

Нейрохимия, 2021, T. 38, № 4, стр. 355-363
Доставка CRISPR–Cas9 в виде рибонуклеопротеиновых комплексов повышает эффективность редактирования EGFP
Я. С. Слесаренко 1, А. Г. Быконя 2, А. А. Анучина 1, М. И. Зайнитдинова 1, Н. А. Евтушенко 3, А. В. Лавров 1, С. А. Смирнихина 1
1 Федеральное государственное бюджетное научное учреждение “Медико-генетический научный центр
имени академика Н.П. Бочкова”
Москва, Россия
2 Первый Московский государственный медицинский университет имени И.М. Сеченова
Москва, Россия
3 Центр высокоточного редактирования и генетических технологий для биомедицины
Российского национального исследовательского медицинского университета им. Н.И. Пирогова
Москва, Россия
Поступила в редакцию 20.05.2021
После доработки 05.07.2021
Принята к публикации 15.07.2021
Аннотация
Стремительное развитие технологии редактирования генома с помощью CRISPR–Cas9 привело к ее широкому использованию во многих областях биологических исследований. Однако применению этой технологии в клинической практике препятствует отсутствие эффективного способа доставки компонентов CRISPR–Cas9 в клетки, а также низкая эффективность и возможность неспецифического действия. Использование прямой доставки в виде рибонуклеопротеинового комплекса (RNP) может позволить избежать многих нежелательных эффектов, а также повысить эффективность геномного редактирования. В этой работе мы показали, что эффективность коррекции однонуклеотидной делеции в гене EGFP в плазмидном локусе достоверно увеличивается до 35% при использовании RNP вместо плазмид. Полученные результаты создают основу для разработки методов коррекции мутаций, приводящих к наследственным заболеваниям человека.
ВВЕДЕНИЕ
Одна из новейших технологий редактирования генома, основанная на системе CRISPR–Cas9, уже привела к прогрессу в области биотехнологии и медицины. В классическом варианте системы используется сконструированная единая направляющая РНК (sgRNA), чтобы направлять нуклеазу Cas9 в таргетную область по принципу комплементарности. Затем Cas9 расщепляет ДНК и образует двухцепочечные разрывы (ДЦР), репарация которых приводит к вставкам или делециям (инделам) в целевых геномных локусах [1]. Этот подход – нокаут гена – редко может быть применен для генной терапии, когда важно восстановить нормальную последовательность гена. В этом случае в процессе репарации важно участие гомологичной матрицы ДНК, роль которой может играть как гомологичная хромосома, так и искусственно добавленная молекула.
Как и для классических подходов генной терапии, для геномного редактирования остро стоит вопрос доставки этого генетического инструмента в клетки. Важно, чтобы компоненты CRISPR–Cas9 с высокой эффективностью попали в таргетные клетки, но в то же время не попали в другие (или при попадании не функционировали); чтобы быстро начинали свое действие внутри клетки и не персистировали в ней.
Наиболее частыми формами доставки CRISPR‒Cas9 являются плазмидная ДНК (пДНК), матричная РНК (мРНК) или рибонуклеопротеиновый комплекс (RNP) [2]. Мировые данные [2–5] свидетельствуют о том, что эффективность доставки CRISPR–Cas9 в клетки можно повысить, используя RNP, который состоит из белка Cas9 в комплексе с синтезированной in vitro sgRNA. Прямая доставка RNP комплекса позволяет избежать многих нежелательных эффектов, связанных с пДНК или мРНК доставкой [5–8]. Одно из преимуществ доставки в форме RNP заключается в том, что редактирование гена начинается сразу, без процесса внутриклеточного синтеза компонентов и сборки системы CRISPR, также RNP достаточно быстро деградирует, что способствует снижению потенциального неспецифического действия CRISPR–Cas9 [7]. Все это в свою очередь позволяет повысить эффективность коррекции мутаций при геномном редактировании.
Целью работы явилось сравнение эффективности внесения инделов в ген EGFP и коррекции путем гомологичной репарации мутации с.337delG в этом гене при доставке CRISPR–Cas9 в виде плазмид и RNP с помощью липидных наночастиц.
МЕТОДЫ ИССЛЕДОВАНИЯ
Этическое одобрение. Для данного исследования этическое одобрение не требовалось, так как в рамках него не проводилось каких-либо исследований с использованием животных или с участием людей в качестве объектов.
