

Нейрохимия, 2021, T. 38, № 4, стр. 298-312
Технология геномного редактирования для исследования и коррекции нейродегенеративных заболеваний
И. А. Гривенников 1, В. З. Тарантул 1
1 Федеральное государственное бюджетное учреждение Институт молекулярной генетики
Национального исследовательского центра “Курчатовский институт”
Москва, Россия
Поступила в редакцию 02.06.2021
После доработки 05.07.2021
Принята к публикации 06.07.2021
Аннотация
Прогресс науки в значительной мере связан с возникновением новых технологий. Одной из таких прорывных технологий, созданной в начале XXI века, являются различные варианты геномного редактирования, среди которых наиболее простой и эффективной является технология, основанная на использовании коротких палиндромных повторов, регулярно расположенных группами (англ. сокр. СRISPR). Другой важнейшей технологией стало получение индуцированных плюрипотентных стволовых клеток (ИПСК, англ. сокр. iPSC). Совместное использование этих двух технологий существенно повысило возможности тонкого манипулирования с геномом для исследования молекулярно-генетических основ различных патологий, поиска генов-мишений и в перспективе для практического применения в терапевтическом воздействии на разнообразные тяжелые наследственные заболевания. Такой подход оказался чрезвычайно полезным для изучения нейропатологий человека, особенно с учетом того, что многочисленные терапевтические средства, которые оказывают свою эффективность в моделях таких болезней у грызунов, не принесли пользы больным людям. В настоящем обзоре суммированы основные результаты последних лет по генетическому и эпигенетическому редактированию генома, полученные главным образом с использованием различных вариантов технологии CRISPR/Сas9 на моделях ИПСК и культивируемых соматических клеток пациентов с такими тяжелыми нейродегенеративными заболеваниями как болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона и боковой амиотрофический склероз.
Принятые сокращения: Aβ – бета-амилоидный пептид; AICD – внутриклеточный домен предшественника амилоида (amyloid precursor protein intracellular domain); АМРА-рецептор – рецептор α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты; APP – предшественник бета-амилоида (amyloid precursor protein); APOЕ – аполипопротеин Е; CRISPR/Сas9 – короткие палиндромные повторы, регулярно расположенные группами/CRISPR-ассоциированный ген 9; ДА-нейроны – дофаминeргические нейроны; dCas9 – деактивированный Cas9; DSB – двухцепочечный разрыв (double-strand break); GMF – фактор созревания глии (glia maturation factor); GSAP – белок, активирующий γ-секретазу (γ-secretase activating protein); HDR – гомологически направленная репарация (homology directed repair); NHEJ – негомологичное соединение концов (non-homologous end joining); NFT – нейрофибриллярные клубки (neurofibrillary tangles); PSEN – пресенилин (presenilin); TREM2 – триггерный ген рецептора, экспрессируемый на миелоидных клетках 2 (triggering receptor expressed on myeloid cells 2); БАС - боковой амиотрофический склероз; БП – болезнь Паркинсона; БА – болезнь Альцгеймера; БГ – болезнь Гантингтона; ДПП – дипептидные повторы; ИПСК – индуцированные плюрипотентные стволовые клетки; ЭР – эндоплазматический ретикулум; Tau – тубулин-ассоциированная единица (tubulin-associated unit); MAPT – ген белка Тау, ассоциированный с микротрубочками (microtubule associated protein tau); ЭСК – эмбриональные стволовые клетки.
ВВЕДЕНИЕ
Нейродегенеративные заболевания – наследственные или приобретенные заболевания нервной системы, сопровождающиеся прогрессирующей гибелью определенных популяций нейронов в центральной нервной системе. К числу таких заболеваний относят болезни Альцгеймера, Паркинсона, Хантингтона, боковой амиотрофический склероз и ряд других более редких нейропатологий. Разные нейродегенеративные заболевания определяются многими общими фундаментальными процессами, такими как протеотоксический стресс и сопутствующие ему нарушения в убиквитин-протеасомной и аутофагосомной/лизосомальной системах, окислительный стресс, митофагия, апоптоз и нейровоспаление. Патофизиологию и молекулярную основу этих заболеваний исследуют уже много десятилетий с использованием разных подходов, однако понимание тонких механизмов этих патологий пока остается малоизученным [1]. Основная сложность таких исследований заключалась в том, что изучение образцов мозга пациентов с нейродегенеративными заболеваниями были возможны только post mortem, но эти исследования ограничены редкостью таких образцов, этическими проблемами, а также потерей наиболее уязвимых нейронов и ограниченным временным окном для их изучения.
Успеху способствовало создание технологии индуцированных плюрипотентных стволовых клеток (ИПСК), которая позволила получать в большом количестве нейрональные предшественники и зрелые нейроны человека, а также разные типы глиальных клеток от пациентов с различными нейропатологиями. Другой качественно новый этап в исследованиях нейродегенеративных заболеваний связан с разработкой подхода функциональной геномики на основе технологии CRISPR/Cas9 [2]. По сравнению с другими методами редактирования генома, система CRISPR/Cas9 проста, эффективна и более специфична. CRISPR/Cas9‑направленное редактирование генов становится перспективным инструментом для нарушения экспрессии генов, вызывающих заболевания, или редактирования патогенных мутаций без изменения имеющегося генетического фона. Генетическое редактирование в ИПСК и дифференцированных из них нейрональных клетках с помощью CRISPR/Cas9 стали критическим подходом к изучению молекулярных механизмов патологий нервной системы и обнаружению терапевтических мишеней для лечения неврологических заболеваний [3, 4] (рис. 1). Обо всем этом и пойдет речь в настоящем обзоре.
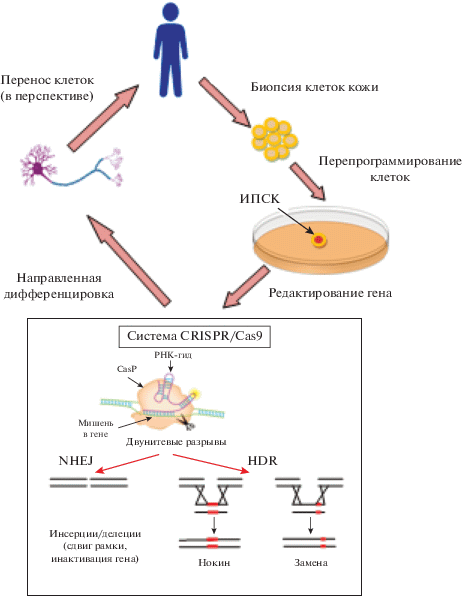
Технология CRISPR/Cas9 основана на использовании направляющей РНК (гидРНК), которая содержит область гомологии с геном-мишенью, и эндонуклеазы Cas9. Комплекс рибонуклеопротеинов гидРНК:Cas9 присоединяется к комплементарному участку ДНК-мишени. После этого Cas9 осуществляет в мишени двухцепочечные разрывы, которые восстанавливаются с помощью негомологичного соединения концов (NHEJ) или гомологически направленной репарации (HDR).
Далее на конкретных примерах рассмотрим, как эта технология в сочетании с технологией получения ИПСК продвинула наше понимание природы основных нейродегенеративных заболеваний человека.
БОЛЕЗНЬ ПАРКИНСОНА
Болезнь Паркинсона (БП) – распространенное прогрессирующее нейродегенеративное заболевание, характеризующееся преимущественной гибелью дофаминергических нейронов (ДА-нейронов) в центральной нервной системе. Это заболевание ассоциировано с возрастом и является одной из значимых причин инвалидизации пожилых лиц в современном обществе [5]. Его частота колеблется от 60 до 140 человек на 100 тысяч населения, при этом число больных значительно увеличивается среди представителей старшей возрастной группы [6].
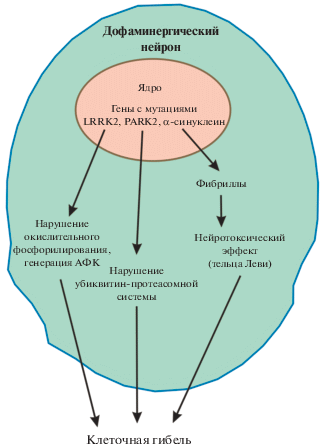
От 10 до 15% случаев БП представлены семейными формами заболевания, связанными с мутациями в генах [7, 8]. На сегодняшний день выявлен ряд таких генов (SNCA, PARK2, LRRK2, PINK1, GBA, DJ1, UCHL1, ATP13A2), вовлеченных в патогенез семейной формы БП [9–11]. Семейные формы БП могут иметь аутосомно-доминантный или аутосомно-рецессивный тип наследования [12]. Мутации в генах SNCA, LRRK2 и UCHL1 вызывают БП с аутосомно-доминантной формой наследования. Аутосомно-рецессивные формы заболевания развиваются при наличии мутаций в генах PINK1, DJ-1, PАRK2. Наиболее часто к развитию заболевания приводят мутации в генах LRRK2, PАRK2 и SNCA (рис. 2).
Рис. 2.
Схематическое представление участия продуктов мутантных генов LRRK2, PАRK2 и SNCA в дегенерации ДА-нейронов при БП.
Редактирование генома позволяет исправить разнообразные мутации в этих генах, что имеет как теоретическое значение, так и сугубо прикладное, в свете использования данного подхода в клеточной терапии нейродегенеративных заболеваний. Кроме того, внесение строго определенных изменений в отдельные участки “нормальных” генов позволяет исследовать функции этих генов и их белковых продуктов.
Так, исправление в ИПСК с помощью технологии “цинк-пальцевых” нуклеаз наиболее часто встречающейся и значимой для развития БП мутации (rs34637584 G > A) в гене LRRK2 (кодирует белок дардарин, обладающий протеинкиназной активностью), позволило установить, что повреждение митохондриальной ДНК является одним из ранних признаков нейрональной дисфункции и в сочетании с нарушенной аутофагией играет важную роль в развитии данной патологии [13].
