

Нейрохимия, 2022, T. 39, № 3, стр. 243-250
Уровень карбонилирования белков и активность протеаз в мозге новорожденных крыс с пренатальной гипергомоцистеинемией
А. В. Яковлев 1, С. А. Дмитриева 2, А. Н. Краснова 1, О. В. Яковлева 1, Г. Ф. Ситдикова 1
1 Казанский Федеральный университет
Казань, Россия
2 Казанский институт биохимии и биофизики, ФИЦ КазНЦ РАН
Казань, Россия
Поступила в редакцию 10.03.2022
После доработки 19.04.2022
Принята к публикации 27.04.2022
- EDN: GORSAZ
- DOI: 10.31857/S1027813322030141
Аннотация
Гомоцистеин – серосодержащая аминокислота, образующаяся из метионина, является фактором риска развития целого ряда патологий. Повышение уровня гомоцистеина (гипергомоцистеинемия, ГГц) во время беременности приводит к различным осложнениям беременности, гипоксии плода и, как следствие, развитию ранних и отсроченных постнатальных патологий. Одним из основных механизмов действия гомоцистеина является окислительный стресс. Целью нашего исследования было проанализировать окислительную модификацию белков и активность протеаз, а также уровень окислительного стресса в ткани головного мозга крыс с пренатальной ГГц в первую неделю после рождения. Экспериментальная модель ГГц создавалась у самок крыс путем использования корма с повышенным содержанием метионина в течение 3 недель до начала и во время беременности. Было показано, что в гомогенате ткани мозга потомства с пренатальной ГГц наблюдалось усиление спонтанного карбонилирования белков, что свидетельствует о падении резервно-адаптационного потенциала клеток мозга и снижении устойчивости ткани к действию свободных радикалов. В мозге крыс с пренатальной ГГц была увеличена активность кислых и нейтральных протеаз, что, по-видимому, обусловлено агрегацией и фрагментацией белковых молекул вследствие карбонилирования аминокислотных остатков. Ткани мозга новорожденных крыс с пренатальной ГГц характеризовались высоким содержанием H2O2, маркера перекисного окисления липидов – малонового диальдегида, а также снижением активности антиоксидантных ферментов. Таким образом, полученные данные свидетельствуют о значительном усилении необратимого процесса окислительной модификации белков в головном мозге новорожденных крыс с пренатальной ГГц в результате развития окислительного стресса. Эти процессы вносят вклад в механизмы нейротоксичности гомоцистеина в критический период развития мозга, когда в условиях повышенной нейропластичности наблюдается интенсивный нейрогенез, синаптогенез и формирование нервных сетей.
ВВЕДЕНИЕ
Мозг плода чрезвычайно пластичен и уязвим к воздействиям как окружающей, так и внутренней среды, которые могут иметь долгосрочные последствия для здоровья и развития потомства [1]. Известно, что ведущая роль в возникновении и развитии пренатального стресса принадлежит окислительным реакциям, которые приводят к увеличению концентрации активных форм кислорода (АФК) и стимуляции процессов свободно-радикального окисления [2]. Избыточная продукция АФК на ранних сроках беременности вызывает повреждение клеточных мембран плода, что во многом определяет долговременные постнатальные изменения в головном мозге, и является одной из причин развития нейродегенеративных заболеваний [3]. Одним из факторов, вызывающих окислительный стресс в тканях плода является гомоцистеин и его продукты [4–9]. Повышение уровня гомоцистеина в крови матери свыше 12 мкМ/л, называемое гипергомоцистеинемией (ГГц), является результатом генетических дефектов ферментов метаболизма метионина, дефицита фолиевой кислоты и других витаминов группы В (В6, B12), приема противоэпилептических препаратов и других факторов [6, 10]. ГГц вызывает эндотелиальные дисфункции и считается фактором риска развития сердечно-сосудистых заболеваний, а также патологий центральной и периферической нервной системы, таких как болезнь Альцгеймера и Паркинсона, шизофрения, эпилепсия, боковой амиотрофический склероз и мигрень [6, 11, 12].
