала, Россия
Поступила в редакцию 05.03.2021
После доработки 12.03.2021
Принята к публикации 16.03.2021
Аннотация
Острая ишемия мозга инициирует каскад реакций, результатом которых может стать развитие окислительного стресса и нарушение сбалансированной работы ряда нейромедиаторных систем мозга. Протекторные эффекты терапевтической гипотермии при ишемии достаточно известны и изучены, однако механизмы ее воздействия на холинэргическую систему мозга, тесно связанную с функционированием ацетилхолинэстеразы (АХЭ), остаются невыясненными. В данной работе исследованы активность и кинетические параметры АХЭ, а также взаимосвязь между каталитическими свойствами фермента и интенсивностью свободно-радикальных процессов в синаптических окончаниях нейронов мозга крыс при окклюзии сонных артерий в условиях нормотермии и умеренной (33°С) гипотермии. Обнаружено, что церебральная ишемия приводит к снижению активности АХЭ, в то время как гипотермия в период окклюзии сонных артерий препятствует этому. При ишемии мозга значение Vmax и эффективность катализа АХЭ снижаются, а Ki увеличивается, что на фоне неизменных значений Km способствует повышению диапазона эффективных концентраций ацетилтиохолина и значений [S]опт. Мягкая гипотермия на фоне ишемии мозга препятствует существенным изменениям кинетических параметров фермента. Ишемия стимулирует процессы окислительной деструкции мембранных белков и липидов. Гипотермия во время ишемии предотвращает повышение уровней продуктов окислительной модификации липидов и белков, а также активности супероксиддисмутазы в синаптосомах. Обнаружена отрицательная корреляция между значениями кинетических параметров АХЭ и уровнями продуктов окислительной модификации липидов и белков синаптосом, что указывает на важную роль активных форм кислорода в модуляции активности ключевого фермента холинергической системы. Полученные результаты позволяют сделать заключение, что гипотермия во время ишемии предотвращает активацию свободнорадикальных процессов в синаптических окончаниях нейронов, что, в свою очередь, препятствует снижению активности и эффективности катализа АХЭ.
ВВЕДЕНИЕ
Церебральная ишемия возникает из-за дефицита кровоснабжения мозга, что, в свою очередь, инициирует каскад событий, приводящих к ряду важных клеточных изменений, которые развиваются во времени и в пространстве. Они включают в себя быстрое снижение уровня АТФ, нарушение ионного гомеостаза, избыточное накопление возбуждающих аминокислот, обладающих нейротоксическим действием, дисфункции митохондрий и эндоплазматического ретикулума, активация гидролитических ферментов и возрастание активных форм кислорода (АФК) и азота (АФА), индуцирующих развитие окислительного стресса [1, 2]. Головной мозг особенно уязвим к окислительному повреждению из-за низкого запаса источников энергии, высокого аэробного метаболизма, относительно низкой активности антиоксидантных ферментов, а также высокого содержания субстратов окисления и металлов с переменной валентностью [3, 4]. Исследования последних лет выявили связь между избыточным образованием АФК и развитием гибели нейронов при острой церебральной ишемии [5]. Чрезмерная генерация АФК и АФА может вызывать функциональное и структурное повреждение нейрональных клеток и играть важную роль в патофизиологии церебральной ишемии [6].
Роль окислительного стресса становится более значимой после возобновления кровообращения после периода ишемии, называемого реперфузией, поскольку восстановление кровотока в ишемическом мозге способствует возрастанию парциального давления кислорода в тканях головного мозга. Образующиеся при этом ${\text{О}}_{{\text{2}}}^{{ \bullet --}}$ и NO• принимают непосредственное участие в отсроченной гибели нейронов периинфарктной области [3, 7, 8].
Известно, что устойчивость мозга к дефициту кровоснабжения может повышаться при снижении температуры тела [9]. Основной причиной защитного эффекта гипотермии при ишемических состояниях головного мозга является снижение потребления кислорода и сохранение запасов глюкозы. Гипотермия переводит организм на низкую метаболическую активность, при которой большинство физиологических и патофизиологических процессов находятся на низком уровне. Особенно эффективным является мягкая гипотермия (33–35°С). Нейропротекторные механизмы гипотермии обычно связывают со снижением скорости обмена веществ и последующего истощения энергии, уменьшением секреции эксайтотоксических нейромедиаторов, предотвращением разрушения гематоэнцефалического барьера и последующего формирования мозгового отека, уменьшением образования свободных кислородных радикалов, подавлением специфических путей гибели клеток [10, 11].
Холинергические нейроны и их проекции широко распространены по всей центральной нервной системе. Их высокая плотность в таламусе, стриатуме, лимбической системе и неокортексе позволяет предположить, что холинергическая передача особенно важна для регулирования таких функций, как обучение, память, корковая организация движения и контроль мозгового кровотока [12, 13]. Ацетилхолинэстераза (АХЭ; КФ 3.1.1.7) является одним из ключевых ферментов этой системы и часто используется в качестве маркера его функционирования. АХЭ представляет собой фермент из группы сериновых эстераз, который осуществляет гидролиз нейромедиатора ацетилхолина (АХ) в холинергических синапсах, обеспечивая прерывистость нервного импульса, ответственного за нейронную связь. Этот фермент широко экспрессируется в мозге и тканях, имеющих холинергическую иннервацию [14, 15]. АХЭ локализована на внешней поверхности синаптических мембран и гидролизует АХ, поступающий в синаптическую щель в ответ на приходящий в терминаль нервный импульс [16]. Следует отметить, что АХЭ вносит существенный вклад в синаптическую передачу не только в холинергических нейронах, но также в дофаминергических и глутаматергических синапсах [17]. АХЭ присутствует в нейронах в мембраносвязанной и растворимой формах [18].
Установлено, что повреждение нейронов, связанное с ишемией, сопровождается изменением в холинергической системе мозга [19, 20]. Например, после окклюзии средней мозговой артерии обнаружено нарушение функции холинергического пути между лобной корой головного мозга и базальным ядром Мейнерта [21]. Показано, что уровень АХ в ткани мозга изменяется как во время ишемии, так и в постишемическом периоде [22]. По данным различных авторов, активность АХЭ в головном мозге в модели экспериментальной церебральной ишемии снижалась [23–26]. По данным Ванга и сотр. [27], через сутки после 45 мин окклюзии среднемозговой артерии активность АХЭ в коре головного мозга крыс достоверно снижалась. Существенное снижение активности АХЭ мозга было обнаружено через 7 сут после 15 мин окклюзии сонных артерий у крыс [28]. В то же время иммуногистохимические данные свидетельствуют о повышении уровня АХЭ в мозге уже через 0.5 ч после реперфузии, пик увеличения которого достигался через 12 ч реперфузии после окклюзии среднемозговой артерии [29]. Ясно, что на молекулярные события, лежащие в основе вызванных ишемией изменений активности АХЭ, может повлиять использованная модель ишемии, ее характер – является ли окклюзия обратимой или постоянной, а также тяжесть ишемии ткани за период исследования. Несмотря на неоднозначность данных, все эти исследования позволяют предположить, что активность АХЭ может изменяться во время и после ишемического повреждения мозга. Однако в этих работах активность АХЭ исследовалась в гомогенатах мозга, а не на уровне синаптических мембран, где, в основном, и локализуется фермент, мало уделено внимания изменениям ее кинетических характеристик, а также не изучено влияние гипотермии на эти параметры фермента при ишемии.