Клеточные культуры. Эксперименты проводили на двух клеточных культурах: HEK293T (предоставлена к. б. н. М.Ю. Скобловым, лаборатория функциональной геномики ФГБНУ “МГНЦ”, Москва) и HEK293T-GFPmut. Последнюю получали путем лентивирусной трансдукции. Ген EGFP с мутацией с.337delG был амплифицирован с плазмиды pEGFP-mut (см. далее) и клонирован в плазмиду pLVT_turboRFP635. Вирионы были получены в HEK293T (60 мм чашка) из трех плазмид: 2 мкг pR8.91, 0.6 мкг pMD.G и (2 мкг) pLVT_turboRFP635-eGFPmut (все плазмиды были любезно предоставлены Didier Trono, https:// www.epfl.ch/labs/tronolab/). Среду с вирусом пропускали через 0.45 мкм фильтр и использовали ее для заражения клеток HEK293T (5–7 MOI). С помощью клеточной сортировки были отобраны позитивные по красной флуоресценции клетки.
За день до трансфекции клетки высевали в 12-луночный планшет по 200 тысяч клеток на лунку и культивировали в DMEM (ПанЭко, Россия) с добавлением 10% эмбриональной бычьей сыворотки (Thermo Fisher Scientific, USA), 100 мкг/мл пенициллин/стрептомицина (ПанЭко, Россия) и 4 мМ L-глутамина (ПанЭко, Россия). В день трансфекции конфлюентность составляла 60–70%.
Клонирование плазмид. Исходная плазмида для CRISPR–Cas9 еSpCas9(1.1) была любезно предоставлена Feng Zhang (Addgene #71814). Направляющие РНК (sgRNA) были подобраны с помощью свободного программного обеспечения, разработанного Broad Institute (США) (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). sgRNA GFP1 (на последовательность EGFP дикого типа) и GFP2 (на последовательность EGFP с мутацией с.337delG) были клонированы в исходную плазмиду – Cas9(1.1)-GFP1 и Cas9(1.1)-GFP2. Плазмида pEGFP-mut была получена из pEGFP-C1 (Clontech, США) путем внесения мутации с.337delG сайт-направленным мутагенезом.
Сборка RNP комплексов. Исходный белок Cas9 (NEB, USA) готовили в буфере того же производителя, добавляли sgRNA, синтезированную набором Guide-it™ sgRNA In Vitro Transcription Kit (Takara Bio, Japan) по протоколу производителя, в соотношении 1 : 5 (мкг) в среде Gibco Opti-MEM (Thermo Fisher Scientific, США). Для in vitro транскрипции использовали праймеры sgGFP1: 5'‑GTCGCCCTCGAACTTCACCT-3' и sgGFP2: 5'-TGTCGCCCTCGAACTTCACT-3'. Далее создавали липидные комплексы с Lipofectamine CRISPRMAX (Thermo Fisher Scientific, США) по протоколу производителя, после чего производили трансфекцию.
Сборка плазмидных комплексов. Для внесения инделов в плазмидный локус EGFP использовали таргетную плазмиду pEGFP-C1 (Clontech, США) в количестве 0.6 мкг на 200 тыс. клеток в сочетании либо с RNP (Cas9 + sgRNA GFP1), либо с плазмидой Cas9(1.1)-GFP1 в количестве 5.8 мкг. Для восстановления правильной последовательности EGFP использовали таргетную плазмиду pEGFP-mut в количестве 0.6 мкг на 200 тыс. клеток в сочетании либо с RNP (Cas9+sgRNA GFP2), либо с плазмидой Cas9(1.1)-GFP2 в количестве 5,8 мкг. При этом использовали донорную молекулу ssODN-GFP длиной 120 нуклеотидов (5'-TTCTTCAAGGACGACGGCAACTACAAGACCC- GCGCCGAGTGAAGTTCGAGGGCGACACCCT- GGTGAACCGCATCGAGCTGAAGGGCATCG-ACTTCAAGGAGGACGGCAACATCCTGGGG-3') в количестве 100 пмоль на 200 тыс. клеток. Все плазмиды и ssODN трансфицировали в клетки с помощью Lipofectamine 2000 (Thermo Fisher Scientific, США) по протоколу производителя.