Гомоцистеин и продукты его метаболизма способны свободно проникать через плацентарный и гематоэнцефалический барьер [13], вызывая нарушения кровообращения плаценты, хроническую фетоплацентарную недостаточность и внутриутробную гипоксию плода [14, 15], а также оказывать нейротоксические эффекты на развивающийся мозг как в эмбриональном, так и в раннем постнатальном периоде развития. Помимо окислительного стресса нейротоксическое действие гомоцистеина обусловлено его способностью активировать ионотропные и метаботропные глутаматные рецепторы [16–20], вызывать нейровоспаление, активацию глиальных клеток, нарушение целостности мембран и, в конечном счете, апоптоз нейронов [21–25]. Эти процессы лежат в основе нарушения формирования нейрональных сетей мозга, поскольку именно в раннем постнатальном онтогенезе происходит миграция и дифференцировка нейронов, созревание синапсов [26]. Одним из последствий ГГц также является снижение активности ферментов, синтезирующих эндогенный газотрансмиттер – сероводород, который проявляет антиоксидантные свойства [8, 9], а также участвует в регуляции плацентарной сосудистой сети [27].
АФК взаимодействуют с функциональными группами аминокислот, вызывая окислительную модификацию белков (ОМБ), включающую процессы карбонилирования аминокислотных остатков, образование дисульфидов, S-нитрозилирование и окисление до сульфопроизводных продуктов и гомоцистеинилирование [28–31]. Сравнительный анализ чувствительности к окислению белков и липидов под действием АФК показал, что белки реагируют раньше на действие радикалов, чем липиды [32]. Модификация белков делает их более чувствительными к протеолизу за счет активации протеаз. Повышение активности нейтральных и кислых протеаз в различных структурах мозга было показано при окислительном стрессе [33], что, по мнению авторов, связано с усилением входа ионов кальция в клетку. С другой стороны, карбонилирование белков может приводить к образованию комплексов, защищённых от действия протеаз и снижению скорости протеолитических реакций [34].
Целью нашей работы было проанализировать окислительную модификацию белков и активность протеаз, а также уровень окислительного стресса в тканях головного мозга крыс в модели пренатальной ГГц в первую неделю после рождения.
МЕТОДЫ ИССЛЕДОВАНИЯ
Объект исследования. Исследование проводили на крысах линии Wistar в течение первой недели после рождения. Эксперименты выполнены с соблюдением принципов Хельсинской декларации о гуманном обращении с животными и одобрены локальным этическим комитетом Казанского федерального университета (протокол № 8 от 05.05.2015). Были приняты меры для использования минимального количества экспериментальных животных. Животные содержались в стандартных условиях вивария и имели постоянный доступ к воде. Крысы получали стандартный комбикорм для лабораторных крыс и мышей “Дельта Фидс”, ДбК 120 С-19 (АО “БиоПро”, Новосибирская обл., Россия). Самки крыс были разделены на две группы. Одна группа находилась весь период на контрольной диете, а другая группа получала корм с повышенным содержанием метионина (7.7 г/кг корма) в течение 3 недель до начала и во время беременности, а также 3 недели после родоразрешения [8, 9, 35].
Определение содержания гомоцистеина. Забор крови у крыс проводили путем пункции сердца или надреза десны животного [36, 37]. Полученные образцы центрифугировалась в течение 15 мин при 1500 g. Гомоцистеин в плазме крови крыс определялся с использованием набора Homocysteine Colorimetric Assay Kit (E-BC-K143, ElabScience, США) спектрофотометрическим методом c использованием ИФА-ридера (Multiskan FS, Thermo Fisher Scientific, США). Концентрация гомоцистеина в плазме у контрольных самок составляла 7.9 ± 0.3 мкМ (n = 12), а у самок, получавших метионин, – 27.3 ± 2.4 мкМ (n = 15, p < 0.05) [8, 9].
Обработка ткани мозга. Для исследований использовали мозг новорожденных крыс в возрасте от 2 до 7 дней после рождения. Ткани мозга после декапитации немедленно замораживались в жидком азоте и хранились при –80°С до начала анализа. Для измерения редокс-метаболизма ткани мозга гомогенизировали в 20 мМ HEPES (pH 7.2) (1 : 9 – масса : объем), центрифугировали при 10 000 g в течение 10 мин при 4°C, и супернатант использовали для анализа. Содержание общего белка оценивали прямым методом по оптической плотности 1 мкл супернатанта при длине волны 280 нм с помощью спектрофотометра NanoDrop 1000 (Thermo Fisher Scientific, США). Полученные значения содержание белка в мг/мл использовали для количественного выражения содержания окисленных белков и активности ферментов.