Целью данной работы является выяснение характера изменения кинетических параметров АХЭ синаптических мембран мозга при ишемии и гипотермии, а также установление связи между активностью фермента и интенсивностью свободнорадикальных процессов в синаптических окончаниях нейронов.
МАТЕРИАЛЫ И МЕТОДЫ
Опыты выполнены на белых крысах-самцах Вистар, массой 220–230 г, полученных из питомника “Столбовая” ГУ НЦ (г. Чехово). Животных содержали в стандартных условиях вивария Дагестанского государственного университета со свободным доступом к воде и пище при окружающей температуре 22 ± 1°С и режимом освещения свет-темнота 12 : 12 ч. Эксперименты выполнены с соблюдением правил надлежащей лабораторной практики (приказ МЗ РФ от 01.04.2016 г. № 199н). Животные были случайным образом разделены на 3 группы по 10 крыс в каждой: 1) контроль (ложно-оперированные животные); 2) ишемия мозга в течение 60 мин; 3) ишемия 60 мин на фоне гипотермии 33°С.
Моделирование неполной ишемии мозга у крыс. Ишемия головного мозга осуществлялась полной перевязкой обеих сонных артерий в течение 60 мин. У животных контрольной группы воспроизводилась наркотизация, кожный разрез и выделение артерий без последующей перевязки сосудов. Все хирургические процедуры проводили под наркозом (внутрибрюшное введение тиопентала натрия в дозе 40–50 мг/кг). В период ишемии температуру тела животного поддерживали на нормальном уровне (37°С), используя тепло настольной лампы.
Условия моделирования гипотермии. Непосредственно перед окклюзией сонных артерий температуру тела животного снижали до 33°С, обкладывая тело животного целлофановым пакетом с мелко колотым льдом. Температуру тела измеряли ректальным цифровым термометром MS6501 (Mastech, Гонконг), датчик которого вводили в прямую кишку на глубину 3–4 см.
Выделение синаптических мембран. После декапитации животного быстро извлекали головной мозг, промывали в ледяном 0.9%-ном растворе NaCl, очищали от мозговых оболочек и выделяли кору больших полушарий мозга. Фракцию синаптосом выделяли методом дифференциального центрифугирования в ступенчатом градиенте плотности сахарозы (AppliСhem, Германия) [30]. Мозг гомогенизировали с помощью стеклянного гомогенизатора Поттера в 10-кратном объеме охлажденного (0–4°С) изотонического раствора, содержащего 0.32 М сахарозу, 0.2 мМ ЭДТА (AppliСhem, Германия) и 10 мМ трис-HCl (рН 7.4). Полученный гомогенат центрифугировали при 1000 g в течение 10 мин. Осадок ресуспендировали в среде гомогенизации и вновь центрифугировали в тех же условиях. Объединенный супернатант центрифугировали при 11 500 g в течение 20 мин на центрифуге MR 23i (Thermo Fisher Scientific, США). Полученный осадок (грубая митохондриальная фракция) суспендировали в 3 мл 0.32 М сахарозы, наслаивали на градиент сахарозы (0.8 и 1.2 М) и центрифугировали при 100 000 g в течение 1 ч на ультрацентрифуге Optima L-90K (Beckman Coulter, США). Фракцию синаптосом собирали на границе 0.8 и 1.2 М сахарозы. Мембраны синаптосом получали после гипоосмотического шока, для чего к осадку синаптосом добавляли 3.0 мл холодной бидистиллированной воды и выдерживали их на холоде в течение 1 ч при периодическом перемешивании. Мембраны осаждали при 32 000 g в течение 10 мин. Надосадочную жидкость – содержимое синаптосом (названную нами в этой работе синаптоплазмой) отбирали и использовали для определения активности антиоксидантных ферментов супероксиддисмутазы (СОД) и каталазы. Мембраны синаптосом дважды промывали в 40 мМ Tris-HCl, pH 7.4, при 32 000 g в течение 10 мин. Получившийся осадок синаптических мембран ресуспендировали в 5 мМ Трис НCI-буфере (рН 7.4) и хранили до использования при –70°С. Все процедуры были выполнены при температуре 4°С.
Определение содержания белка. Белок во фракции синаптических мембран определяли по методу Лоури [31].
Определение кинетических характеристик АХЭ. Активность АХЭ определяли методом Эллмана с сотр. [32] с ацетилтиохолином (АТХ) (Koch Light, Великобритания) в качестве субстрата. В термостатируемую при 37°С кювету спектрофотометра ПЭ 3000УФ (Шанхай, Китай) вносили 1.45 мл 0.1 М фосфатного буфера, рН 8.0, 0.05 мл суспензии мембран синаптосом (40–50 мкг белка) и 0.05 мл 0.01 М раствора 5,5'-дитиобис-(2-нитробензойной кислоты) (Koch Light, Великобритания). Реакцию запускали добавлением 0.02 мл раствора АТХ. Изменение оптической плотности раствора в ходе гидролиза АТХ, катализируемой АХЭ, регистрировали при 412 нм в течение 5 мин с интервалом 20 с. Параллельно регистрировали изменение оптической плотности контрольного раствора, в который вместо суспензии мембран вносили соответствующий объем 0.1 М фосфатного буфера. Активность фермента рассчитывали, используя коэффициент молярного поглощения желтого аниона 5-тио-2-нитро-бензоата, который равен 1.36 × 104 M−1 см−1 и выражали в мкмоль/мин мг белка.
Для определения кинетических характеристик АХЭ измеряли зависимость скорости гидролиза от концентрации АТХ в диапазоне 1.56 × 10–5–6.4 × × 10–2 М. В этих исследованиях использовали 0.036 М фосфатный буфер, рН 8.0. При изменении концентрации АТХ ионную силу реакционной среды поддерживали на уровне 0.1 М путем добавления в пробу соответствующего количества NaCl. При концентрациях АТХ, превышающих 1.0 мМ, ферментативная активность была скорректирована с учетом спонтанного неферментативного гидролиза субстрата.