Флуоресцентная микроскопия. Через 72 ч после трансфекции оценивали эффективность редактирования с помощью флуоресцентной микроскопии на микроскопе ZEISS Axio Vert.A1 (Carl Zeiss Microscopy, Германия).
Проточная цитофлуориметрия и сортировка клеток. Эффективность трансфекции оценивали в лунках, трансфицированных комбинацией исходной плазмиды еSpCas9(1.1) и pEGFP-C1 в соотношении 10 : 1 (5.8 мкг и 0.6 мкг, соответственно) с помощью проточной цитофлуориметрии на приборе Partec FloMax через 72 ч после трансфекции. Эффективность редактирования оценивали в экспериментальных лунках так же. GFP-положительные клетки были отсортированы на клеточном сортере S3e (Bio-Rad Laboratories, США).
Полимеразная цепная реакция (ПЦР). Амплификацию проводили методом ПЦР на амплификаторе “Терцик” (ДНК-технология, Москва). Последовательность праймеров:
eGFP65_f – 5'-ACGTAAACGGCCACAAGTTCA-3';
eGFP527_r – 5'-CTGCCGTCCTCGATGTTGT-3'.
Условия ПЦР: начальная денатурация – 95°С, 5 мин; 35 циклов амплификации со следующими параметрами: 1) денатурация – 95°С, 30 с; 2) отжиг – 61°С, 20 с; 3) синтез – 72°С, 30 с; 4) инкубация при 72°С в течение 3 мин.
Секвенирование по Сэнгеру. Амликоны секвенированили по Сэнгеру с использованием праймеров для ПЦР в качестве праймеров для секвенирования на приборе ABI Prism 3130XL genetic analyzer (Applied Biosystems, США) по протоколу производителя.
TIDE/TIDER. Полученные хроматограммы после секвенирования были проанализированы с помощью программы Tracking Indels for Decomposition (https://tide.nki.nl). Приложение позволяет оценить уровни гомологичной репарации или негомологичного соединения концов ДНК. Вариант программы TIDER позволяет статистически оценить процент правильно восстановленных последовательностей и весь процент восстановленных последовательностей.
Статистический анализ. Для статистической обработки данных использовали критерий Манна–Уитни и t-тест. Статистическую обработку проводили в программе STATISTICA версия 10.0 (StatSoft, США). Различие считалось значимым, если значение p было меньше 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Внесение инделов в EGFP. На первом этапе вносили инделы в EGFP (схема эксперимента приведена на рис. 1a) с помощью CRISPR–Cas9. Эффективность разрушения (нокаутирования) EGFP оценивали проточной цитофлуориметрией с нормализацией на эффективность трансфекции. Контрольное (исходное) количество флуоресцентных клеток принимали за 100%. При нокаутировании EGFP наблюдали уменьшение доли флуоресцентных клеток (рис. 1в). На этом этапе были апробированы несколько протоколов (разное количество Cas9 в RNP, одновременное введение RNP и pEGFP-C1, а также отсроченное на 6 и 24 ч введение pEGFP-C1 после RNP). Различия во времени внесения плазмид и RNP обусловлены тем, что комплексу CRISPR–Cas9 необходимо время для синтеза и сборки внутри клетки (не менее 6 ч для RNP и 24 ч для редактирующей плазмиды). При одновременной трансфекции компонентов в клетку белок EGFP успевает накопиться до начала работы CRISPR–Cas9. В этом случае не исключены ложноотрицательные результаты редактирования и, как следствие, недостаточное снижение флуоресценции. Если разнести по времени внесение Cas9(1.1)-sgGFP_1 или RNP и pEGFP-C1wt, то комплекс CRISPR–Cas9 успевает собраться и сразу начинает вносить разрезы в рEGFP-С1wt.
Рис. 1.
а – схема эксперимента. Внесение инделов в гене EGFP в результате двухцепочечного разрыва и последующий нокаут гена. б – сравнение эффективности разрушения (нокаутирования) EGFP. Данные представлены как среднее значение ± стандартная ошибка среднего (*p < 0.05 – доставка с помощью RNP по сравнению с плазмидной доставкой). в – микроскопия экспериментальных лунок через 72 ч после трансфекции.