Анализ спонтанной и металл-зависимой (индуцированной) окислительной модификации белков (ОМБ). Уровень спонтанной ОМБ определяется количеством присутствующих в пробе карбонильных производных белков. Метод основан на реакции взаимодействия карбонильных производных окисленных аминокислотных остатков с 2,4-динитрофенилгидразином (2,4-ДНФГ) с образованием окрашенных 2,4-динитрофенилгидразонов [28]. Реакцию проводили в 100 мкл образца с добавлением 1 мл 0.01 М 2,4-ДНФГ, растворенного в 2 М НCl. Пробы инкубировали в темноте при комнатной температуре в течение 1 ч, затем центрифугировали в течение 15 мин при 10 000 g. Полученный осадок промывали смесью этанол : этилацетат (1 : 1) для удаления липидов и 2,4-ДНФГ, не прореагировавшего с карбонильными группами окисленных белков, и растворяли в 3 мл 8 М раствора мочевины; для лучшего растворения к осадку добавляли 2 М НСl. Оценка металл-зависимой ОМБ проводилась после предварительной инкубации с реактивом Фентона, содержащем 0.4 мМ FeSO4 и 0.1 мМ Н2О2 в течение часа. Оптическую плотность образовавшихся производных 2,4-ДНФГ определяли при длине волны 363 нм (ε = 22 мМ–1 см–1) с помощью спектрофотометра Lambda-25 (Perkin Elmer, США). Содержание окисленных белков выражали в нг/мг белка.
Для оценки резервно-адаптационного потенциала ткани мозга использовали соотношение концентрации спонтанно окисленных к уровню металл-индуцированных карбонильных производных белков, принимая последний за 100%. Чем больше доля спонтанной ОМБ, тем меньше резервно-адаптационный потенциал исследуемого образца [26, 28].
Общую протеолитическую активность измеряли с использованием специфического протеазного субстрата – азоказеина (Sigma Aldrich, США) [38]. Для исследования активности кислых протеаз в пробирку вносили 400 мкл 1 мМ ацетатного буфера (pH 5.5), а цитоплазматических протеаз – 400 мкл 1 мМ HEPES буфера (pH 7.2). Полученные растворы смешивали с 10 мкл 0.1% раствора Triton X-100, затем в пробирку добавляли 400 мкл 0.4% азоказеина, 200 мкл супернатанта, 5 мкл 250 мМ β-меркаптоэтанола и инкубировали в течение 12 ч при 30°С. Реакцию останавливали путем добавления 100 мкл 50% ТХУ, затем центрифугировали 10 мин при 10 000 g. Оптическую плотность супернатанта измеряли при длине волны 330 нм c использованием спектрофотометра Lambda-25 (Perkin Elmer, США). Протеазную активность рассчитывали по изменению оптической плотности азоказеина и представляли в виде удельной активности в единицу времени, пересчитанной на количество белка в пробе (UPr/мин мкг).
Интенсивность перекисного окисления липидов определяли в растворимой фракции гомогената ткани мозга по содержанию малонового диальдегида (МДА), реагирующего с тиобарбитуровой кислотой. Образцы смешивали с реактивом, содержащим 0.3% Тритона Х-100, 0.1 М НCl и 0.03 М 2-тиобарбитурата. Смесь инкубировали в течение 45 мин при 95°С, затем центрифугировали в течение 10 мин при 10 000 g. Оптическую плотность измеряли при длине волны 532 нм c использованием спектрофотометра Lambda-25 (Perkin Elmer, США). Концентрация МДА в пробах рассчитывалась с использованием молярного коэффициента экстинкции (ε = 1.55 мМ–1 см–1) и выражалась в мкг/г ткани.
Содержание перекиси водорода (Н2О2) определяли методом, основанным на реакции окисления пероксидами Fe(II) до Fe(III) с реагентом FOX1 и последующим образованием красно-фиолетового ферроксиленолового комплекса с максимумом поглощения при длине волны 560 нм c использованием спектрофотометра Lambda-25 (Perkin Elmer, США). В состав реагента входили: 0.5 мМ FeSO4, 0.5 мМ (NH4)2SO4, 50 мМ H2SO4, 0.2 мМ ксиленол оранжевый, 200 мМ сорбитол (Sigma Aldrich, США). Супернатант и реагент смешивали в соотношении 1 : 1 при комнатной температуре, через 30 мин измеряли оптическую плотность. Содержание H2O2 рассчитывали по стандартной калибровочной кривой c известными концентрациями Н2О2 и выражали в мкг/г ткани.