Особенностью катализа АХЭ является наличие субстратного ингибирования, для объяснения и описания которого были предложены различные кинетические модели [33–35]. В модели, предложенной Стояном с сотр. [33], механизм ингибирования АХЭ зависит от концентрации субстрата: умеренно высокие ([S] < 100 мМ) или высокие ([S] > 100 мМ). В настоящем исследовании были использованы умеренно высокие концентрации субстрата (<64 мМ). Для описания этой модели наиболее приемлемым является уравнение Холдейна–Радича [36]. Однако из-за высокой скорости катализа фермента не всегда удается экспериментально вычислить константы скорости образования и распада всех интермедиатов реакции, особенно когда исследуется не очищенный фермент, а фермент в составе нативных мембран. В этом случае при расчете кинетических характеристик АХЭ целесообразным считают использование упрощенного уравнения Холдейна [34]:
(1)
${v} = \frac{{{{V}_{{\max }}}[{\text{S}}]}}{{{{K}_{{\text{m}}}} + [{\text{S}}] + {{{{{[{\text{S}}]}}^{2}}} \mathord{\left/ {\vphantom {{{{{[{\text{S}}]}}^{2}}} {{{K}_{{\text{i}}}}}}} \right. \kern-0em} {{{K}_{{\text{i}}}}}}}},$где v – скорость реакции, Vmax – максимальная скорость, Km – константа Михаэлиса, Ki – константа субстратного ингибирования.
Данное уравнение нами было использовано для расчета кинетических характеристик фермента с помощью многомерного нелинейного регрессионного анализа.
Определение веществ, реагирующих с тиобарбитуровой кислотой (ТБК) в синаптосомах. Перекисное окисление липидов (ПОЛ) анализировали путем измерения реактивных веществ, реагирующих с ТБК (ТБКРВ), в результате которого измеряется, в основном, образование малонового диальдегида (МДА) [37]. При этом определяли исходный уровень ТБКРВ и их накопление in vitro в присутствии инициаторов ПОЛ – Fe2+-аскорбата. Для определения исходного уровня ТБКРВ, к 1.8 мл буферно-солевой среды (БСС), содержащей 132 ммоль/л NaCl, 5 ммоль/л KCl, 1.3 ммоль/л MgCl2, 1.2 ммоль/л NaH2PO4, 1.0 ммоль/л CaCl2 и 10 ммоль/л трис-НСl буфер, рН 7.4, последовательно добавляли 0.2 мл суспензии синаптосом (1–2 мг/мл белка), 1 мл 10%-ной ТХУ (Acros Оrganics, Бельгия) и 1 мл 0.67%-ной ТБК (Sigma, США). Пробы нагревали до 100°С в течение 15 мин, а затем центрифугировали при 600 g в течение 10 мин. В супернатанте на спектрофотометре ПЭ 3000УФ (Шанхай, Китай) определяли оптическую плотность при 532 и 560 нм против контрольной пробы, не содержащей суспензию синаптосом. При определении ПОЛ, индуцированного системой Fe2+-аскорбат, к 1.4 мл БСС, добавляли 0.2 мл суспензии синаптосом (1–2 мг/мл белка), 0.2 мл 0.5 мМ аскорбата и 0.2 мл 12 мкМ соли Мора (Fe(NH4)2(SO4)2·6H2O). Пробы инкубировали при 37°С в течение 10 мин. Дальнейшие измерения проводили как при определении исходного уровня ТБКРВ. Разность оптических плотностей (Е532–Е560) использовали для определения концентрации ТБКРВ, используя коэффициент молярной экстинкции МДА, равный 1.56 × 105 л/М см.
Определение карбонильных групп в белках мембран синаптосом. Содержание карбонильных групп в белках мембран синаптосом измеряли в соответствии с методом, описанным Venditti et al. [38] с некоторыми изменениями. При этом определяли исходный уровень карбонильных групп, а также их накопление под действием генерируемых в среде инкубации оксидантов. Для определения исходного уровня карбонильных групп, 0.2 мл суспензии мембран (2.5 мг/мл белка) смешивали с 0.2 мл 20%-ной ТХУ. Для запуска процессов окислительной модификации белков in vitro с образование карбонильных групп, суспензию мембран синаптосом (~2.5 мг/мл белка) инкубировали с 10–3 М Fe2+, 10–3 М ЭДТА и 3 × 10–4 М Н2О2 в течение 15 мин при 37°С, после чего смешивали с 0.2 мл 20%-ной ТХУ. Во всех пробах белки осаждали центрифугированием при 1500 g в течение 10 минут. К осадку белков (~0.5 мг белка), после растворения в 100 мкл 0.1 М NaOH, добавляли 1 мл 10 мМ 2,4-динитрофенилгидразина (ДНФГ) (Sigma, США) в 2 М HCl, смесь инкубировали при комнатной температуре в течение 1 часа, время от времени встряхивая. В контрольный образец добавляли только 2 М HCl. Затем во все пробы добавляли 20%-ную TХУ и инкубировали в течение 10 мин на ледяной бане. Затем пробы центрифугировали при 1500 g в течение 15 мин для осаждения белков. Для удаления непрореагировавших ДНФГ и остатка липидов осажденные белки трижды промывали смесью этанол : этилацетат (1 : 1, об./об.). Конечный осадок белков растворяли в 6 М растворе гуанидин гидрохлорида. Оптическую плотность полученного белкового раствора регистрировали при 370 нм. Определяли разницу в оптической плотности пробы по отношению к контролю и рассчитывали количество карбонильных групп в нмоль на мг белка, используя молярный коэффициент экстинкции 22 000 М–1 см–1.
Определение активности СОД. Суммарную активность СОД в синаптоплазме определяли адренохромовым методом, основанным на спонтанном автоокислении адреналина с образованием диформазана, имеющего максимум поглощения при 560 нм [39]. Скорость реакции образования диформазана оценивали по изменению оптической плотности в единицу времени. В опытную пробу вносили 3.67 мл 0.2 М бикарбонатного буфера, рН 9.7, 0.1 мл 2.4 мМ нитросинего тетразолия (НСТ) (Sigma, США), 0.03 мл синаптоплазмы и 0.2 мл 0.1%-ного раствора адреналина. В контрольную пробу добавляли 3.87 мл 0.2 М бикарбонатного буфера (рН 9.7), 0.1 мл 2.4 мМ НСТ и 0.03 мл синаптоплазмы. Количество фермента, которое вызывало 50%-ное ингибирование автоокисления адреналина, принимали за 1 условную единицу (у. е.). Активность СОД была выражена в у. е. на мг белка.