Интересные данные были получены группой Reinhardt с соавт. [14]. С использованием технологии “цинк-пальцевых” нуклеаз в двух линиях ИПСК от пациентов с БП, несущих точковую мутацию G2019S в гене LRRK2, была проведена коррекция этой мутации. Кроме того, в контрольной линии ИПСК, полученной от здорового донора, было произведено внедрение мутации G2019S. Дальнейший анализ показал, что длина нейритов в клетках генетически исправленной мутантной линии ИПСК была сходной с таковой в контроле и намного превышала длину нейритов в мутантных линиях. Кроме того, во всех линиях, содержащих мутации G2019S, но не в контрольных линиях, была повышена экспрессия α-синуклеина и тау-белка, ассоциированного с микротрубочками. Таким образом, авторам удалось в значительной степени обратить патологические фенотипы, ассоциированные с мутацией G2019S в гене LRRK2.
В последние годы для геномного редактирования все большее распространение получает нуклеазная система CRISPR/Cas9 [2]. Qing с соавт. [15] с помощью этой технологии осуществили введение мутации G2019S в ген LRRK2 в ИПСК человека. При последующей дифференцировке мутантных ИПСК в дофаминергические нейроны (ДА-нейроны) у них происходило существенное снижение ветвления отростков, а также значительное уменьшение их длины по сравнению с линиями контрольных клеток. Известно, что данная мутация приводит к увеличению киназной активности белкового продукта дардарина. Обработка таких клеток ингибитором киназы дардарина приводила к восстановлению нормального фенотипа ДА-нейронов. В итоге проведенной работы авторы показали возможность использования генетического редактирования для эффективного и стандартизованного создания мутантных линий ИПСК для моделирования БП.
С помощью системы CRISPR/Cas9 также была осуществлена коррекция мутации G2019S в гене LRRK2 в ИПСК человека, полученных из фибробластов кожи пациента с диагностированной формой БП [16]. Было показано, что в различных клонах нейрональных предшественников, дифференцированных из ИПСК после геномного редактирования, наблюдалась тетраплоидия 92 XXYY/46,XY (24–43% клеток в разных клонах). Кроме того, были обнаружены одиночные транслокации и разрывы хромосом. Полученные данные свидетельствуют о необходимости тщательного морфологического анализа получаемых культур клеток и разработки улучшенных методов геномного редактирования, обеспечивающих стабильность генома в таких клетках.
Известно, что приматы являются наиболее близкими к человеку млекопитающими по своим физиологическим свойствам. Поэтому использование их в качестве моделей различных тяжелых заболеваний человека является перспективной задачей. Vermilyea с соавт. [17] использовали эмбриональные стволовые клетки (ЭСК) и ИПСК от обыкновенной мартышки для моделирования молекулярных и клеточных механизмов патогенеза БП. С помощью технологии CRISPR/Cas9 авторы ввели в клетки ген LRRK2 с мутацией G2019S, а также укороченный вариант этого гена. Также, как и в клетках человека, введение мутантного гена приводило к увеличению киназной активности дардарина. После дифференцировки трансфицированных мутантных клеток в нейрональном направлении в них было обнаружено увеличение уровня активных форм кислорода, снижение выживаемости ДА-нейронов и уменьшение длины и ветвления нейритов у этих клеток. Причем в клетках с введенным укороченным геном таких фенотипических изменений не происходило. Полученные результаты свидетельствуют о перспективности использования соответствующих клеток приматов для создания моделей БП.
Система CRISPR/Cas9 была использована также для поиска генов, осуществляющих физиологическую регуляцию гена PARK2, который кодирует белок паркин, обладающий Е3-убиквитин-лигазной активностью. Мутации в этом гене ведут к нарушению функций данного фермента, что сопровождается накоплением аномальных белковых субстратов в клетке, индукцией апоптоза и гибелью нейронов [18]. Широкомасштабный геномный нокаут, проведенный с помощью технологии CRISPR/Cas9 в клетках, экспрессирующих трансген GFP-PARK2, позволил идентифицировать 53 регулятора экспрессии этого гена, включая ген-репрессор THAP11 [19].
Ген SNCA (PARK1) кодирует α-синуклеин – белок, который является основным компонентом так называемых телец Леви. Нарушение синтеза или процессинга данного белка, вследствие соответствующих мутаций, является центральным звеном молекулярного патогенетического каскада, ведущего к накоплению в клетке нерастворимых белковых комплексов и прогрессирующей гибели нейронов при БП [12]. Arias-Fuenzalida с сотр. [20] разработали эффективный метод точного введения мутаций в интересующий ген и быстрый отбор соответствующих клонов, с применением комбинации CRISPR/Cas9 системы и FACS анализа. С помощью этого метода в ген SNCA ИПСК от здорового человека были введены А30Р и А53Т мутации, ответственные за развитие симптомов БП. Анализ дифференцированных в нейрональном направлении мутантных клеток выявил у них нарушения в клеточном дыхании и синтезе АТФ. Разработанный метод, по мнению авторов, позволит в дальнейшем перейти к автоматизации процесса редактирования генома с быстрой оценкой фенотипа получаемых клеток.
С использованием технологии CRISPR/Cas9 Chen с соавт. [21] делетировали ген SNCA в ЭСК человека и получили соответствующие гетеро и гомозиготные (SNCA+/− and SNCA−/−) линии клеток. Затем эти клетки дифференцировали в ДА-нейроны и индуцировали в них процесс синуклеинопатии добавлением экзогенно сформированных фибрилл α-синуклеина. Авторы показали, что SNCA+/− и SNCA−/− нейроны были более устойчивы к образованию патологических агрегатов по сравнению с исходными нейронами. Таким образом, частичное или полное удаление аллелей гена SNCA отражает степень устойчивости клеток к патологии, связанной с формированием телец Леви.
Технология CRISPR/Cas9 была использована также для изучения нейровоспалительных путей, вовлеченных в патогенез БП, в частности, сигнального пути протеинкиназы Cδ (PKCδ). Подавление в ДА-нейронах экспрессии гена, кодирующего PKCδ, сильно снижало индуцируемый эндосульфаном апоптоз в этих клетках [22].
В последние годы появилась определенная надежда на новые терапевтические стратегии, направленные на эпигенетические изменения в геноме клеток человека. Так, известно, что повышенные уровни SNCA, связанные с трипликацией гена, вовлечены в патогенез БП. В то же время для поддержания функции нейронов необходим нормальный физиологический уровень SNCA. Поскольку метилирование ДНК в 1-ом интроне гена SNCA регулирует транскрипцию этого гена, для точной регуляции уровней его экспрессии была использована система, которая основана на CRISPR-деактивированном Cas9 (dCas9) слитым с каталитическим доменом ДНК-метилтрансферазы 3A (DNMT3A) [23]. С помощью такого подхода в нейронах, с трипликацией SNCA, полученных из ИПСК пациента с БП, было осуществлено направленное метилирование ДНК в 1-ом интроне гена SNCA. Это вызывало снижение транскрипции мРНК и уровня синтеза белка α-синуклеина в нейронах. В итоге происходило ревертирование патологических свойств фенотипа ДА-нейронов к норме. Полученные результаты позволяют предположить, что технология CRISPR-dCas9-DNMT3A имеет определенный потенциал для лечения БП в качестве нового терапевтического подхода на основе эпигенетики. Модификации гистонов, такие как H3K4me3 и H3K27ac, способны увеличивать транскрипцию гена SNCA, внося таким образом вклад в патогенез БП. Guhathakurta c сотр. [24], используя уникальную систему, основанную на технологии CRISPR/dCas9 (CRISPR/dCas9 SunTag-JARID1A), существенным образом снизили уровень H3K4me3 в районе промотора гена SNCA, что привело к снижению уровня экспрессии этого гена как в линии клеток SH-SY5Y, так и в ДА-нейронах, дифференцированных из ИПСК, полученных от пациентов с БП.
Следует отметить, что при исследовании патогенеза БП с использованием ИПСК человека и технологии CRISPR/Cas9 особое внимание надо обратить на создании моделей таких как 3D культуры (органоиды), а также моделей, включающих совместное культивирование нейронов, клеток глиального ряда и микроглии [25].
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА
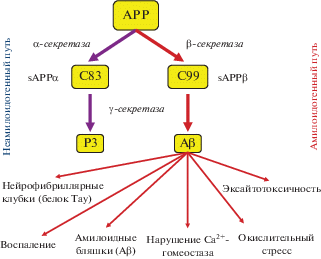
Болезнь Альцгеймера (БА) – прогрессирующее нейродегенеративное заболевание, обычно поражающее взрослых людей, которое является наиболее распространенной причиной деменции и в конечном итоге приводящая к смерти. В настоящее время около 40 млн людей в мире подвержены этому заболеванию [26]. БА характеризуется обширной потерей холинергических нейронов и накоплением в головном мозге внутриклеточных нейрофибриллярных клубков, состоящих из гиперфосфорилированного Тау-белка, и внеклеточных амилоидных бляшек, содержащих бета-амилоидный пептид (Aβ). Хотя и Аβ, и Тау могут проявлять токсичность независимо друг от друга, между ними существует тесная взаимосвязь: накопление Аβ может индуцировать гиперфосфорилирование Тау. На протяжении длительного времени проводятся многочисленные исследования этой патологии, однако патофизиология и молекулярные механизмы развития БА до сих пор остаются предметом споров. Существует множество предположений относительно основной причины БА. В 1970-х годах, когда был продемонстрирован холинергический дефицит в мозге пациентов с БА, опосредованный дефицитом фермента холинацетилтрансферазы, возникла “холинергическая гипотеза” [27]. Продолжает существовать так называемая “пресенилиновая гипотеза”, согласно которой потеря нормальной функции мутантным геном PSEN1 (входит в комплекс фермента γ-секретазы) вызывает БА через доминирующее негативное влияние на аллель дикого типа [28]. Структурные и функциональные различия митохондрий у больных БА привели к “гипотезе митохондриального каскада” БА [29]. Однако преобладающая на сегодняшний день точка зрения о причине этого заболевания заключается в накоплении Aβ и называется “гипотеза амилоидного каскада” [30, 31]. Схема функционирования этого каскада изображена на рис. 3.