Активность супероксиддисмутазы (SOD) определяли с помощью нитросинего тетразолия (NBT) в системе ксантин – ксантиноксидаза [39]. Реакционная смесь объемом 500 мкл содержала 100 мМ натрий-фосфатного буфера (рН 7.4), 0.1 мМ ЭДТА, 1 мМ цитохрома C, 1 мМ ксантина, 0.04 мМ NBT и 150 мкл образца. Реакция инициировалась добавлением 50 мкл 0.5 ед. ксантиноксидазы. Процентное ингибирование восстановления NBT, пропорциональное активности SOD, присутствующей в образце, определяли на спектрофотометре Lambda-25 (Perkin Elmer, США) при длине волны 560 нм. За единицу активности SOD принимали количество фермента, способного подавить на 50% реакцию восстановления NBT, а удельная активность выражали в единицах на миллиграмм белка (USOD/мин мг).
Активность каталазы (CAT) определяли по скорости разложения H2O2 (ε = 0.44 мМ–1 см–1) при длине волны 240 нм [39] с помощью спектрофотометра Lambda-25 (Perkin Elmer, США). Реакционная смесь (500 мкл) содержала 50 мМ HEPES (pH 7.0), 40 мМ H2O2 и 150 мл образца. Реакцию инициировали добавлением H2O2. За единицу активности каталазы принимали количество субстрата (H2O2) в мкМ, преобразуемого ферментом в единицу времени (мин), рассчитанное на мг сырой ткани в пробе (UCAT/мин мг).
Активность глутатионпероксидазы (GPх) определяли по методу Вейдера и Каллена [40]. В присутствии глутатионредуктазы и НАДФН окисленный глутатион (GSH) превращается в восстановленную форму (GSSG) с сопутствующим окислением НАДФН в НАДФ. Скорость реакции, катализируемой GPх, оценивали с помощью спектрофотометра Lambda-25 (Perkin Elmer, США) по уменьшению оптической плотности раствора при длине волны 340 нм (ε = 6.22 мМ–1 см–1). Реакционная смесь (400 мкл) состояла из 50 мМ Na2HPO4 буфера (рН 7.2), 1 мМ восстановленного глутатиона, 0.5 единицы глутатионредуктазы, 0.15 мМ НАДФН, 1 мМ ЭДТА и 150 мкл образца. Одна единица GPх определяется как 1 мкМ глутатиона, потребляемого в минуту, а удельная активность указывается в единицах на мг белка в минуту (UGPx/мин мг).
Активность глутатионредуктазы (GR), которая катализирует НАДФН-зависимое восстановление окисленного глутатиона до восстановленного, определяли с помощью спектрофотометра Lambda-25 (Perkin Elmer, США) при длине волны 412 нм. Предварительно пробы разводили в HEPES (pH 8.0) до конечного объема 500 мкл в соотношении 1 : 9. Тиоловые группы (–SH) в пробе восстанавливали в течение 5 мин с использованием 10 мкл 3% H2O2, затем образцы инкубировали в течение 5 мин с 10 мкл свежеприготовленного раствора каталазы (Sigma Aldrich, США). Реакцию инициировали внесением в пробу НАДФН в объеме 10 мкл до конечной концентрации 0.1 мМ. Активность глутатионредуктазы оценивали по изменению содержания глутатиона с помощью 5,5'-дитиобис-2-нитробензойной кислоты и выражали в единицах на мг белка (UGR нмоль/мин мг).
Статистическая оценка различий в сравниваемых выборках оценивалась для 5% уровня значимости. Нормальность распределения выборки определяли при помощи F-теста Фишера и критерия Шапиро–Уилка с использованием программы OriginPro 8.5 (OriginLab Corporation, Northampton, MA, США). Достоверность различий оценивали с помощью критерия Манна-Уитни для непараметрических выборок (OriginPro 8.5, OriginLab Corporation, Northampton, MA, США), где n – количество животных. Все измерения проводили не менее, чем в 3 параллельных пробах, используя для повторения животных из разных помeтов. Экспериментальные данные в тексте представлены как среднее значение ± стандартная ошибка среднего.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Результаты исследования показали, что содержание продуктов спонтанной ОМБ в тканях мозга крыс с пренатальной ГГц в течение первой недели постнатального развития существенно выше, чем в контрольной группе. Средний уровень карбонильных производных в гомогенате мозга крыс (Р2–Р7) в контрольной группе составлял 0.28 ± ± 0.02 нг/мг белка (n = 5), а у крыс с пренатальной ГГц – 0.36 ± 0.03 нг/мг белка (n = 5, p < 0.05, рис. 1а).