Определение активности каталазы (КАТ). Активность КАТ определяли в синаптоплазме при 37°C с помощью спектрофотометра ПЭ-3000УФ (Шанхай, Китай) по методу Аеби [40]. Синаптоплазма (0.03 мг белка) добавляли к 1 мл реакционной смеси, содержащей 100 мМ натрий-фосфатного буфер, pH 7.4, и 8.8 мМ H2O2. Уменьшение поглощения при 240 нм контролировали в течение 3 мин при 37°С. Активность КАТ выражали в мкМ/мин мг белка.
Статистическая обработка. Обработка данных произведена с помощью однофакторного дисперсионного анализа (ANOVA) с использованием пакета прикладных программ Statistica 8.0 (StatSoft, Inc., США). Нормальность распределения оценивали критерием Шапиро–Уилка. Достоверность различий между нормально распределенными данными определяли с помощью критерия Фишера на уровне значимости p ≤ 0.05. Для оценки взаимосвязи между нормально распределенными переменными был использован корреляционный анализ Пирсона. Данные в таблицах приведены в виде: среднее ± ошибка среднего. Каждая кривая на графиках концентрационной зависимости скорости гидролиза АТХ – среднее 10-ти независимых экспериментов.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Как видно на рис. 1, график концентрационной зависимости активности АХЭ у контрольных животных имеет характерную колоколообразную форму, т.е. скорость гидролиза растет до определенного оптимального значения концентрации субстрата ([S]опт), затем по мере дальнейшего увеличения концентрации АТХ падает. Таким образом, кинетика ацетилхолинэстеразной реакции характеризуется наличием субстратного ингибирования, что согласуется с литературными данными [35]. Характер концентрационной зависимости активности АХЭ не изменяется при ишемии, а также на фоне гипотермии (рис. 1).
Рис. 1.
Концентрационная зависимость активности АХЭ синаптических мембран коры головного мозга крыс: 1 – контроль; 2 – ишемия 60 мин; 3 – ишемия 60 мин на фоне гипотермии 33°С (n = 10).
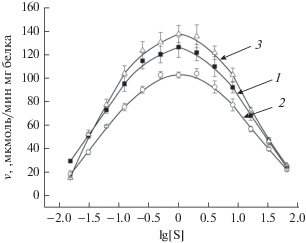
При всех исследованных состояниях наиболее высокая активность АХЭ соответствует концентрации АТХ, приблизительно равной 1 мМ (рис. 1). Абсолютное значение оптимальной концентрации субстрата ([S]опт), вычисленное из уравнения (2):
существенно не отличаются от значений, найденных по графику (табл. 1).
Таблица 1.
Кинетические характеристики АХЭ мембран синаптосом коры головного мозга крыс при ишемии/реперфузии и гипотермии (М ± m, n = 10)
| Состояние животного |
Vmax, мкмоль/мин мг белка |
Km, мМ |
Vmax/Km | Ki, мМ |
[S]опт, мМ |
|---|---|---|---|---|---|
| Контроль | 144.5 ± 8.6 | 0.063 ± 0.002 | 2293.6 ± 102.4 | 12.76 ± 1.02 | 0.890 ± 0.039 |
| Ишемия 60 мин | 118.7 ± 3.8* | 0.069 ± 0.005 | 1721.1 ± 94.5* | 15.20 ± 0.34* | 1.024 ± 0.063 |
| Ишемия 60 мин + гипотермия | 158.9 ± 10.7# | 0.071 ± 0.003 | 2225.3 ± 154.2# | 13.62 ± 0.68 | 0.983 ± 0.029 |
Церебральная ишемия приводит к снижению активности АХЭ при всех исследованных концентрациях субстрата (рис. 1, табл. 1). Так, например, при концентрации субстрата, равной 1 мМ, уменьшение активности АХЭ составляет 18.8% относительно контроля (в контроле 126.16 ± ± 8.79 мкмоль/мин мг, при ишемии – 102.55 ± ± 2.54 мкмоль/мин мг; р < 0.05). При этом наблюдается незначительное смещение [S]опт, но ее величина достоверно не отличается от значения в контроле (табл. 1).
Снижение температуры тела до 33°С в период окклюзии сонных артерий мозга крыс способствует сохранению активности АХЭ на уровне контроля, а в диапазоне концентраций 1.25 × 10–4–0.8 × 10–2 M АТХ-активность фермента становится незначительно выше по сравнению с контролем (рис. 1).
Анализ кинетических характеристик АХЭ синаптических мембран мозга показал, что значение Vmax при ишемии снижается на 18.1% относительно контроля (табл. 1), а изменения константы Михаэлиса несущественны. Мы рассчитали отношение Vmax/Km, которое отражает относительную эффективность катализа при физиологических концентрациях субстрата. Оказалось, что при ишемии эффективность катализа становится на 25% ниже уровня контроля (табл. 1).
Из табл. 1 видно, что константа ингибирования (Ki), которая является мерой сродства субстрата к периферическому месту связывания, при ишемии увеличивается на 19%, что на фоне неизменных значений Km способствует повышению диапазона эффективных концентраций АТХ и теоретических значений [S]опт АХЭ, вычисленных по формуле (2).
Корреляционный анализ показал слабую связь между значениями Km и Ki при исследованных состояниях животных (r = 0.59; p > 0.05). С биофизической точки зрения это вполне возможно, так как периферическое место связывания, ответственное за субстратное ингибирование, находится на периферии и никак не связано с активным центром и конформационной динамикой молекулы АХЭ. Достоверное повышение Ki при ишемии свидетельствует об ослаблении прочности тройного комплекса SES в схеме субстратного ингибирования фермента [35].
Мягкая гипотермия на фоне субтотальной ишемии мозга препятствует снижению Vmax фермента (табл. 1). При этом значения Km достоверно не отличается от контроля, что способствует нормализации уровня эффективности катализа. Кроме того, наблюдается нормализация и таких параметров, как Ki и [S]opt.
АХЭ заякорена в липидной матрице мембраны. Обнаруженные изменения активности и кинетических параметров АХЭ могут быть индуцированы окислительными модификациями молекул как самого фермента, так и белков и липидов в его микроокружении. Для того, чтобы оценить уровень окислительной модификации липидов и белков мембран синаптосом, обусловленный АФК, мы определили количество ТБКРВ и карбонильных групп. Анализ интенсивности ПОЛ в синаптосомах после острой ишемии не выявил изменения исходного уровня ТБКРВ, но установил достоверный рост их накопления в инкубируемых in vitro в среде Fe2+-аскорбат пробах (рис. 2).
Рис. 2.
Содержание ТБКРВ (а) и скорость их накопления в присутствии прооксидантов (б) в синаптосомах из коры головного мозга крыс при ишемии и гипотермии (n = 10): 1 – контроль, 2 – ишемия, 3 – ишемия + гипотермия. Достоверные различия: * – относительно контроля, # – относительно ишемии.