Рис. 3.
Схема функционирования амилоидного каскада. Предшественник бета-амилоида (АРР) разрезается ферментами α-секретазой (неамилоидогенный путь) или β-секретазой (амилоидогенный путь) с образованием секретируемых форм пептида С83 (sAPPα) и С99 (sAPPβ) соответственно. Затем трансмембранный фрагмент С99 разрезается γ-секретазой (включает субъединицу PSEN) с образованием β-амилоидных пептидов (Аβ), состоящих из 39–43 аминокислот. Секреция длинных Аβ (особенно Aβ42) ассоциирована с различными нарушениями в нервной системе, приводящими к БА.
К настоящему времени у пациентов с семейной формой БА описано более 300 вариантов мутаций в генах, кодирующих белок-предшественник аполипопротеина (APP), пресенилин 1 (PSEN1) и пресенилин 2 (PSEN2) (https://www.alzforum.org/mutations). Кроме этого, в спонтанных формах БА идентифицировано более 30 генетических локусов, мутации в которых ассоциированы с БА через пути, участвующие в продукции бета-амилоида (Аβ), и его клиренса [32]. При этом многочисленные дополнительные гены риска в спонтанных формах экспрессируются преимущественно в ненейрональных клетках головного мозга [4]. Эти новые данные начинают смещать фокус исследований БА в сторону лучшего понимания роли и функций ненейрональных клеток во время нейродегенерации.
Чтобы разобраться с причинно-следственной взаимосвязью между мутациями и заболеванием, было использовано редактирование генов с помощью системы CRISPR/Cas9. Такой подход дает теперь возможность устанавливать, какие из ранее известных и вновь идентифицированных мутаций действительно непосредственно ответственны за фенотипы БА [33].
Давно известны доминантные мутации либо в субстрате (APP), либо в протеазе (PSEN), которые ассоциированы с раннем началом БА [34, 35]. Большинство из них усиливают синтез β-амилоидного пептида, состоящего из 42 аминокислот (Aβ42), основного компонента сенильных бляшек. Дисбаланс между продукцией и клиренсом патогенного Аβ42 является очень ранним, часто инициирующим фактором БА [36].
Для изучения роли этих мутаций в развитии БА было применено геномное редактирование. Например, с помощью системы CRISPR/Cas9 осуществляли коррекцию доминантной мутации N141I в гене PSEN2, используя для этого в качестве модели базальные холинергические нейроны человека, полученные из ИПСК и несущие эту точечную мутацию [37]. Коррекция этой мутации в гене PSEN2 приводила к устранению электрофизиологического дефицита в клетках и уменьшала в них соотношение Aβ42/40. Полученные данные подтверждают существование тесной взаимосвязи между мутацией N141I в гене PSEN2 и увеличением соотношении Aβ42/40, а также нарушением электрофизиологии нейронов у пациентов с БА.
Система CRISPR/Cas9 была также использована в фибробластах пациента с семейной формой БА, содержащих мутацию APPswe [38]. Селективное нарушение экспрессии мутантного аллеля вызывало снижение в клетках секретируемого Аβ на 60%. Авторы предполагают, что использованный подход в перспективе может быть применен в терапевтических целях.
Для подавления патогенного амилоидогенного пути в нейронах, полученных из ИПСК от пациента с семейной формой БА, несущего мутацию V717I в APP, с помощью системы CRISPR/Cas9 редактировали ген АРР на крайнем С-конце [39]. Были подобраны условия, при которых в клетках ослаблялись как расщепление АРР до APP-β, так и продукция Aβ (амилоидогенный путь). Одновременно повышалась нейропротекторное расщепление АРР с образованием APP-α (неамилоидогенный путь). При этом N-концевые и компенсаторные гомологи APP оставались нетронутыми, без видимого влияния на нейрофизиологию in vitro.
Недавно описан интересный вариант терапии in vitro с помощью модифицированной системы CRISPR/Cas9. Ранее в скандинавской популяции была обнаружена исландская мутация APP (A673T). У людей, несущих эту мутацию, почти не наблюдается накопления пептидов Аβ в мозге до глубокой старости. Основываясь на этих данных, мутацию A673T ввели в ген APP клеток HEK293T, рядом с уже содержавшейся в этом гене патогенной мутации E674K [40]. В результате этой процедуры в клетках происходило снижение синтеза патогенных пептидов Аβ. Таким образом, можно через введение дополнительной мутации останавливать патологические процессы, ведущие к БА.
В случаях, когда обнаруживаемая у пациентов с БА мутация является новой и нет возможности в получении семейного анамнеза и клинических образцов для исследований сегрегации мутаций, сложно приписать клинические проявления БА наличию этой мутации. Недавно описан один из подходов для решения этой задачи применительно к генам PSEN1 и PSEN2. С помощью CRISPR/Cas9 был осуществлен двойной нокаут генов PSEN1 и PSEN2 в клетках нейробластомы мыши N2A, что приводило к полному исчезновению в них Аβ40 и Аβ42 [41]. Трансфекция в эти клетки экзогенного дикого типа гена PSEN1 или PSEN2, обеспечивала полное восстановление продукции Aβ40 и Aβ42 до уровней дикого типа. На этой модели было также протестировано несколько известных и новых мутаций в гене PSEN1 с неопределенной патогенностью и влиянием на функцию γ-секретазы. Все они показали увеличение соотношения Aß42/Aß40, а также влияние на стабильность γ-секретазы.
Поскольку β-секретаза необходима для производства пептидов Aβ, нацеливание на кодирующий ее ген BACE1 является потенциальной стратегией лечения БА. Благодаря использованию нанокомплекса, состоящего из гидРНК, Cas9 и амфифильного пептида R7L10, Park с соавт. [42] удалось подавить экспрессию этого гена, что привело к ингибированию Aβ-ассоциированной патологии в моделях БА, созданных на мышах.
Другой потенциальной мишенью для генной терапии при БА является внутримембранный комплекс, содержащий γ-секретазу и белок, активирующий γ-секретазу (GSAP). Нокаут гена GSAP c помощью системы CRISPR/Cas9 в клетках HEK293, которые экспрессируют АPP, снижал активность γ-секретазы и, следовательно, генерацию Аβ. При этом прекращение экспрессии гена GSAP приводило к изменению конформации белка PSEN1 и снижению секреции Aβ. Полученные результаты открывают еще один путь к изучению модуляции γ-секретазы и, в конечном итоге, к совершенствованию терапевтических средств при БА [43].
Одним из подходов к изучению причин БА является раскрытие роли патологического белка Тау в прогрессировании заболевания. Этот белок кодируется геном MAPT, им обогащены аксоны развивающихся и зрелых нейронов. На важность гиперфосфорилирования Тау в развитии БА указывают клинические исследованиями, установившие тесную корреляцию между развитием БА с Тау-положительными нейрофибриллярными клубками (neurofibrillary tangles) (NFTs) [44]. Недавно с целью установления генетических модуляторов множественных морфологических и функциональных клеточных фенотипов, связанных с БА, на клеточной модели Тау-патологии был проведен широкомасштабный скрининг с помощью автоматизированной системы CRISPR/Cas9 в сочетании с мультиплексными гидРНК [45]. В результате было получено несколько интересных результатов. В частности, была обнаружена сильная связь между лизосомами и агрегацией Тау. Установлено также, что агрегацию Тау модулирует сеть генов, включая регуляторный путь NF-kB и комплекс LKB1, которые участвует в регуляции иммунного ответа.
Считается, что основными факторами риска позднего начала БА является наличие аллели E4 (R112/R158) в гене аполипопротеина (APOЕ4) [46]. При этом аллель E3 (C112/R158) гена APOЕ не оказывала негативных эффектов. Нейроны человека, полученные из ИПСК, экспрессирующих белок APOE4, характеризуются более высоким уровнем фосфорилирования белка Тау и повышенной дегенерацией ГАМК-ергических нейронов. APOЕ4 связывается с Аβ и вместе c ним образует сенильные бляшки. С помощью двух подходов осуществляли замещение в геноме ИПСК человека аллеля APOЕ4 на аллель APOE3. Комоr и соавт. [47] использовали для этой цели редактирование пар оснований с помощью системы, состоящей из CRISPR/Cas9 и фермента цитидиндезаминазы, способного осуществлять прямое превращение цитидина в уридин, тем самым осуществляя замену C → T (или G → A). Ванг с соавт. [48] проводили редактирования путем замены в аллеле APOЕ4 фрагмента ДНК, кодирующего аминокислотные остатки 112-158, на подобный фрагмент из аллеля APOЕ3. Применение обоих подходов приводило к реверсии фенотипов нейронов к норме, что указывает на специфические патогенные эффекты APOЕ4. Полученные результаты позволили авторам заключить, что коррекция патогенной конформации APOЕ4 может стать терапевтическим подходом для лечения APOЕ4-связанной БА.
Lin с соавт. [49] использовали CRISPR/Cas9 для получения из клеточной линии с APOЕ3 изогенных гомозиготных ИПСК с аллелем APOЕ4. Из этих клеток были сформированы нейроны, астрациты и микроглия-подобные клетки. Переключение с APOE3 на APOE4 резко изменило транскриптомы как нейронов, так и глии. В нейронах, содержащих аллель APOE4, происходило нарушение метаболических путей, связанных с формированием синапсов. Кокультивирование модифицированных микроглия-подобных клеток с органоидами, полученными из нервных клеток, несущих дупликацию гена APP, показало, что APOE4 отрицательно влияет на некоторые аспекты функции микроглии, которые потенциально препятствуют способности микроглии очищать внеклеточный Аβ из мозга БА.