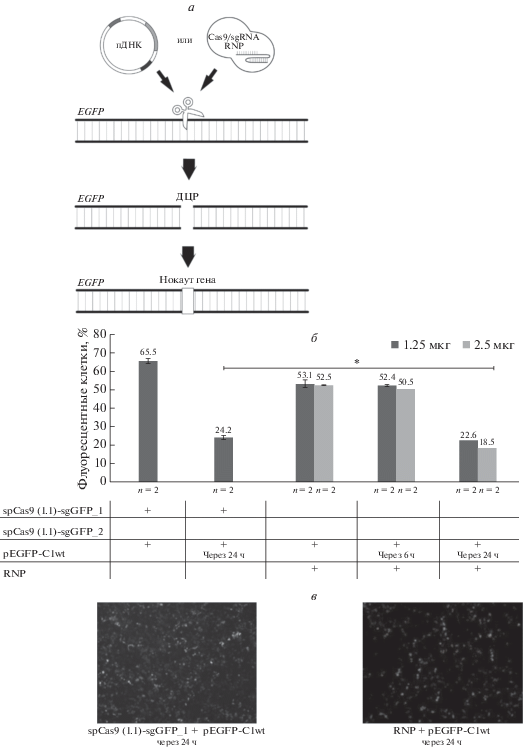
Как видно из гистограммы (рис. 1б), наиболее эффективное внесение инделов было достигнуто при добавлении 2.5 мкг Cas9 и трансфекции плазмиды pEGFP-C1 через 24 ч после RNP (p < 0.05 по сравнению с плазмидной доставкой). Доля флуоресцентных клеток при этих условиях уменьшилась в 5 раз. На следующих этапах работы использовали 2.5 мкг Cas9.
Восстановление последовательности EGFP. Восстановление правильной последовательности EGFP проводили в двух клеточных культурах: в культуре HEK293T, в которую трансфицировали плазмиду EGFP-mut, и в культуре HEK293T-GFPmut, в геном которой была встроена последовательность EGFP с той же мутацией с.337delG. Для восстановления последовательности использовали донорную молекулу ssODN. В случае восстановления правильной последовательности наблюдали флуоресценцию.
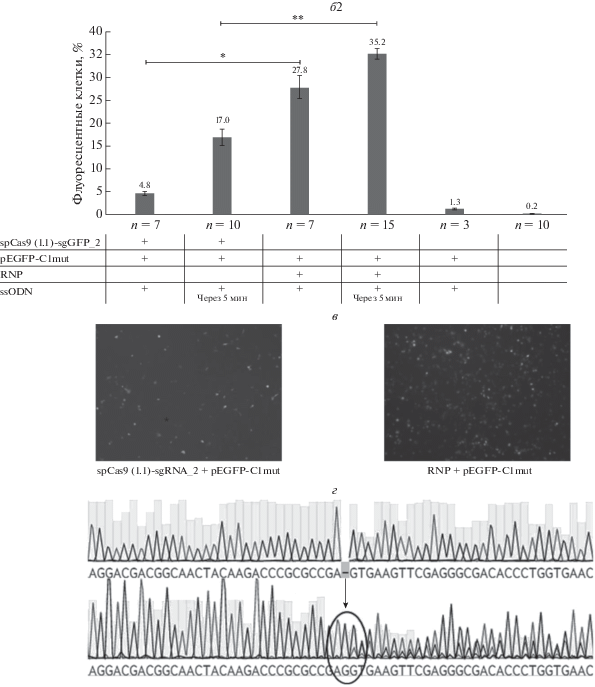
В культуре HEK293T эффективность коррекции мутации составила почти 28% при использовании RNP, по сравнению с плазмидной доставкой (5%, p < 0.05) (рис. 2б. 1). Последующая отработка протокола трансфекции плазмид и RNP комплексов позволила еще больше увеличить эффективность коррекции мутации в EGFP – практически до 17% для плазмид и 35% для RNP (p < 0.05) (рис. 2б. 2).
Рис. 2.
а – схема эксперимента. Восстановление последовательности EGFP с использованием донорной молекулы ssODN. б. 1 и 2 – сравнение эффективности восстановления EGFP. Данные представлены как среднее значение ± стандартная ошибка среднего (*p < 0.05, **p < 0.05 – доставка с помощью RNP по сравнению с плазмидной доставкой). в – микроскопия экспериментальных лунок через 72 ч после трансфекции. г – секвенограмма восстановленного EGFP.