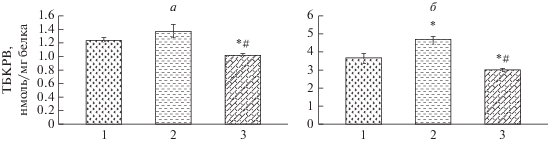
Снижение температуры тела во время ишемии предотвращает активацию процессов пероксидации липидов мембран синаптосом. При этом как исходный, так и индуцированный уровень ТБКРВ в синаптосомах снижается не только относительно значений у ишемированных животных, но и значений контроля. Обнаружена корреляция между кинетическими параметрами АХЭ и содержанием продуктов ПОЛ в синаптосомах. Так, между значениями Vmax, и уровнями ТБКРВ обнаружены отрицательные корреляции (r = –0.95 для исходных и r = –1 для индуцированных процессов, р < 0.05). Отрицательные корреляции обнаружены также между значениями показателя эффективности катализа АХЭ и ТБКРВ (r = –0.70 для исходных и r = –0.87 для индуцированных процессов, р < 0.05).
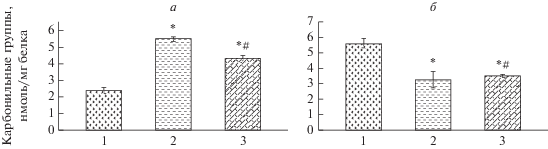
АФК и продукты ПОЛ, образующиеся в мозге при ишемии, могут способствовать окислительной модификации отдельных аминокислотных остатков в мембранных белках, что будет сопровождаться глубокими изменениями их структурной организации [41]. Результаты нашего исследования показали, что в белках мембран синаптосом при ишемии уровень карбонильных групп на 128% выше по сравнению с контролем (рис. 3). При ишемии, вызванной на фоне гипотермии, уровень карбонильных групп также существенно выше контроля (на 79.6%), но достоверно ниже по сравнению с их уровнем при ишемии. Вместе с тем при ишемии и ишемии на фоне гипотермии в инкубируемых in vitro в присутствии оксидантов пробах количество карбонильных групп накапливается значительно меньше по сравнению с контролем. Между кинетическими характеристиками АХЭ и уровнями карбонильных групп обнаружены существенные корреляционные связи. Так, между концентрацией исходных карбонильных групп и значениями Vmax/Km, Ki имеет место отрицательная корреляция (r = –0.85, р < 0.05), а значениями Ki – положительная корреляция (r = +0.95, р < 0.05).
Рис. 3.
Содержание карбонильных групп и их прирост за 15 мин в присутствии Fe2+-H2O2 в белках мембран при ишемии и гипотермии: 1 – контроль, 2 – ишемия, 3 – ишемия + гипотермия (n = 10). Достоверные различия: * – относительно контроля, # – относительно ишемии.
Интенсификация свободнорадикальных процессов при ишемии может быть связана не только с избыточной генерацией АФК и АФА, реакциями окислительной модификации липидов, но и с изменениями в активности компонентов антиоксидантной системы с последующим смещением окислительно-восстановительного баланса клетки в сторону прооксидантов. Анализ СОД выявил трехкратное повышение ее активности в синаптосомах при ишемии (табл. 2). При этом активность КАТ снижается на 58% относительно контроля, что привело к значительному повышению отношения СОД/КАТ. При ишемии на фоне гипотермии активность СОД остается на уровне контроля, а активность каталазы снижается как при ишемии. В результате отношение СОД/КАТ возрастет в меньшей степени, чем при ишемии. При этом между активностью СОД и уровнями МДА обнаружена положительная корреляция (r = +0.82, р < 0.05 для исходного уровня и r = = +0.95, р < 0.05 для индуцированного). Положительная корреляция обнаружена также между активностью СОД, отношением СОД/КАТ и уровнями карбонильных групп (r = +0.74 и r = +0.86).
Таблица 2.
Активность СОД и КАТ в синаптосомах из коры головного мозга крыс при ишемии и гипотермии (М ± m, n = 10)
| Состояние животного | СОД, усл. ед./мг белка | КАТ, мкмоль/мин мг белка | СОД/КАТ |
|---|---|---|---|
| Ложная операция | 2.79 ± 0.16 | 0.99 ± 0.01 | 2.81 ± 0.17 |
| Ишемия 60 мин | 8.36 ± 0.54* | 0.42 ± 0.02* | 19.9 ± 1.39* |
| Ишемия 60 мин + гипотермия | 2.24 ± 0.24# | 0.43 ± 0.03* | 5.21 ± 0.39*# |
Между кинетическими параметрами АХЭ и активностью антиоксидантных ферментов синаптосом также имеют место высокие уровни корреляции. Так, коэффициенты корреляции между Vmax,Vmax/Km, Ki и отношением СОД/КАТ составляют –0.9 (р < 0.05), –1 (р < 0.05) и +1 (р < < 0.05) соответственно.
ОБСУЖДЕНИЕ ПОЛУЧЕННЫХ РЕЗУЛЬТАТОВ
Полученные нами данные свидетельствуют о том, что церебральная ишемия существенно влияет на активность и кинетические характеристики АХЭ мембран синаптических окончаний нейронов. Снижение температуры тела в период окклюзии сонных артерий мозга крыс предотвращает изменение активности и кинетических характеристик фермента.
Обнаруженные разнонаправленные изменения активности АХЭ при ишемии и ишемии–г-ипотермии обусловлены модуляциями механизмов функционирования фермента, чувствительными индикаторами которых являются кинетические характеристики. Исследование показало, что снижение эффективности катализа АХЭ при ишемии происходит исключительно за счет Vmax, поскольку значения Km при этом не изменяются. Известно, что изменение Vmax может происходить за счет изменения концентрации фермента или же его каталитической константы, в соответствии с уравнением:

где kcat – каталитическая константа (константа скорости образования продукта реакции), [E] – концентрация фермента.
Концентрация фермента в клетке определяется балансом между его биосинтезом и деградацией.
Известно, что АХЭ кодируется одним геном, но может существовать в нескольких молекулярных формах [42]. Соотношения молекулярных форм определяются их тканевым, клеточным и субклеточным распределением и изменяются соответственно развитию или патогенетическому состоянию данных тканей. У позвоночных 3'-конец пре-мРНК AChE альтернативно сплайсирован для генерации трех разных изоформ, которые несут идентичный каталитический домен, но различный C-терминальный конец. В мозге экспрессируются две изформы: тетрамерная AChET, которая закрепляется на богатый пролином якорь мембраны PRiMA [42], и растворимая мономерная форма AChER, синтез которой активируется при остром физиологическом стрессе в мозге [43].