Геномное редактирование было использовано для изучения еще одного фактора риска спорадической БА – белка FERMT2. Мутагенез в гене, кодирующем этот белок, проведенный с помощью системы CRISPR/Cas9 в нейронах и астроцитах человека, показал, что уменьшение экспрессии этого гена в изученных клетках снижает секрецию Аβ и фосфорилирование Тау, т.е. способствует реверсии к нормальному фенотипу [50].
Известно, что дефекты эндосомальной сети являются еще одним из ранних событий в патогенезе БА [51]. Среди генов, которые могут влиять на развитие этой патологии, является ген SORL1 [52]. Недавно Кнупп с соавт. [53], используя систему СRISPR/Cas9 показали, что при удалении гена SORL1 в нейронах, полученных из ИПСК, происходит изменение трафика и процессинга APP в эндосомальной сети. Эти данные свидетельствуют о том, что цитопатологию при БА обеспечивают не только расщепление АРР, но и транспорт этого белка.
В связи с важной ролью хронического нейровоспаления в патогенезе БА внимание было обращено также на провоспалительный фактор созревания глии (GMF), содержание которого значительно повышается в различных частях мозга при БА. Редактирование гена GMF в иммортализованных микроглиальных клетках с помощью CRISPR/Cas9 вызывало ингибирование воспалительного сигнального пути, что связано с подавлением фосфорилирования р38 MAPK, которая обычно повышается у пациентов с БА [54]. Авторы полагают, что такое редактирование может стать новой терапевтической стратегией при БА.
С генетикой и нейропатологией позднего начала БА тесно связана микроглия, которая в качестве первичной иммунной клетки мозга выполняет ключевые функции макрофагов, такие как фагоцитоз остатков мертвых клеток и белковых агрегатов, сигнализация цитокинов/хемокинов, а также иммунный надзор и ответ [55]. Она обеспечивает трофическую поддержку нейронов, стимулирование дифференцировки олигодендроцитов и модуляции синаптической активности и пластичности. Среди специфичных локусов риска БА в микроглии обнаружены варианты триггерного гена рецептора, экспрессируемого на миелоидных клетках 2 (TREM2), которые вызывают такое же увеличение риска, как и аллель APOE ε4. Мутации, обнаруженные в этом гене, обычно приводят к частичной или полной потере его функции. Для выяснения механизмов действия таких мутаций были использованы изогенные линии клеток глии, полученные их ИПСК, в которых с помощью системы CRISPR/Cas9 ген TREM2 был полностью инактивирован [56]. У генетически модифицированных клеток снижалась выживаемость, ухудшался фагоцитоз ключевых субстратов, включая APOE, подавлялся SDF-1α/CXCR4-опосредованный хемотаксис и сильно повышалась чувствительность к вызываемой стрессом гибели клеток. Все это в модельной системе БА (трансгенных мыши) приводило к нарушению кластеризации клеток глии вокруг амилоидных бляшек и ухудшало их миграцию к культурам, продуцирующим βА. Полученные результаты раскрывают новые аспекты биологии ТРЕМ2 человека и важной роли глии в развитии и прогрессировании БА. В настоящее время преобладающая терапевтическая стратегия в лечении БА основана на восстановлении холинергической функции за счет использования соединений, которые блокируют ферменты, расщепляющие ацетилхолин [57]. Однако такой подход пока мало эффективен.
Приведенные выше примеры позволяют заключить, что совместное использование системы CRISPR/Cas9 и технологии ИПСК существенно продвинуло исследователей к пониманию механизмов возникновения и развития БА и указало на новые пути поиска подходов для лечения этого заболевания. Результаты, полученные в последние годы с помощью редактирования генов, связанных с амилоидогенным путем, позволяют ожидать серьезных изменений в стратегиях лечения БА.
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Боковой амиотрофический склероз (БАС) является смертельно опасным нейродегенеративным заболеванием человека. БАС ассоциируется с прогрессирующей потерей двигательных нейронов головного и спинного мозга, приводящей к мышечной слабости, параличу и в конечном итоге к смерти. Средняя выживаемость пациентов с БАС составляет 2–5 лет после его диагностирования.
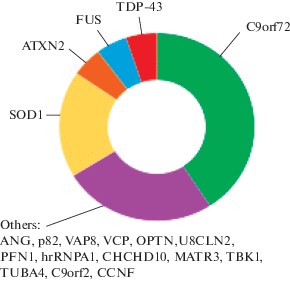
Исследователи пока не пришли к согласию относительно патофизиологии этого заболевания. Одни из них склонны считать правильной гипотезу, которая подразумевает, что истоки дегенерации нейронов лежат в мышечных клетках или нейромышечных синапсах. При этом дегенерация двигательных нейронов начинается с нервных окончаний и прогрессирует к клеточным телам в спинном мозге. Другая гипотеза предполагает, что первичное повреждение происходит в кортикальных моторных нейронах и впоследствии распространяется антероградным образом на кортикоспинальные проекции [58]. Это заболевание связано с различными генетическими изменениями. В 5–10% случаев БАС наследуются по законам Менделя. К настоящему времени открыто около трех десятков генов и локусов, существенно влияющих на вероятность возникновения данного заболевания (рис. 4). Большая часть из них связана с миссенс мутациями в таких генах как FUS, SOD1 и TARDBP или экспансией повторов в генах C9orf72 и ATXN2. Эти мутации составляют до 60% семейных и 10% спорадических форм БАС; причина остальных случаев остается неясной.
Тонкие молекулярно-генетические механизмы, приводящие к БАС, долгое время оставались малоизученными пока на помощь не пришла система редактирования генов CRISPR/Cas9 [60]. Далее будут приведены примеры использования этой системы для изучения роли различных мутаций в патогенезе БАС.
Одним из признаков БАС, характерным для более 97% пациентов, является наличие убиквитинированных цитоплазматических включений, состоящих в основном из Tar-DNА-связывающего белка 43 кДа (TDP-43) [61]. Недавно установлено участие TDP-43 в ответе на повреждение ДНК как ключевого компонента негомологичного соединения концов (NHEJ) при репарации двухцепочечных разрывов ДНК (DSBs) в постмитотических нейронах. С помощью кондиционного нокаута гена TARDBP, кодирующего TDP-43, по индуцибельной технологии CRISPR/Cas9 в нейрональной линии клеток человека было показано, что истощение TDP-43 коррелирует с накоплением DSB, устойчивой активацией ответа на повреждение ДНК и последующей апоптотической гибелью клеток [62]. Таким образом, TDP-43 играет важную роль в поддержании геномной целостности нейронов.
Однако это, по-видимому, не единственная роль TDP-43. Эксперименты на нейронах, полученных из ИПСК пациентов с мутацией M337V в TDP-43, показали, что в этих клетках нарушен сплайсинг мРНК сортилина, что отражается в снижении уровня секреции BDNF, который необходим для выживания нейронов и синаптической пластичности [63]. В результате коррекции гена с помощью технологии CRISPR/Cas9 мутации M337V в TDP-43 в трех линиях iPSCs и дифференцировки их в нейроны установили, что наблюдавшиеся нарушения в нейронах исчезали. Авторы предположили, что основные изменения в нервных клетках, вызванные аберрантной активностью TDP-43, могут быть объяснены аномальной функцией нескольких критических белков, включая BDNF.
Еще одной из частых причин семейных форм БАС является экспансия гексануклеотидных повторов (GGGGCC) в первом интроне гена C9orf72. Экспансии этих повторов приводит к образованию дипептидных повторов (ДПП) в белке, что способствует его агрегации и может приводить к нейродегенерации [64]. Для установления роли этих повторов в развитии БАС в нейронах, полученных из ИПСК, проводили удаление области экспансии повторов в гене C9orf7 с помощью технологии CRISPR/Cas9, с использованием двух гидРНК [65]. Делеция экспансии повтора приводила к уменьшению образования в ядре фокусов транскрибированной с повторов РНК и уменьшала метилирование островков CpG вблизи повторов G4C2, что характерно для клеток с мутантным геном C9orf7. При этом изменялась экспрессия генов, связанных с процессингом РНК, ядерным транспортом, синаптической передачей и регуляцией фосфатного обмена.
Сходные результаты были получены еще в одном исследовании с использованием ИПСК, несущих от 638 до 960 повторов в гене C9orf72 [66]. Удаление области экспансии ДПП с использованием CRISPR/Cas9 приводило к спасению АМРА-индуцированной эксайтотоксичности и изменению экспрессии тех же генов, что и в описанном выше исследовании Прибади с соавт. [65]. На модели клеток линии K562 лейкемии человека с помощью системы CRISPR/Cas9 был проведен скрининг с целью выявления супрессоров и усилителей токсичности синтетических ДПП [67]. Такой подход позволил обнаружить мощные модификаторы токсичности этих ДПП, генные продукты которых функционируют в нуклеоцитоплазматическом транспорте, эндоплазматическом ретикулуме (ЭР), протеасомах, процессинге РНК и модификации хроматина. Один из таких модификаторов, TMX2, модулировал генную сигнатуру стресса ЭР, вызываемого дипептидами C9orfF72 в нейронах, и улучшал выживаемость индуцированных моторных нейронов у пациентов с БАС, содержащих мутантный C9orf72.