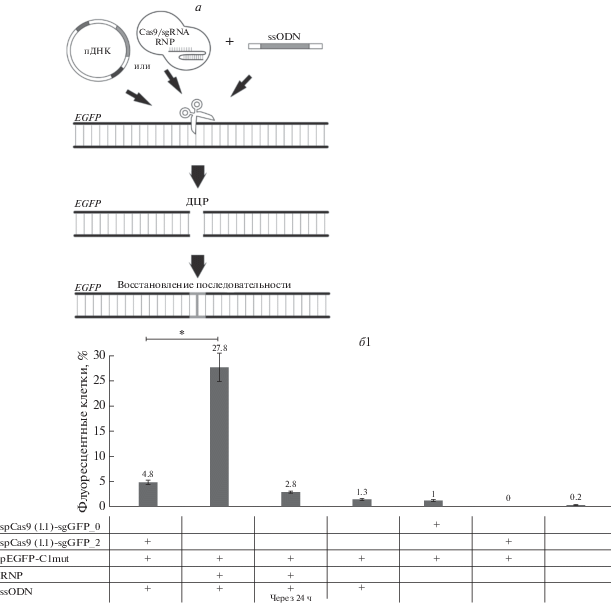
Рис. 2.
Окончание
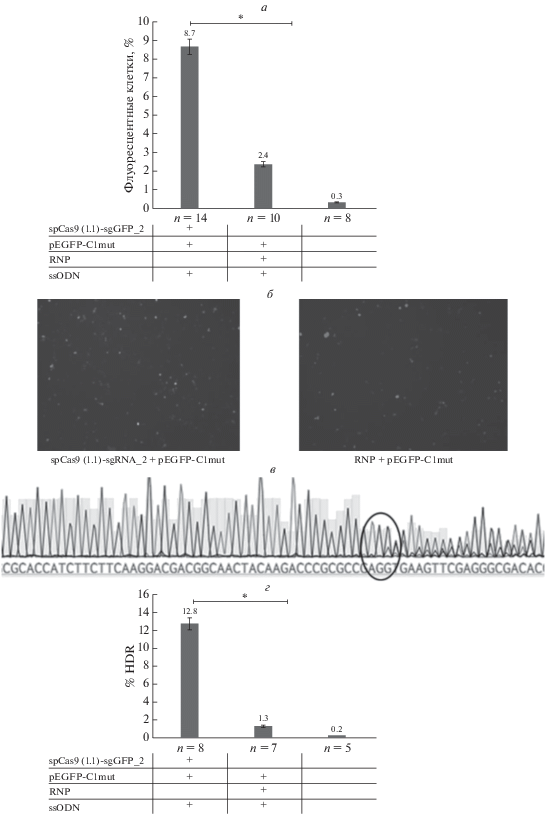
На последнем этапе работы были проведены эксперименты по восстановлению EGFP в клеточной культуре HEK293T-GFPmut. Вопреки ожиданиям, редактирование геномного локуса EGFP было менее эффективным, как с доставкой CRISPR–Cas9 в виде RNP, так и в виде плазмид. При этом доля флуоресцентных клеток при использовании RNP была в 3.6 раза ниже, чем при плазмидной доставке (p < 0.05) (рис. 3).
Рис. 3.
а – сравнение эффективности восстановления EGFP в HEK293T-GFPmut. Данные представлены как среднее значение ± стандартная ошибка среднего (*p < 0.05 – доставка с помощью RNP по сравнению с плазмидной доставкой). б – микроскопия экспериментальных лунок через 72 ч после трансфекции. в – секвенограмма GFP-положительных клеток после сортировки. г – результаты, обработанные с помощью TIDER (*p < 0.05 – доставка с помощью RNP по сравнению с плазмидной доставкой).