В работе [24] было установлено, что содержание АХЭ в мозге при ишемии существенно снижается. Поскольку ишемия сопровождается развитием стрессорной реакции, гипотетически она может снизить концентрацию АХЭ в синаптической мембране за счет модуляции альтернативного сплайсинга, в частности, переключения синтеза AChET на AChER.
Установлено, что при глобальной ишемии существенно подавляется синтез белков в мозге [44], поэтому высказано предположение [24], что снижение содержания АХЭ в мозге при ишемии связано с подавлением экспрессии фермента.
Действительно, при ишемии снижение содержание АХЭ в синаптических мембранах может зависеть и от скорости синтеза фермента. Известно, что АХЭ сначала синтезируется в виде каталитически неактивной молекулы, которая затем быстро созревает в каталитически активную форму. Однако большая часть неактивного фермента остается в этой незрелой форме. Эти молекулы имеют очень короткую продолжительность жизни и разрушаются за счет деградации, связанной с эндоплазматическим ретикулумом с периодом полураспада около 45 минут. Молекулы, созревшие до каталитически активных форм, являются промежуточными между вновь синтезированным каталитически неактивным ферментом и полностью зрелыми и стабильными олигомерными формами. Эти формы способны стабилизироваться прямым взаимодействием с некаталитическими субъединицами PRiMA [45]. Следовательно, экспрессия PRiMA тоже является ключевым фактором в организации G4 АХЭ и нацеливании ее на клеточную мембрану [46]. Однако в нашем случае моделирование ишемии производилось за достаточно короткий промежуток времени (1 ч), что может оказаться недостаточным для того, чтобы существенно изменить концентрацию АХЭ в синаптической мембране, поскольку период полужизни АХЭ в мозге взрослой крысы составляет 2.84 ± 0.13 дней [47].
Снижение концентрации АХЭ при церебральной ишемии могло происходить, вследствие деградации мембраносвязанного фермента в результате его протеолиза. Известно, что мембранные белки деградируют либо посредством эндолизосомного механизма, либо аутофагии [48, 49]. Гу и сотр. [50] с помощью трансмиссионного электронного микроскопа через 4 часа после ишемического повреждения головного мозга обнаружили в нейронах заметное накопление аутофагических тел и лизофом аутофагии.
Полученные нами данные свидетельствуют о том, что при ишемии активность и каталитические характеристики АХЭ коррелируют с уровнем продуктов окислительной модификации белков и липидов мембран синаптосом. Это указывает на то, что окислительный стресс может способствовать снижению активности АХЭ при ишемии. Считается, что АФК играет важную роль в развитии окислительного стресса, вызывающего повреждение клеток мозга во время ишемии. АФК в синаптических окончаниях нейронов могут образоваться преимущественно при нарушении дыхательной цепи митохондрий [51], при функционировании моноаминоксидазы [52], а также поступать из активированной микроглии [53]. ${\text{O}}_{{\text{2}}}^{{ \bullet --}},$ поступающийся из этих источников, конвертируется (преобразовывается) с помощью СОД в H2O2, которая затем превращается в безвредные H2O и O2 под действием КАТ. СОД и КАТ является ключевыми антиоксидантными ферментами, играющими жизненно важную роль в устранении АФК. Некоторые исследования показали, что активность СОД при церебральной ишемии увеличилась непропорционально изменению активности КАТ [54, 55]. В нашем исследовании также обнаружен существенный дисбаланс в работе этих ферментов в синаптосомах при ишемии. Различия в модуляции активности СОД и КАТ при ишемии могут происходить по различным причинам. Активность СОД увеличивается, вероятно, как реакция на окислительный стресс, вызванный ишемией. Активация СОД во время ишемии может быть компенсаторной реакцией на перепроизводство ${\text{O}}_{{\text{2}}}^{{ \bullet --}}$ [56]. Кроме того, повышение уровня интерлейкина-1 (ИЛ-1) [55], фактора некроза опухолей (TNF) и липополисахаридов [57] во время ишемии также может активировать СОД [58]. В отличие от СОД, избыток ${\text{O}}_{{\text{2}}}^{{ \bullet --}}$ или H2O2 может вызывать повреждения активного центра КАТ, что приводит к инактивации фермента [59]. Дисбаланс между СОД и КАТ может привести к накоплению чрезмерного количества H2O2 в синаптических окончаниях нейронов. По данным микродиализного исследования Лей и др. [60], базальный уровень H2O2 в гиппокампе песчанок составлял примерно 1 мкМ. В стриатуме реперфузируемого мозга крыс после ишемического инсульта концентрация H2O2 достигала 100 мкМ [61].
Данилович [62] на сарколемме миометрия свиньи показал, что H2O2 в физиологических концентрациях (от 1.6 до 6.4 мкМ) способен ингибировать АХЭ. В присутствии дитиотреитола ингибиторный эффект Н2О2 частично снижался, что, по мнению автора, свидетельствует о вовлеченности тиоловых групп в ингибирование фермента. Накопление H2O2 в миллимолярном диапазоне концентраций может снижать экспрессию эпидермальной АХЭ и ингибировать активность рекомбинантной АХЭ человека [63, 64], а также растворимой и мембраносвязанной АХЭ эритроцитов человека [65].
H2O2 является мягким окислителем, способным окислять тиоловые группы белков, однако в присутствии металлов переменной валентности пероксид водорода может образовать •OH в реакции Фентона. Было подтверждено, что •ОН ингибирует АХЭ мозга крысы и активность рекомбинантной АХЭ человека [66]. Кинетический анализ с использованием очищенной рекомбинантной АХЭ человека и молекулярное моделирование трехмерной структуры человеческого АХЭ подтвердили, что АФК-опосредованное окисление Trp432, Trp435 и Met436 вызывает перемещение и дезориентацию His440 в активном центре АХЭ и, как результат, дезактивацию белка [63, 64].
Известно, что ПОЛ может изменить физиологические функции клеточных мембран, модифицируя такие свойства мембранного бислоя, как мембранный потенциал, текучесть, проницаемость для различных веществ и/или посттрансляционные химические модификации [67, 68]. При окислительном стрессе изменения в липидной матрице мембраны могут быть решающим фактором в модификации конформационного состояния некоторых молекул, в том числе АХЭ [69]. Хотя уровень ТБКРВ в синаптосомах при ишемии не изменяется, достоверно увеличивается их накопление при генерации радикалов в условиях in vitro. Это говорит о повышении доступности жирнокислотных остатков фосфолипидов для оксидантов в условиях ишемии из-за нарушений структуры липидного бислоя.