Давно существуют свидетельства того, что глиальные клетки могут влиять на гибель моторных нейронов при БАС [68, 69]. В частности, установлено, что мутация в гене SOD1 индуцирует внеклеточную автономную гибель моторных нейронов через токсическое действие ближайшего клеточного окружения [68]. Для изучения последствий экспрессии мутанта C9orf72 в астроцитах, которые являются важными автономными участниками патогенеза БАС, были использованы система CRISPR/Cas9 и ИПСК человека [70]. Астроциты, полученные из ИПСК пациентов с БАС, содержали внутриядерные фокусы РНК, характерные для БАС. После коррекции экспансии повторов G4C2 в мутантном гене C9orf72 эти фокусы РНК в астроцитах исчезали. Электрофизиологический анализ показал, что ко-культивирование астроцитов, несущих мутацию, с нормальными мотонейронами приводит к потере потенциала действия в мотонейронах. В тоже время после удаления экспансии повторов в астроцитах их совместное культивирование с мотонейронах не изменяло свойств последних. Полученные данные свидетельствуют о том, что мутация гена C9orf72 в астроцитах ответственна как за клеточно-автономную астроцитарную патологию, так и за патофизиологию неклеточных автономных двигательных нейронов. Первый ген, мутации в котором приводят к БАС, был идентифицирован как ген супероксиддисмутазы Cu/Zn 1 (SOD1) [71]. В этом гене было описано около 30 мутаций, ассоциированных с БАС. Эти мутации оказывали влияние на нейрональную возбудимость, стресс ЭР, митохондриальную дисфункцию, окислительный стресс, нарушение транспорта белков и накопление неправильно свернутого белка SOD1 [72].
Для исследования роли мутаций в гене SOD1 в качестве модели были использованы ИПСК, полученные от пациентов с БАС. Целенаправленная генная коррекция мутации A272C в SOD1 в ИПСК и последующий анализ дифференциированных из них моторных нейронов, показали значительные изменения в экспрессии генов, участвующих в активности нервной системы, передаче сигналов и гомеостазе ЭР [73].
Подобного рода исследование было проведено на ИПСК, полученных от пациента с БАС, которые содержали другую мутацию гена SOD1 (E100G) [74]. Коррекция этой мутации в гене SOD1 в изогенной клеточной линии с помощью системы CRISPR/Cas9 увеличивала долю моторных нейронов, размер сомы и длину нейритов и уменьшала гибель клеток. Транскрипционные изменения, обнаруженные при секвенировании РНК в генно-модифицированных нейронах, идентифицировали изменения в нескольких метаболических путях, включая те, которые связаны с AP1, окислительным фосфорилированием и функционированием ионных каналов. Ингибирование в модифицированных клетках ERK, MAPK, JNK, WNT, TP53 и CDK-киназ, уменьшало дегенерацию моторных нейронов. Полученные данные позволили авторам предположить, что эти метаболические пути имеют решающее значение при SOD1-ассоциированном БАС.
Несколько иную стратегию применили Imamura с соавт. [75]. В ИПСК, полученных от пациента с БАС, несущего мутацию L144FVX в гене SOD1, эту мутацию исправляли путем вставки с помощью CRISPR/Cas9 последовательностей гена SOD1 дикого типа. В скорректированной клеточной линии происходила реверсия патологического фенотипа, связанного с SOD1, которая включает увеличение выживаемости моторных нейронов и деградацию неправильно свернутого белка SOD1.
Исследования механизмов возникновения и развития БАС с помощью редактирования генома проводились и на модели лабораторных животных. Система CRISPR/Cas9 позволяет легко вводить патогенные мутации в различные гены, проверять гипотезы о совместном появлении аллелей в начале и прогрессировании заболевания и исследовать влияние разных модификаторов в присутствии известного причинного аллеля [76]. У трансгенных мышей со сверхэкспрессирующимся мутантным геном SOD1 наблюдали значительную потерю двигательных нейронов, аксональную денервации, прогрессирующий паралич, агрегацию белков и сокращение продолжительности жизни [77]. Удаление гена SOD1 с помощью редактирования генома значительно улучшало продолжительность жизни трансгенных мышей и существенно восстанавливало область мышечной дистрофии [78].
Недавно было проведено CRISPR/Cas9-опосредованное редактирование SOD1 с мутацией G93A, связанных с БАС в двух трансгенных моделях [79]. Эта модификация полностью предотвратила развитие каких-либо признаков БАС. Трансгенные мыши оказались здоровыми без явных признаков других заболеваний, таких как злокачественное перерождение клеток или воспалительные заболевания, даже после наблюдения до 2-летнего возраста. Вместе с тем, авторы обнаружили у трансгенных мышей с редактированным геномом высокую частоту больших делеций ДНК. Эти данные указывают на необходимость оптимизации терапевтического таргетинга для предотвращения или минимизации непреднамеренных и потенциально вредных событий. Тем не менее, редактирование генов в сочетании с использованием ИПСК и трансгенных животных дает все основания утверждать о существовании тесной причинно-следственной связи между различными мутациями в гене SOD1 и развитием БАС.
Еще одним геном, ассоциированным с развитием БАС, является ген FUS. Этот ген кодирует РНК-связывающий белок, локализующийся преимущественно в ядре. Считается, что в норме он участвует в процессах репарации повреждений ДНК, транскрипции, сплайсинга, а также аксонального транспорта. [80]. C мутациями в гене FUS связано большое разнообразие клинических фенотипов, начиная от общих проявлений заболевания и заканчивая наиболее агрессивными, ювенильными формами заболевания. Для выяснения влияния этих мутаций на развитие БАС и механизмов их действия также использовали технологию редактирования с помощью CRISPR/Cas9 применительно к ИПСК пациентов с БАС. Редактирование ДНК в клетках, содержащих миссенс-мутации G1566A в гене FUS, показало [73], что в мотонейронах, полученных из ИПСК, восстанавливались типичная цитоплазматическая локализация FUS, возбудимость и аксональный транспорт. В моторных нейронах с мутацией R521H в гене FUS наблюдали электрофизиологические изменения и дефекты аксонального транспорта. CRISPR/Cas9-опосредованная коррекция гена в ИПСК, полученных от пациентов с этой мутацией, восстанавливала возникшие дефекты в нейронах [81]. Систему CRISPR/Cas9 использовали также для коррекции рецессивной мутации FUS H517Q в ИПСК от пациентов с БАС [73]. При этом в мотонейронах, полученных из ИПСК, активировались р38 и ERK-киназа, что свидетельствует о ключевом пути активации сигнализации MAPK в FUS-опосредованной нейродегенерации при БАС.
Приведенные выше данные указывают на важную роль технологии геномного редактирования ИПСК для установления новых молекулярных механизмов, приводящих к БАС при разных генных мутациях, и поиска на разных моделях подходов к лечению этого заболевания.
БОЛЕЗНЬ ГЕНТИНГТОНА
Болезнь Гентингтона (БГ) – это аутосомно-наследственное неврологическое расстройство, характеризующееся деменцией и моторными нарушениями. Основной причиной этого заболевания считается генетическое нарушение, вызываемое экспансией триплетов CAG (кодируют аминокислоту глутамин) в первом экзоне гена хантингтина (HTT), структура которого хорошо изучена (рис. 5), но его функции остаются неизвестными [82, 83].
Рис. 5.
Структура гена гентингтина (НТТ). Схематически показана локализация CAG-повторов в первом экзоне этого гена.

В гене дикого типа у разных здоровых людей присутствует разное количество CAG-повторов, однако, БГ развивается только когда их число превышает 36. Причем чем большее количество CAG-повторов присутствует в гене, тем тяжелее и стремительнее протекает болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3–7 на 100 000, среди других рас она ниже [84].
Относительной “простотой” данного заболевания при всей тяжести течения является то, что за его развитие отвечают мутации (разное количество CAG-повторов) в одном единственном гене. Поэтому были предприняты попытки коррекции длинных CAG-повторов с помощью CRISPR/Cas9 системы в клетках, полученных от пациентов с диагностированной БГ. В принципе работы по редактированию гена НТТ развивались по двум направлениям. Первое - исправление мутаций в этом гене у пациентов с БГ и, второе, введение мутаций в здоровые клетки.
Впервые коррекция гена НТТ с помощью CRISPR/Cas9 метода была продемонстрирована в работах An с соавт. [85, 86]. Однако им не удалось удалить из полученных линий клеток генетический вектор, содержащий ген селекции, который потенциально мог бы оказывать существенное влияние на регуляцию экспрессии гена НТТ. Позднее это осуществили Xu с сотр. [87] с использованием CRISPR/Cas9 и piggyBac технологий. Полученные из ИПСК линии клеток сохраняли плюрипотентность и нормальный кариотип, а также способность к дифференцировке в возбудимые и синаптически активные нейроны. Кроме того и, что особенно важно, клетки с редактированным геномом имели фенотип присущий нормальным клеткам, тогда как ИПСК, полученные от пациента с БГ, демонстрировали нарушения в образовании розеток, повышенную чувствительность к присутствию нейротрофических факторов и нарушения в митохондриальном дыхании.
Ooi с сотр. [88], используя ЭСК, получили панель изогенных клеточных линий с разным количеством CAG–повторов в гене НТТ. Такой подход позволил авторам оценить тяжесть течения данной патологии в зависимости от длины этих повторов.
Сходная по идеологии работа была также выполнена другой группой исследователей [89]. С использованием технологий CRISPR/Cas9 авторы ввели в первый экзон гена НТТ эмбриональных фибробластов человека 69 CAG-повторов. Причем соответствующий вектор для гомологичной рекомбинации не содержал вектор с геном селекции. Такой подход обеспечивал получение изогенных линий клеток и позволял проводить сравнение эффектов нормального и мутантного генов НТТ на клеточном уровне. В дальнейшем авторы путем репрограммирования получили линии ИПСК, которые были дифференцированы в ГАМК-ергические шипиковые нейроны полосатого тела. “Мутантные” нейроны проявляли существенные отличия от “нормальных” клеток при образовании розеток in vitro и были более чувствительны к удалению из ростовой среды нейротрофических факторов. Кроме того, в “мутантных” нейронах были обнаружены ультраструктурные дефекты, не связанные с агрегатами белка гентингтина, что указывало на появление дефектов на более ранних стадиях развития БГ [89].