Рис. 1.
Уровень ОМБ и протеолитической активности ферментов в головном мозге крыс с пренатальной ГГц. (а) Уровень спонтанной ОМБ в контроле и в условиях пренатальной ГГц (пГГц). (б) Вклад спонтанной ОМБ в металл-катализируемое окисление, принятое за 100% в контроле и в условиях пренатальной ГГц. (в) Общая протеолитическая активность кислых (pH 5.5) и нейтральных (pH 7.2) протеаз в тканях мозга животных из контрольной (белый) и ГГц (серый) групп. Боксплот – 25–75% проценталь, усы – минимальное и максимальное значения, поперечная линия – медиана, квадрат – среднее значение. * p < 0.05 относительно контроля.
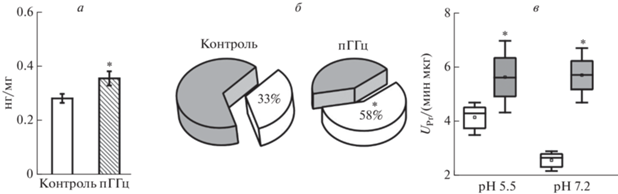
Индуцированная ОМБ отражает количество всех имеющихся на данный момент в ткани молекул, которые могут подвергаться карбонилированию [26]. У животных контрольной группы уровень металл-катализируемой ОМБ составил 0.86 ± 0.03 нг/мг белка (n = 5), тогда как у крыс с пренатальной ГГц количество индуцированной ОМБ было меньше – 0.61 ± 0.07 нг/мг белка (n = 5, p < 0.05). Далее был проведен анализ вклада спонтанной ОМБ в металл-индуцируемое окисление. Оказалось, что доля спонтанной ОМБ в группе ГГц составляла 58% (n = 5, p < 0.05), тогда в контрольной группе – 33% (n = 5; рис. 1б).
Известно, что продукты ОМБ способны вызывать повышение проницаемости мембран лизосом и усиливать активность лизосомальных ферментов [41]. В следующей серии экспериментов была проанализирована активность протеаз в тканях мозга крыс с пренатальной ГГц. Общая протеолитическая активность кислых протеаз при pH 5.5 составляла в контроле 4.16 ± 0.23 UPr/мин мкг и в группе ГГц – 5.65 ± 0.54 UPr/мин мкг (n = 4, p < 0.05, рис. 1в). Кроме того, наблюдали двукратное увеличение активности нейтральных протеаз в тканях мозга крыс с пренатальной ГГц (5.72 ± 0.41 UPr/мин мкг, n = 4, p < 0.05, рис. 1в) по сравнению с группой контроля (2.58 ± 0.14 UPr/мин мкг, n = 5).
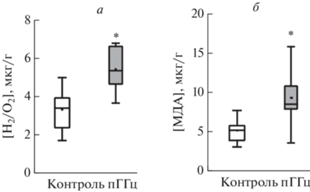
АФК являются основными индукторами ОМБ [31]. Для оценки степени окислительного стресса исследовали уровень H2O2 и МДА, продукта перекисного окисления липидов в тканях головного мозга животных. Уровень H2О2 в контрольной группе составил 3.4 ± 0.4 мкг/г (n = 15), а в группе ГГц – 5.4 ± 0.5 мкг/г (n = 6, р < 0.05; рис. 2а). Уровень МДА в контрольной группе составил 5.2 ± 0.3 мкг/г (n = 15), а у животных группы ГГц – 9.4 ± 0.9 мкг/г (n = 13, p < 0.05; рис. 2б).
Рис. 2.
Концентрация Н2О2 (а) и маркера перекисного окисления липидов – МДА (б) в тканях головного мозга крыс с пренатальной ГГц. Боксплот – 25–75% проценталь, усы – минимальное и максимальное значения, поперечная линия – медиана, квадрат – среднее значение. * p < 0.05 относительно контроля.
Активность антиоксидантных ферментов – SOD, CAT, GPx и GR была достоверно ниже у животных с пренатальной ГГц. Активность SOD в контроле составила 1.31 ± 0.11 USOD/мин мг (n = 13) и в группе ГГц – 0.99 ± 0.14 USOD/мин мг (n = 12, p < 0.05; рис. 3а). Активность CAT в контроле составила 39.31 ± 5.35 Uсat/мин мг (n = 7), а у крыс с пренатальной ГГц – 21.91 ± 4.54 Uсat/мин мг (n = 7, p < 0.05; рис. 3б).