То, что АФК ответственны за ингибирование АХЭ, свидетельствуют данные, полученные при гипотермии. Результаты наших исследований показали, что гипотермия, вызванная во время ишемии, не только предотвращает снижение активности и изменение кинетических характеристик АХЭ, но одновременно снижает уровень продуктов окислительной модификации белков и липидов и дисбаланс антиоксидантных ферментов синаптосом. Ранее Лей и коллеги [70] обнаружили, что гипотермия (30–32°С) снижает ПОЛ в мозге после глобальной ишемии. В последующем было установлено, что умеренная гипотермия, моделированная во время ишемии, снижает уровни NO и •OH после глобальной и фокальной церебральной ишемии [71, 72]. Снижение температуры тела крыс до 33°С при ишемии мозга, вызванной окклюзией среднемозговой артерии, способствовало значительному уменьшению образования супероксид-аниона как в преинфарктной зоне, так и контралатеральной области [73]. Эти данные позволяют с уверенностью предположить, что умеренная гипотермия, снижая генерацию АФК и АФА, предотвращает изменение активности и кинетических характеристик АХЭ мембран синаптосом.
ЗАКЛЮЧЕНИЕ
Полученные в работе новые данные, во-первых, свидетельствуют, что острая церебральная субтотальная ишемия снижает активность и эффективность катализа АХЭ синаптических мембран мозга крыс, главным образом посредством снижения Vmax. Во-вторых, впервые продемонстрировано, что мягкая гипотермия на фоне ишемии предотвращает изменения кинетических констант (Vmax и Ki), в результате чего эффективность катализа АХЭ сохраняется на уровне контроля. Установлено, что при церебральной ишемии существенно увеличивается уровень маркеров окислительной модификации белков и липидов мембран синаптосом, происходит дисбаланс ключевых антиоксидантных ферментов. Гипотермия на фоне ишемии препятствует развитию окислительного стресса в синаптосомах. Полученные результаты вносят вклад в понимание участия АФК в модуляции активности синаптической АХЭ при ишемии мозга и путей защиты фермента при умеренной гипотермии.
Список литературы
Lin L, Wang X., Yu Z. // Biochem. Pharmacol. 2016. V. 5. № 4. https://doi.org/10.4172/2167-0501.1000213
Awooda H.A. // J. Biosci. Med. 2019. V. 7. P. 20–28.
Chan P.H. // J. Cereb. Blood Flow Metab. 2001. V. 21. P. 2–14.
Sayre L.M., Perry G., Smith M.A. // Chem. Res. Toxicol. 2008. V. 21. № 1. P. 172–188.
Valko M., Leibfritz D., Moncol J., Cronin M.T.D., Mazur M., Telser J. // Int. J. Biochem. Cell Biol. 2007. V. 39. P. 44–84.
Awooda H.A., Lutfi M.F., Saeed A.M. // Int. J. Health Sci. 2015. V. 9. P. 17–24.
Chen H., Yoshioka H., Kim G.S., Jung J.E., Okami N., Sakata H., Chan P.H. // Antioxid. Redox Signal. 2011. V. 14. № 8. P. 1505–1517.
Sun M.S., Jin H., Sun X., Huang S., Zhang F.L., Guo Z.N., Yang, Y. // Oxid. Med. Cell. Longev. 2018. https://doi.org/10.1155/2018/3804979
van der Worp H.B., Macleod M.R., Kollmar R. // J. Cereb. Blood Flow. Metab. 2010. V. 30. P. 1079–1093.
Olsen T.S., Weber U.J., Kammersgaard L.P. // The Lancet Neurology. 2003. V. 2. № 7. P. 410–416.
Sun Y.-J., Zhang Z.-Y., Fan B., Li G.-Y. // Front. Neurosci. 2019. V. 13. Article 586. https://doi.org/10.3389/fnins.2019.00586
Schliebs R., Arendt T. // J. Neural Transm. 2006. V. 113. № 11. P. 1625–1644.
Solari N., Hangya B. // Eur. J. Neurosci. 2018. V. 48. № 5. P. 2199–2230.
Taylor P. // Goodman and Gilman’s Pharmacological Basis of Therapeutics / Ed.: Brunton L. N.Y.: McGraw-Hill, 2006. P. 201–217.
Orta-Salazar E., Cuellar-Lemus C.A., Díaz-Cintra S., Feria-Velasco A.I. // Neurología. 2014. V. 29. № 8. P. 497–503.
Hicks D., John D., Makova N.Z., Henderson Z., Nalivaeva N.N., Turner A.J. // J. Neurochem. 2011. V. 116. № 5. P. 742–746.
Zimmerman G., Soreq H. // Cell Tiss. Res. 2006. V. 326. № 2. P. 655–669.
Thullbery M.D., Cox H.D., Schule T., Thompson C.M., George K.M. // J. Cell Biochem. 2005. V. 96. № 3. V. 599–610.
Sakuma M., Hyakawa N., Kato H., Araki T. // J. Neural Trans. 2008. V. 115. № 3. P. 413–422.
Katsuki H., Matsumoto K. // Nicotinic acetylcholine receptor signaling in neuroprotection / Ed.: Akaike A., Shimohama S., Misu Y. Singapore: Springer, 2018. P. 113–136.
Kataoka K., Hayakawa T., Kuroda R., Yuguchi T., Yamada K. // Stroke. 1991. V. 22. P. 1291–1296.
Beley A., Bertrand N., Beley P. // Neurochem. Res. 1991. V. 16. № 5. P. 555–561.
Malatová Z., Gottlieb M., Marsala J. // Gen. Physiol. Biophys. 1999. V. 18. № 1. P. 57–71.
Saez-Valero J., Gonzalez-Garcia C., Cena V. // Mol. Brain Res. 2003. V. 117. P. 240–244.
Мохаммед М.Т., Кличханов Н.К. // Изв. Даг. гос. пед ун-та. Естеств. и точн. науки. 2010. № 2. С. 69–73.
Ojo O.B., Amoo Z.A., Saliu I.O., Olaleye M.T., Farombi E.O., Akinmoladun A.C. // Biomed. Pharmacother. 2019. V. 111. P. 859–872.
Wang Z.-F., Wang J., Zhang H.Y., Tang X.C. // J. Neurochem. 2008. V. 106. P. 1594–1603.
Deb B., Sreenath C., Kumar M.S., Elango K. // J. Appl. Pharm. Sci. 2012. V. 2. № 3. P. 106–111.
Hu T., Fu Q., Liu X., Zhang H., Dong M. // J. Neuroimmunol. 2009. V. 211. P. 84–91.
Lapetina E.G., Soto E.F., De Robertis E. // Biochem. Biophis. Acta. 1967. V. 135. P. 33–43.
Lowry D.H., Rosebrough H.J., Farr A.L., Randall R.J. // J. Biol. Chem. 1951. V. 193. № 1. P. 265–275.