Таким образом, использование технологии CRISPR/Cas9 позволяет создавать разнообразные клеточные модели БГ, которые можно использовать как для исследований молекулярных механизмов данной патологии, так и для скрининга потенциальных лекарственных средств. В перспективе возможно использование “исправленных” клеток для клеточной терапии этого тяжелого и неизлечимого в настоящее время заболевания [90–92].
ЗАКЛЮЧЕНИЕ
Фундаментальные исследования по редактированию генома показали целесообразность использования этой мощной технология в качестве интервенционного инструмента для лечения нейродегенеративных заболеваний. Учитывая, что многочисленные усилия по поиску лекарств от нейродегенеративных заболеваний, хотя и показали многообещающие результаты в исследованиях на лабораторных животных, однако они часто терпели неудачу в испытаниях на людях. Кроме того, хотя эти модели обеспечили лучшее понимание механизмов патогенеза нейродегенеративных заболеваний in vivo, они часто ограничены отсутствием специфической дегенерации нейронов и не могут воспроизвести прогрессирование заболевания и особенности двигательных расстройств. Более перспективными для этой цели могут служить крупные животные, особенно похожие на людей нечеловекообразные приматы. Эти животные позволяют воспроизводить основные нейропатологические проявления, присущие человеку, которые могут не наблюдаться при использование в качестве моделей грызунов. Из-за сходства физиологии, анатомии и поведения нечеловекообразных приматов с людьми они полезны для поиска эффективных терапевтических мишеней и терапевтических средств для лечения нейродегенеративных расстройств [93].
Вместе с тем, существует настоятельная необходимость изучения нейродегенеративных процессов в модельных системах на основе клеток человека. Появление технологии ИПСК имеет большие перспективы для преодоления существующих ограничений. Эта технология совместно с технологией CRISPR/Cas9, уже начала давать принципиально новые данные о патологических механизмах, лежащих в основе нейродегенеративных заболеваний, и обещает продвинуть вперед исследования как в области нейродегенерации, так и за ее пределами. На сегодняшний день основные достижения и надежды, возлагаемые на эти технологии, связаны главным образом с углублением нашего понимания ключевых нейропатологических процессов и с поиском генов-мишеней для терапевтического воздействия на них.
Следует, однако, заметить, что современные подходы к редактированию генов имеют существенные ограничения по практическому применению. Во-первых, удаление или полная инактивация отдельных генов, скорее всего, окажет пагубное воздействие на физиологию клеток, поскольку большинство генов играют в организме определенную роль. Во-вторых, стратегии, направленные на коррекцию мутаций, применимы только к небольшой доле (<10%) наследственных нейродегенеративных заболеваний. Серьезной проблемой остается также частая нецелевая модификация генома CRISPR-Cas9. Кроме того, для каждой мутации в генах необходим свой подход к редактированию, что еще больше усложняет его практическое использование. Очевидно, что потребуются еще немалые усилия, чтобы сделать технологию редактирования генома безопасной и эффективной для терапевтического применения. В этом отношении может оказаться полезным программируемое редактирование эпигенома, которое является настраиваемым, обратимым, не требует разрывов ДНК и не вызывает клеточную токсичность, связанную со стандартным редактированием генов [94].
Несмотря на важные преимущества, предоставляемые использованием ИПСК человека для исследований нейродегенеративных заболеваний, следует учитывать некоторые недостатки этой модели. Необходимо помнить о существовании соматического мозаицизма, который возникает как in vivo, так и в моделях на основе ИПСК [95]. Кроме того, следует иметь в виду, что нейроны, полученные из ИПСК, больше напоминают клетки мозга плода [96]. Поэтому они, по-видимому, лучше моделируют различные аспекты предрасположенности к заболеванию, чем само состояние заболевания. Нейрональные предшественники и нейроны, полученные из ИПСК пациента с нейродегенеративными заболеваниями, скорее всего, могут служить моделями самых ранних форм этих нейропатологий.
Большие перспективы в моделировании нейродегенеративных заболеваний связывают сегодня также с использованием органоидов – самоорганизующихся моделей нервных тканей in vitro [97]. В отличие от 2D модели на основе ИПСК 3D модель церебральных органоидов может быть более эффективной для изучения механизмов возникновения различных нейропатологий и для скрининга лекарств. Предполагается, что в дальнейшем эта модель будет усовершенствована в результате увеличения клеточного разнообразия в органоидах за счет включения в нее нескольких типов глиальных клеток и сосудистой сети.
Список литературы
Dugger B.N., Dickson D.W. // Cold Spring Harb. Perspect. Biol. 2017. V. 9. № 7. P. a028035. https://doi.org/10.1101/cshperspect.a028035
Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. // Science. 2013. V. 339. № 6121. P. 819-823. https://doi.org/10.1126/science.1231143
Kampmann M. // Nat. Rev. Neurol. 2020. V. 16. № 9. P. 465–480. https://doi.org/10.1038/s41582-020-0373-z
Penney J., Ralvenius W.T., Tsai L.-H. // Mol. Psychiatry. 2020. V. 25. № 1. P. 148–167. https://doi.org/10.1038/s41380-019-0468-3
de Lau L.M., Breteler M.M. // Lancet Neurol. 2006. V. 5. № 6. P. 525–535. https://doi.org/10.1016/S1474-4422(06)70471-9
Jankovic J. // J. Neurol. Neurosurg. Psychiatry. 2008. V. 79. № 4. P. 368–376.
Elbaz A., Carcaillon L., Kab S., Moisan F. // Rev. Neurol. (Paris). 2016. V. 172. № 1. P. 14–26. https://doi.org/1016/j.neurol.2015.09.012.
Левин О.С., Федорова Н.В. // Болезнь Паркинсона. 7-е изд. М.: МЕДпресс-информ, 2017. 384 с.
Shulman J.M., De Jager P.L., Feany M.B. // Annu. Rev. Pathol. 2011. V. 6. P. 193–222.
Иллариошкин С.Н., Хаспеков Л.Г., Гривенников И.А. // Моделирование болезни Паркинсона с использованием индуцированных плюрипотентных стволовых клеток. Монография. М.: Соверо-пресс, 2016. 183 с.
Blauwendraat C., Nalls M.A., Singleton A.B. // Lancet Neurol. 2020. V. 19. P. 170–178.
Shadrina M.I., Slominsky P.A., Limborska S.A. // International Review of Cell and Molecular Biology. 2010. V. 281. P. 229–266. https://doi.org/10.1016/S1937-6448(10)81006-8
Sanders L.H., Laganiere J., Cooper O., Mak S.K., Vu B.J., Huang Y.A., Paschon D.E., Vangipuram M., Sundararajan R.,Urnov F.D., Langston J.W., Gregory P.D., Zhang H.S., Greenamyre J.T., Isacson O., Schüle B. // Neurobiol. Dis. 2014. V. 62. P. 381–386. https://doi.org/10.1016/j.nbd.2013.10.013
Reinhardt P., Schmid B., Burbulla L.F., Schöndorf D.C., Wagner L., Glatza M., Höing S., Hargus G., Heck S. A., Dhingra A., Wu G., Müller S., Brockmann K., Kluba T., Maisel M., Krüger R., Berg D., Tsytsyura Y., Thiel C.S., Psathaki O.-E., Klingauf J.,Kuhlmann T., Klewin M., Müller H., Gasser T., Schöler H.R., Sterneckert J. // Cell Stem Cell. 2013. V. 12. P. 354–367. https://doi.org/10.1016/j.stem.2013.01.008
Qing X., Walter J., Jarazo J., Arias-Fuenzalida J., Hillje A.-L., Schwamborn J.C. // Stem Cell Res. 2017. V. 24. P. 44–50. https://doi.org/10.1016/j.scr.2017.08.013
Ветчинова А.С., Иллариошкин С.Н., Новосадова Е.В., Абрамычева Н.Ю., Хаспеков Л.Г., Гривенников И.А. // Сибирское медицинское обозрение. 2017. V 4. P. 53–58. https://doi.org/10.20333/2500136-2017-4-53-58
Vermilyea S.C., Babinski A., Tran N., To S., Guthrie S., Kluss J.H., Schmidt J.K., Wiepz G.J., Meyer M.G., Murphy M.E., Cookson M.R., Emborg M.E., Golos T. G. // Sci. Reps. 2020. V. 10. P. 3447–3461. https://doi.org/10.1038/s41598-020-60273-2
Funayama M., Hasegawa K., Kowa H. // Ann. Neurol. 2002. V. 51. P. 296–301.
Potting C., Crochemore C., Moretti F., Nigsch F., Schmidt I., Manneville C., Carbone W., Knehr J., De Jesus R., Lindeman A., Maher R., Russ C., McAllister G., Reece-Hoyes J.S., Hoffman G.R., Roma G., Müller M., Sailer A.W., Helliwell S.B. // Proc. Natl. Acad. Sci. USA. 2018. V. 115. P. E180–E189. https://doi.org/10.1073/pnas.1711023115
Arias-Fuenzalida J., Jarazo J., Qing X., Walter J., Gomez-Giro G., Nickels S.L., Zaehres H., Schöler H.R., Schwamborn J.C. // Stem Cell Reports. 2017. V. 9. P. 1423–1431. https://doi.org/10.1016/j.stemcr.2017.08.026
Chen Y., Dolt K.S., Kriek M., Baker T., Downey P., Drummond N.J., Canham M.A., Natalwala A., Rosser S., Kunath T. // Eur. J. Neurosci. 2019. V. 49. P. 510–524. https://doi.org/10.1111/ejn.14286
Song C., Charli A.,Luo J., Zainab Riaz Z., Jin H., Anantharam V., Kanthasamy A., Kanthasamy A.G. // Toxicol. Sci. 2019. V. 169. № 2. P. 333–352. https://doi.org/10.1093/toxsci/kfz049
Kantor B., Tagliafierro L., Gu J., Zamora M.E., Ilich E., Grenier C., Huang Z.Y., Murphy S., Chiba-Falek O. // Mol. Ther. 2018. V. 26. P. 2638–2649. https://doi.org/10.1016/j.ymthe.2018.08.019
Guhathakurta S., Kim J., Adams L., Basu S., Song M.K., Adler E., Je G., Fiadeiro M.B., Kim Y.S. // EMBO Mol. Med. 2021. V. 13. P. e12188. https://doi.org/10.15252/emmm.202012188
Coccia E., Ahfeldt T. // Stem Cell Res. Ther. 2021. V. 12. P. 253–264. https://doi.org/10.1186/s13287-021-02326-5
Alzheimer’s Association (2019) // Alzheimers Dement. 2019. V. 15. P. 321–387. https://doi.org/10.1016/j.jalz.2019.01.010
Drachman D.A., Leavitt J. // Arch. Neurol. 1974. V. 30. № 2. P. 113–121. https://doi.org/10.1001/archneur.1974.00490320001001
Heilig E.A., Gutti U., Tai T., Shen J., Kelleher R.J. // J. Neurosci. 2013. V. 33. № 28. P. 11606–11617.