ОБСУЖДЕНИЕ
Классический вариант CRISPR–Cas9 с использованием нуклеазы считается довольно эффективным методом, особенно при нокаутировании генов или вырезания фрагментов генов. Однако нуклеаза способна создавать двуцепочечные разрывы не только в таргетном локусе, но и в других, тем самым потенциально внося дополнительные мутации в геном. Часто нецелевые эффекты наблюдаются в исследованиях с использованием плазмидной формы доставки [9, 10]. В свою очередь эти нецелевые эффекты могут приводить к нарушениям функций генов, что усложняет переход технологии редактирования генома в клиническую практику. Разработка безопасных и эффективных способов доставки системы CRISPR–Cas9 к тканям или клеткам-мишеням в организме человека отчасти может решить эту проблему. RNP представляет собой комплекс белка Cas9 с sgRNA, который формируется в пробирке и вносится в клетку уже готовым. Время жизни такого комплекса в клетке меньше, чем плазмидной ДНК, что позволяет снизить частоту неспецифического действия CRISPR–Cas9.
Кроме того, в своих работах исследователи используют не только разные способы доставки, но и разные нуклеазы [11–13]. Тогда можно предположить, что эффективность редактирования и снижение неспецифического действия варьируются именно по этой причине, независимо от способа доставки. Однако, согласно литературным данным, специфическая активность модифицированных форм нуклеаз (eSpCas9 с “повышенной специфичностью” –1.0, 1.1, HF) сохраняется на таком же уровне, что и WT-Cas9 (дикого типа) [13]. Поэтому мы считаем, что полученные достоверные отличия при использовании как RNP, так и плазмид связаны как раз со способом доставки.
Несмотря на все достоинства RNP комплексов, существуют и особые требования в выборе материалов и стратегий для их внутриклеточной доставки. Главным образом это зависит от свойства заряда системы – необходимо учитывать ее положительный заряд, в отличие от мРНК и пДНК. Хотя RNP могут быть доставлены в клетки млекопитающих с относительно высокой эффективностью с помощью электропорации, тем не менее липид-опосредованная трансфекция пользуется большей популярностью благодаря простоте использования, низкой стоимости и адаптации к системе с высокой пропускной способностью.
Геномное редактирование в нейронах и других неделящихся клетках имеет ряд ограничений, связанных, не только с доставкой CRISPR–Cas9, а также с невозможностью рекомбинации эндогенной ДНК с донорной молекулой. Известно, что направленная гомологичная репарация, один из механизмов репарации ДЦР, происходит лишь в фазы клеточного цикла G2/M [14]. Отсутствие митозов в клетках приводит к тому, что все ДЦР репарируются по другому механизму – негомологичному соединению концов, который не позволяет встроить правильную последовательность ДНК в место разрыва. Таким образом, в нейронах можно внести инделы в таргетную область генома, что может быть использовано для нокаутирования генов. Однако коррекцию мутаций и восстановление правильной последовательности гена с помощью классического CRISPR–Cas9 в этом случае сделать не получится. Для этих целей можно использовать появившиеся недавно модификации CRISPR–Cas9 – редакторы оснований [15, 16] и праймированное редактирование [17].
Проблема эффективной доставки CRISPR–Cas9 в нейроны остается такой же актуальной, как и для других типов клеток. Данные опубликованных исследований показывают возможность и успешность доставки CRISPR–Cas9 в виде RNP в клетки мозга in vivo. Для этих целей RNP доставляли с помощью различных наночастиц, например, с помощью наноклубков ДНК [18], нанолистов фосфора [19], карбоксилированного разветвленного поли(β-аминоэфира) [20], а также коммерчески доступного Lipofectamine CRISPRMAX, Thermo Fisher Scientific [21]. Для этого исследования был выбран последний упомянутый способ доставки.
В своей работе мы использовали рибонуклеиновые комплексы и плазмидные векторы для трансфекции белка Cas9 и направляющей молекулы sgRNA. В результате можно увидеть, что эффективность внесения инделов в ген EGFP и коррекции мутации с.337delG в этом гене существенно увеличиваются при доставке CRISPR–Cas9 в виде RNP комплексов. Однако это справедливо только в случае с использованием клеточной культуры HEK293T и плазмиды для редактирования pEGFP-C1mut. Повторение эксперимента, но уже с клеточной линией HEK293T-GFPmut не показало нам значительного повышения эффективности коррекции мутации.