Ellman Y.L., Courtney K.D., Andres V.J., Feathestone R.M. // Biochem. Pharmacol. 1961. V. 7. № 1. P. 88–95.
Stojan J., Brochier L., Alies C., Colletier J.P., Fournier D. // Eur. J. Biochem. 2004. V. 271. P. 1364–1371.
Reed M.C., Lieb A., Nijhout H.F. // Bioessays. 2010. V. 32. P. 422–429.
Rosenberry T.L. // J. Mol. Neurosci. 2010. V. 40. P. 32–39.
Putz M.V. // Molecules. 2011. V.16. P. 3128–3145.
Лысакова Т.И., Аксенцев С.Л., Федорович С.В., Левко А.В., Ракович А.А., Самойленко С.Г., Федулов А.С., Конев С.В. // Биофизика. 1997. Т. 42. Вып. 2. С. 408–411.
Venditti P., Rosa R.D., Meo S.D. // Free Rad. Biol. Med. 2004. V. 36. № 3. P. 348–358.
Сирота Т.В. // Биомед. химия. 2013. Т. 59. № 4. С. 399–410.
Aebi H. // Methods of Enzymatic Analysis. Academic Press. 1974. P. 673–684.
Дубинина Е.Е. // Продукты метаболизма кислорода в функциональной активности клеток. Жизнь и смерть, созидание и разрушение: монография. С.-Петербург, 2006. 400 с.
Massoulie J. // Neurosignals. 2002. V. 11. P. 130–143.
Meshorer E., Soreq H. // Trends Neurosci. 2006. V. 29. № 4. P. 216–224.
Bodsch W., Takahashi K., Barbier A., Ophoff B.G., Hossmann K.A. // Prog. Brain Res. 1985. V. 63. P. 197–210.
Rotundo R.L. // J. Neurochem. 2017. V. 142. P. 52–58.
Chen V.P., Choi R.C., Chan W.K., Leung K.W., Guo A.J., Chan G.K., Tsim, K.W. // J. Biol. Chem. 2011. V. 286. № 38. P. 32948–32961.
Wenthold R.J., Mahler H.R., Moore, W.J. // J. Neurochem. 1974. V. 22. № 6. P. 941–943.
Huber L.A., Teis D. // Curr. Opin. Cell Biol. 2016. V. 39. P. 8–14.
Galluzzi L., Baehrecke E.H., Ballabio A., Boya P., Bravo-San Pedro J.M., Cecconi F., Kroemer G. // EMBO J. 2017. V. 36. № 13. P. 1811–1836.
Gu Z., Sun Y., Liu K., Wang F., Zhang T., Li Q., Sun X. // Neural Reg. Res. 2013. V. 8. № 23. P. 2117–2125.
Starkov A. A., Chinopoulos C., Fiskum G. // Cell Calcium. 2004. V. 36. № 3–4. P. 257–264.
Seif-El-Nasr M., Atia A.S., Abdelsalam R.M. // Arzneimittelforschung. 2008. V. 58. № 4. P. 160–167.
Weinstein J.R., Koerner I.P., Möller T. // Future Neurol. 2010. V. 5. № 2. P. 227–246.
Işlekel S., Işlekel H., Güner G., Özdamar N. // Res. Exp. Med. 1999. V. 199. № 3. P. 167–176.
Девятов А.А., Федорова Т.Н., Стволинский С.Л., Рыжков И.Н., Ригер Н.А., Тутельян В.А. // Биомед. химия. 2018. V. 64. № 4. P. 344–348.
Maier C.M., Sun G.H., Cheng D., Yenari M.A., Chan P.H., Steinberg G.K. // Neurobiol. Dis. 2002. V. 11. № 1. P. 28–42.
Al-Amin M.M., Choudhury M.F.R., Chowdhury A.S., Chowdhury T.R., Jain P., Kazi M., Alkholief M., Alshehri S.M, Reza H.M. // Front. Neurosci. 2018. V. 12. P. 384. https://doi.org/10.3389/fnins.2018.00384
Marikovsky M., Ziv V., Nevo N., Harris-Cerruti C., Mahler O. // J. Immunol. 2003. 170. № 6. P. 2993–3001.
Pigeolet E., Corbisier P., Houbion A., Lambert D., Michiels C., Raes M., Remacle J. // Mech. Ageing Dev. 1990. V. 51. № 3. P. 283–297.
Lei B., Adachi N., Arai T. // Brain Res. Prot. 1998. V. 3. № 1. P. 33–36.
Hyslop P.A., Zhang Z., Pearson D.V., Phebus L.A. // Brain Res. 1995. V. 671. P. 181–186.
Danylovych Iu.V. // Ukr. Biokhim. Zh. 2009. V. 81. № 4. P. 32–38.
Schallreuter K.U., Elwary S.M.A., Gibbons N.C.J., Rokos H., Wood J.M. // Biochem. Biophys. Res. Commun. 2004. V. 315. № 2. P. 502–508.
Schallreuter K.U., Elwary S. // Life Sci. 2007. V. 80. № 24–25. P. 2221–2226.
Молочкина Е.М., Озерова И.Б. // Радиац. биология. Радиоэкология. 2003. Т. 43. № 3. С. 294–300.
Méndez-Garrido A., Hernández-Rodríguez M., Zamorano-Ulloa R., Correa-Basurto J., Mendieta-Wejebe J.E., Ramírez-Rosales D., Rosales-Hernández M.C. // Neurochem. Res. 2014. V. 39. № 11. P. 2093–2104.
Catalá A., Díaz M. // Front. Physiol. 2016. V. 7. P. 423. doi.org/https://doi.org/10.3389/fphys.2016.00423
Gaschler M.M., Stockwell B.R. // Biochem. Biophys. Res. Com. 2017. V. 482. № 3. P. 419–425.
Gonçalves J.F., Fiorenza A.M., Spanevello R.M., Mazzanti C.M., Bochi G.V., Antes F.G., Schetinger M.R.C. // Chem.-Biol. Interact. 2010. 186. № 1. P. 53–60.
Lei B., Tan X., Cai H., Xu Q., Guo Q. // Stroke. 1994. V. 25. P. 147–152.
Kil H.Y., Zhang J., Piantadosi C.A. // J. Cereb. Blood Flow Metab. 1996. V. 16. P. 100–106.
Kumura E., Yoshimine T., Takaoka M., Hayakawa T., Shiga T., Kosaka H. // Neurosci. Lett. 1996. V. 220. P. 45–48.
Maier C.M., Sun G.H., Cheng D., Yenari M.A., Chan P.H., Steinberg G.K. // Neurobiol. Dis. 2002. V. 11. № 1. P. 28–42.
Дополнительные материалы отсутствуют.