Swerdlow R.H. // J. Alzheimers Dis. 2018. 62. № 3. P. 1403–1416.
Татарникова О.Г., Орлов М.А., Бобкова Н.В. // Успехи биологической химии. 2015. Т. 55. С. 351–390.
Soria Lopez J.A., González H.M., Léger G.C. // Handb. Clin. Neurol. 2019. V. 167. P. 231–255. https://doi.org/10.1016/B978-0-12-804766-8.00013-3
Karch C.M., Goate A.M. // Biol. Psychiatry. 2015. V. 77. № 1. P. 43–51.
Barman N.C., Khan N.M., Islam M., Nain Z., Roy R.K., Haque A., Barman S.K. // Neurol. Ther. 2020. V. 9. № 2. P. 419–434. https://doi.org/10.1007/s40120-020-00218-z
Chartier-Harlin M.C., Crawford F., Houlden H., Warren A., Hughes D., Fidani L., Goate A., Rossor M., Roques P., Hardy J. // Nature. 1991. V. 353. № 6347. P. 844–846. https://doi.org/10.1038/353844a0
Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. // Nat. Genet. 1992. V. 1. № 5. P. 345–347. https://doi.org/10.1038/ng0892-345
Selkoe D.J., Hardy J. // EMBO Mol. Med. 2016. V. 8. № 6. P. 595–608. https://doi.org/10.15252/emmm.201606210
Ortiz-Virumbrales M., Moreno C.L., Kruglikov I., Marazuela P., Sproul A., Jacob S., Zimmer M., Paull D., Zhang B., Schadt E.E., Ehrlich M.E., Tanzi R.E., Arancio O., Noggle S., Gandy S. // Acta Neuropathol. Commun. 2017. V. 5. № 1. P. 1–20. https://doi.org/10.1186/s40478-017-0475-z
György B., Lööv C., Zaborowski M.P., Takeda S., Kleinstiver B.P., Commins C., Kastanenka K., Mu D., Volak A., Giedraitis V., Lannfelt L., Maguire C.A., Joung J.K., Hyman B.T., Breakefield X.O., Ingelsson M. // Mol. Ther. Nucleic Acids. 2018. V. 11. P. 429–440. https://doi.org/10.1016/j.omtn.2018.03.007
Sun J., Carlson-Stevermer J., Das U., Shen M., Delenclos M., Snead A.M., Koo S.Y., Wang L., Qiao D., Loi J., Petersen A.J., Stockton M., Bhattacharyya A., Jones M.V., Zhao X., McLean P.J., Sproul A.A., Saha K., Roy S. // Nat. Commun. 2019. V. 10. № 1. P. 53. https://doi.org/10.1038/s41467-018-07971-8
Guyon A., Rousseau J., Bégin F.G., Bertin T., Lamothe G., Tremblay J.P. // Mol. Ther. Nucleic Acids. 2021. V. 24. P. 253–263. https://doi.org/10.1016/j.omtn.2021.02.032
Pimenova A.A., Goate A.M. // Neurobiol. Dis. 2020. V. 138. P. 104785. https://doi.org/10.1016/j.nbd.2020.104785
Park H., Oh J., Shim G., Cho B., Chang Y., Kim S., Baek S., Kim H., Shin J., Choi H. // Nat. Neurosci. 2019. V. 22. № 4. P. 524–528.
Wong E., Liao G.P., Chang J.C., Xu P., Li Y.M., Greengard P. // Proc. Natl. Acad. Sci. USA. 2019. V. 116. № 13. P. 6385–6390. https://doi.org/10.1073/pnas.1820160116
Naseri N.N., Wang H., Guo J., Sharma M., Luo W. // Neurosci. Lett. 2019. V. 705. P. 183–194. https://doi.org/10.1016/j.neulet.2019.04.022
Duan L., Hu M., Tamm J.A., Grinberg Y.Y., Shen F., Chai Y., Xi H., Gibilisco L., Ravikumar B., Gautam V., Karran E., Townsend M., Talanian R.V. // Sci. Rep. 2021. V. 11. № 1. P. 2879. https://doi.org/10.1038/s41598-021-82658-7
Eisenstein M. // Nature. 2011. V. 475. № 7355. P. 20–22. https://doi.org/10.1038/475S20a
Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. // Nature. 2016. V. 533. № 7603. P. 420–424. https://doi.org/10.1038/nature17946
Wang C., Najm R., Xu Q., Jeong D.E., Walker D., Balestra M.E., Yoon S.Y., Yuan H., Li G., Miller Z.A., Miller B.L., Malloy M.J., Huang Y. // Nat. Med. 2018. V. 24. № 5. P. 647–657. https://doi.org/10.1038/s41591-018-0004-z
Lin Y.-T., Seo J., Gao F., Feldman H.M., Wen H.-L., Penney J., Cam H.P., Gjoneska E., Raja W.K., Cheng J., Rueda R., Kritskiy O., Abdurrob F., Peng Z., Milo B., Yu C.J., Elmsaouri S., Dey D., Ko T., Yankner B.A., Tsai L.-H. // Neuron. 2018. V. 98. № 6. P. 1141–1154. https://doi.org/10.1016/j.neuron.2018.05.008е7
Sullivan S.E., Liao M., Smith R.V., White C., Lagomarsino V.N., Xu J. // Hum. Mol. Genet. 2019. V. 28. № 5. P. 718–735.
Van Acker Z.P., Bretou M., Annaert W. // Mol. Neurodegener. 2019. V. 14. № 1. P. 20–39. https://doi.org/10.1186/s13024-019-0323-7
Rogaev E., Meng Y., Lee J.H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C.T., Cheng R., Hasegawa H., Chen F., Shibata N., Lunetta K.L., Pardossi-Piquard R., Bohm C., Wakutani Y., Cupples L.A., Cuenco K.T., Green R.C., Pinessi L., Rainero I., Sorbi S., Bruni A., Duara R., Friedland R.P., Inzelberg R., Hampe W., Bujo H., Song Y.Q., Andersen O.M., Willnow T.E., Graff-Radford N., Petersen R.C., Dickson D., Der S.D., Fraser P.E., Schmitt-Ulms G., Younkin S., Mayeux R., Farrer L.A., St George-Hyslop P. // Nat. Genet. 2007. V. 39. № 2. P. 168–177. https://doi.org/10.1038/ng1943
Knupp A., Mishra S., Martinez R., Braggin J.E., Szabo M., Kinoshita C., Hailey D.W., Small S.A., Jayadev S., Young J.E. // Cell Rep. 2020. V. 31. № 9. P. 107719. https://doi.org/10.1016/j.celrep.2020.107719
Raikwar S.P., Thangavel R., Dubova I., Selvakumar G.P., Ahmed M.E., Kempuraj D., Zaheer S.A., Iyer S.S., Zaheer A. // Mol. Neurobiol. 2019. V. 56. № 1. P. 378–393. https://doi.org/10.1007/s12035-018-1068-y
McQuade A., Blurton-Jones M. // J. Mol. Biol. 2019. V. 431. № 9. P. 1805–1817. https://doi.org/10.1016/j.jmb.2019.01.045
McQuade A., Kang Y.J., Hasselmann J., Jairaman A., Sotelo A., Coburn M., Shabestari S.K., Chadarevian J.P., Fote G., Tu C.H., Danhash E., Silva J., Martinez E., Cotman C., Prieto G.A., Thompson L.M., Steffan J.S., Smith I., Davtyan H., Cahalan M., Cho H., Blurton-Jones M. // Nat. Commun. 2020. V. 11. № 1. P. 5370–5386. https://doi.org/10.1038/s41467-020-19227-5
Hampel H., Mesulam M.M., Cuello A.C., Farlow M.R., Giacobini E., Grossberg G.T., Khachaturian A.S., Vergallo A., Cavedo E., Snyder P.J., Khachaturian Z.S. // Brain. 2018. V. 141. № 7. P. 1917–1933. https://doi.org/10.1093/brain/awy132
Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., Burrel J.R., Zoing, M.C. // The Lancet. 2011. V. 377. № 9769. P. 942–955. https://doi.org/10.1016/s0140-6736(10)61156-7
Nguyen H.P., Broeckhoven C.V., van der Zee J. // Trends Genet. 2018. V. 34. P. 404–423. https://doi.org/10.1016/j.tig.2018.03.001
Yun Y., Ha Y. // Int. J. Mol. Sci. 2020. V. 21. № 11. P. 3801–3815. https://doi.org/10.3390/ijms21113801
Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., McCluskey L.F., Miller B.L., Masliah E., Mackenzie I.R., Feldman H., Feiden W., Kretzschmar H.A., Trojanowski J.Q., Lee V.M.-Y. // Science. 2006. V. 314. № 5796. P. 130–133.