Подобная разница в эффективности на разных линиях создает почву для размышлений. С одной стороны, возможно влияние числа копий гена, которые редактируются: в случае с плазмидами мы имеем большое число копий на клетку и тогда здесь эффективность должна быть ниже, поскольку необходимо исправить как можно больше копий. Но при этом, чем больше копий, тем чаще происходит их “встреча” с компонентами CRISPR–Cas9. В случае исправления геномного локуса ситуация обратная – у нас нет такого большого числа копий – а значит и эффективность должна быть выше. Однако система редактирования может сталкиваться со сложной пространственной организацией геномного локуса, в отличие от более доступного локуса плазмиды. Кроме того, качество получаемых рибонуклеиновых комплексов во много определяет результат редактирования. И тогда менее эффективное редактирование геномного локуса EGFP может быть связано с тем, что в работе использовалась sgRNA, синтезированная в результате in vitro транскрипции. Таким образом необходима дальнейшая проработка этого вопроса и оптимизация протокола трансфекции с учетом всех нюансов.
ЗАКЛЮЧЕНИЕ
Эффективность коррекции однонуклеотидной делеции в гене EGFP в плазмидном локусе достоверно увеличивается до 35% при использовании доставки CRISPR-Cas9 в виде RNP. Полученные результаты создают основу для разработки методов коррекции мутаций, приводящих к наследственным заболеваниям человека.
Список литературы
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. // Science. 2012. V. 337(6096). P. 816–821. https://doi.org/10.1126/science.1225829
Wang M., Glass Z.A., Xu Q. // Gene Ther. 2017. V. 24. P. 144–150.
Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M. // J Biotechnol. 2015. V. 208. P. 44–53.
Yu X., Liang X., Xie H., Kumar S., Ravinder N., Potter J. // Biotechnol Lett. 2016. V. 38. P. 919–929.
Mout R., Ray M., Yesilbag Tonga G., Lee Y.W., Tay T., Sasaki K. // ACS Nano. 2017. V. 11. P. 2452–2458.
Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H. // Nat. Biotechnol. 2015. V. 3. P. 73–80.
Liu C., Wan T., Wang H., Zhang S., Ping Y., Cheng Y. // Sci. Adv. 2019. V. 5.
Guan X., Luo Z., Sun W. // J. Biol. Chem. 2018. V. 293. P. 17306–17307.
Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. // Nat. Biotechnol. 2013. V. 31(9). P. 822–826. https://doi.org/10.1038/nbt.2623
Cradick T.J., Fine E.J., Antico C.J., Bao G. // Nucleic Acids Res. 2013. V. 41(20). P.9584–8592. https://doi.org/10.1093/nar/gkt714
Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. // Nature. 2016. V. 529(7587). P. 490–495. https://doi.org/10.1038/nature16526
Casini A., Olivieri M., Petris G., Montagna C., Reginato G. // Nat Biotechnol. 2018. V. 36(3). P. 265–271. https://doi.org/10.1038/nbt.4066
Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. // Science. 2016. V. 351(6268). P. 84–85. https://doi.org/10.1126/science.aad5227
Smirnikhina S.A., Anuchina A.A., Lavrov A.V. // Hum Genet. 2019. V. 138(1). P. 1–19. https://doi.org/10.1007/s00439-018-1953-5
Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. // Nature. 2016. V. 533. P. 420–424.
Nishida K. // Science. 2016. V. 353.
Anzalone A.V., Randolph P.B., Davis J.R. // Nature. 2019. V. 576(7785). P. 149–157. https://doi.org/10.1038/s41586-019-1711-4
Sun W., Ji W., Hall J.M., Hu Q., Wang C., Beisel C.L., Gu Z. // Angewandte Chemie – International Edition. 2015. V. 54(41). P. 12029–12033. https://doi.org/10.1002/anie.201506030
Zhou W., Cui H., Ying L., Yu X.F. // Angewandte Chemie – International Edition. 2018. V. 57(32). P. 10268–10272. https://doi.org/10.1002/anie.201806941
Rui Y., Wilson D.R., Choi J., Varanasi M., Sanders K., Karlsson J., Lim M., Green J.J. // Science Advances. 2019. V. 5(12). https://doi.org/10.1126/sciadv.aay3255
Noureddine A., Maestas-Olguin A., Saada E.A., LaBauve A.E., Agola J.O., Baty K.E., Howard T., Sabo J.K., Espinoza C.R.S., Doudna J.A. // Acta Biomaterialia. 202. V. 114. P. 358–368. https://doi.org/10.1016/j.actbio.2020.07.027
Дополнительные материалы отсутствуют.