Mitra J., Guerrero E.N., Hegde P.M., Liachko N.F., Wang H., Vasquez V., Gao J., Pandey A., Taylor J.P., Kraemer B.C., Wu P., Boldogh I., Garruto R.M., Mitra S., Rao K.S., Hegde M.L. // Proc. Natl. Acad. Sci. U.S.A. 2019. V. 116. № 10. P. 4696–4705. https://doi.org/10.1073/pnas.1818415116
Tann J.Y., Wong L.W., Sajikumar S., Ibáñez C.F. // EMBO J. 2019. V. 38. № 5. P. e100989. https://doi.org/10.15252/embj.2018100989
Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., Haass C., Edbauer D. // Science. 2013. V. 339. P. 1335–1338.
Pribadi M., Yang Z., Kim T.S., Swartz E.W., Huang A.Y., Chen J.A., Dokuru D., Baek J., Gao F., Fua A.T. // J. bioRxiv. 2016. P. 051193. https://doi.org/10.1101/051193
Selvaraj B.T., Livesey M.R., Zhao C., Gregory J.M., James O.T., Cleary E.M., Chouhan A.K., Gane A.B., Perkins E.M., Dando O., Lillico S.G., Lee Y.B., Nishimura A.L., Poreci U., Thankamony S., Pray M., Vasistha N.A., Magnani D., Borooah S., Burr K., Story D., McCampbell A., Shaw C.E., Kind P.C., Aitman T.J., Whitelaw C.B.A., Wilmut I., Smith C., Miles G.B., Hardingham G.E., Wyllie D.J.A., Chandran S. // Nat. Commun. 2018. V. 9. P. 347–360. https://doi.org/10.1038/s41467-017-02729-0
Kramer N.J., Haney M.S., Morgens D.W., Jovičić A., Couthouis J., Li A., Ousey J., Ma R., Bieri G., Tsui C.K., Shi Y., Hertz N.T., Tessier-Lavigne M., Ichida J.K., Bassik M.C., Gitler A.D. // Nat. Genet. 2018. V. 50. № 4. P. 603–612. https://doi.org/10.1038/s41588-018-0070-7
Boillée S., Velde C.V., Cleveland D.V. // Neuron. 2006. V. 52. № 1. P. 39–59. https://doi.org/10.1016/j.neuron.2006.09.018
Ilieva H., Polymenidou M., Cleveland D.W. // J. Cell Biol. 2009. V. 187. № 6. P. 761–772. https://doi.org/10.1083/jcb.200908164
Zhao C., Devlin A.C., Chouhan A.K., Selvaraj B.T., Stavrou M., Burr K., Brivio V., He X., Mehta A.R., Story D., Shaw C.E., Dando O., Hardingham G.E., Miles G.B., Chandran S. // Glia. 2020. V. 68. № 5. P. 1046–1064. https://doi.org/10.1002/glia.23761
Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O’Regan J.P., Deng H.X. // Nature. 1993. V. 362. № 6415. P. 59–62. https://doi.org/10.1038/362059a0
Kaur S.J., McKeown S.R., Rashid S. // Gene. 2016. V. 577. № 2. P. 109–118. https://doi.org/10.1016/j.gene.2015.11.049
Wang L., Yi F., Fu L., Yang J., Wang S., Wang Z., Suzuki K., Sun L., Xu X., Yu Y.// Protein Cell. 2017. V. 8. P. 365–378. https://doi.org/10.1007/s13238-017-0397-3
Bhinge A., Namboori S.C., Zhang X., VanDongen A.M.J., Stanton L.W. // Stem Cell Rep. 2017. V. 8. P. 856–869. https://doi.org/10.1016/j.stemcr.2017.02.019
Imamura K., Izumi Y., Watanabe A., Tsukita K., Woltjen K., Yamamoto T., Hotta A., Kondo T., Kitaoka S., Ohta A.// Sci. Transl. Med. 2017. V. 9. P. eaaf3962. https://doi.org/10.1126/scitranslmed.aaf3962
Lutz C. // Brain Research. 2018. V. 1693. P. 1–10. https://doi.org/10.1016/j.brainres.2018.03.024
Philips T., Rothstein J.D. // Curr. Protoc. Pharmacol. 2015. V. 69. P. 5.67.1-5.67.21. https://doi.org/10.1002/0471141755.ph0567s69
Duan W., Guo M., Yi L., Liu Y., Li Z., Ma Y., Zhang G., Liu Y., Bu H, Song X., Li C. // Gene Ther. 2020. V. 27. № 3–4. P. 157–169. https://doi.org/10.1038/s41434-019-0116-1
Deng H.X., Zhai H., Shi Y., Liu G., Lowry J., Liu B., Ryan É.B., Yan J., Yang Y., Zhang N., Yang Z., Liu E., Ma Y.C., Siddique T. // Commun. Biol. 2021. V. 4. № 1. P. 396–406. https://doi.org/10.1038/s42003-021-01942-4
Sharma A., Lyashchenko A.K., Lu L., Nasrabady S.E., Elmaleh M., Mendelsohn M., Nemes A., Tapia J.C., Mentis G.Z., Shneider N.A. // Nat. Commun. 2016. V. 7. P. 10465–10478. https://doi.org/10.1038/ncomms10465
Guo W., Naujock M., Fumagalli L., Vandoorne T., Baatsen P., Boon R., Ordovás L., Patel A., Welters M., Vanwelden T., Geens N., Tricot T., Benoy V., Steyaert J., Lefebvre-Omar C., Boesmans W., Jarpe M., Sterneckert J., Wegner F., Petri S., Bohl D., Vanden Berghe P., Robberecht W., Van Damme P., Verfaillie C, Van Den Bosch L. // Nat. Commun. 2017. V. 8. № 1. P. 861–875. https://doi.org/10.1038/s41467-017-00911-y
Walker F.O. // Lancet 2007; 369 (9557), 218–228. https://doi.org/10.1016/S0140-6736(07)60111-1
Bates G.P., Dorsey R., Gusella J.F., Hayden M.R., Kay C., Leavitt B.R., Nance M., Ross C.A., Scahill R.I., Wetzel R., Wild E.J., Tabrizi S.J. // Nat. Rev. Dis. Primers. 2015. V. 1. P. 15005. https://doi.org/10.1038/nrdp.2015.5
Kay C., Collins J.A., Miedzybrodzka Z., Madore S.J., Gordon E.S., Gerry N., Davidson M., Slama R.A., Hayden M.R. // Neurology. 2016. V. 87. № 3. P. 282–288. https://doi.org/10.1212/WNL.0000000000002858
An M.C., Zhang N., Scott G., Montoro D.,Wittkop T., Mooney S., Melov S., Ellerby L.M. // Cell Stem Cell. 2012. V. 11. P. 253–263.
An M.C., O’Brien R.N., Zhang N., Patra B.N., Cruz La, De M., Ray, A., Ellerby L.M. // PLoS Curr. 2014. V. 6. https://doi.org/10.1371/currents.hd.0242d2e7ad72225-efa72f6964589369a
Xu X., Tay Y., Sim B., Yoon Su-In, Huang Y., Ooi J., Utami K.H., Ziaei A., Ng B., Radulescu C., Low D., Ng A.Y.J., Loh M., Venkatesh B., Ginhoux F., Augustine G.J., Pouladi M.A. // Stem Cell Reports. 2017. V. 8. P. 1–15. https://doi.org/10.1016/j.stemcr.2017.01.022
Ooi J., Langley S.R., Xu X., Utami K.H., Sim B., Huang Y., Harmston N.P., Tay Y.L., Ziaei A., Zeng R. // Cell Rep. 2019. V. 26. № 9. P. 2494–2508.
Malankhanova T., Suldina L., Grigor’eva E., Medvedev S., Minina J., Morozova K., Kiseleva E., Zakian S., Malakhova A. // J. Pers. Med. 2020. V. 10. № 4. P. 215. https://doi.org/10.3390/jpm10040215
Shin J.W., Kim K.H., Chao M.J., Atwal R.S., Gillis T., MacDonald M.E., Gusella J.F., Lee J.-M. // Hum. Mol. Genet. 2016. V. 25. № 20. P. 4566–4576. https://doi.org/10.1093/hmg/ddw286
Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. // Mol. Ther. 2017. V. 25. P. 12–23. https://doi.org/10.1016/j.ymthe.2016.11.010
Dabrowska M., Juzwa W., Krzyzosiak W.J., Olejniczak M. // ront. // Neurosci. 2018. V. 12. P.75. https://doi.org/10.3389/fnins.2018.00075
Barazesh M., Mohammadi S., Bahrami Y., Mokarram P., Morowvat M.H., Saidijam M., Karimipoor M., Kavousipour S., Vosoughi A.R., Khanaki K. // Current Gene Therapy. 2021. V. 21. № 2. P. 130–148. https://doi.org/10.2174/1566523220666201214115024
Nuñez J.K., Chen J., Pommier G.C., Cogan J.Z., Replogle J.M., Adriaens C., Ramadoss G.N., Shi Q., Hung K.L., Samelson A.J., Pogson A.N., Kim J.Y.S., Chung A., Leonetti M.D., Chang H.Y., Kampmann M., Bernstein B.E., Hovestadt V., Gilbert L.A., Weissman J.S. // Cell. 2021. V. 184. № 9. P. 2503–2519. https://doi.org/10.1016/j.cell.2021.03.025
McConnell M.J., Lindberg M.R., Brennand K.J., Piper J.C., Voet T., Cowing-Zitron C., Shumilina S., Lasken R.S., Vermeesch J.R., Hall I.M., Gage F.H. // Science. 2013. V. 342. № 6158. P. 632–637. https://doi.org/10.1126/science.1243472
Vera E., Studer L. // Development. 2015. V. 142. № 18. P. 3085–3089. https://doi.org/10.1242/dev.1206670
Wray S. // Semin. Cell. Dev. Biol. 2021. V. 111. P. 60–66. https://doi.org/10.1016/j.semcdb.2020.05.012
Дополнительные материалы отсутствуют.