Рис. 3.
Активность антиоксидантных систем супероксиддисмутазы (а), каталазы (б), глутатионпероксидазы (в) и глутатионредуктазы (г) в тканях мозга крыс с пренатальной ГГц. Боксплот – 25–75% проценталь, усы – минимальное и максимальное значения, поперечная линия – медиана, квадрат – среднее значение. * p < 0.05 относительно контроля.
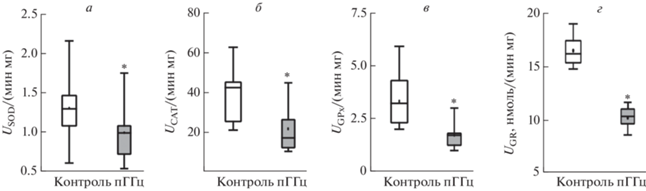
В контроле показатели активности GPx и GR составляли 3.41 ± 0.53 UGPx/мин мг (n = 11) и 16.55 ± ± 0.65 UGRнмоль/мин мг (n = 6), соответственно. У крыс с пренатальной ГГц активность GPx составляла 1.8 ± 0.3 UGPx/мин ∙ мг (n = 10, p < 0.05) и GR – 10.17 ± 0.41 UGRнмоль/мин мг (n = 6, p < 0.05), соответственно (рис. 3в, г).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В настоящем исследовании было продемонстрировано, что высокий уровень гомоцистеина у самок во время беременности вызывает следующие изменения в ткани мозга потомства на первой неделе постнатального развития: повышение уровня карбонильных производных белков, увеличение доли спонтанного окисления в металл-индуцированной модификации белков, одновременно с выраженным снижением антиоксидантной защиты и усилением продукции перекиси водорода и перекисного окисления липидов, и повышение общей протеолитической активности.
Нервная ткань характеризуется высокой чувствительностью к повреждающему действию свободных радикалов из-за повышенного содержания субстратов перекисного окисления (полиненасыщенных жирных кислот и ионов металлов) и низкой активностью антиоксидантных ферментов в сочетании с высокой интенсивностью обменных процессов [42, 43]. Особенно это характерно для раннего периода онтогенеза, когда благодаря высокому уровню нейропластичности происходит интенсивный нейрогенез, синаптогенез, что сопровождается повышением синтеза белков и липидов, нейроспецифических ростовых факторов и медиаторов [26].
Накопление гомоцистеина в ранний период онтогенеза в различных отделах головного мозга приводит к усилению перекисного окисления липидов, снижению общей антиокислительной активности и экспрессии адгезивных белков, участвующих в процессах синаптической пластичности [44], окислительному повреждению ДНК и белков [5], повышению активности каспазы-3 [21], что нарушает созревание нейрональных сетей в первые недели постнатального развития крыс [45]. Действительно, увеличение возбудимости нейронов наряду со снижением частоты гигантских деполяризующих потенциалов, необходимых для установления межнейронных связей, было показано в гиппокампе новорожденных крыс с пренатальной ГГц [46].
ОМБ является одним из ранних и наиболее надежных маркеров окислительного стресса [47, 48], затрагивает рецепторы и каналы мембраны, белки цитоскелета, факторы транскрипции [49]. Ряд исследователей полагают, что ОМБ является превалирующим механизмом токсического действия гомоцистеина, приводящим к ингибированию Na+/K+-АТФазы в нейронах гиппокампа и миндалины [50, 51], а также ферментов антиоксидантной защиты [23, 25, 52, 53]. Действительно, в наших экспериментах уровень карбонильных производных белков, являющихся одним из типов необратимой ОМБ в тканях мозга крыс с пренатальной ГГц, был выше на 30% по сравнению с контролем.
Известно, что в физиологических условиях интенсивность ОМБ повышается в течение раннего постнатального онтогенеза и зависит от процессов созревания нервной системы, в том числе миелинизации нервных волокон и формирования новых синапсов [48]. Максимальный пик ОМБ регистрировался к 2–3-м неделям постнатального развития мозга крыс. В то же время патологические факторы в период эмбриогенеза, включая пренатальный стресс, усиливают спонтанный и индуцированный ОМБ в гиппокампе, стриатуме и гипоталамусе крыс в первый месяц постнатального развития [26]. Нами было отмечено увеличение доли спонтанной ОМБ у животных с пренатальной ГГц с 33% до 58%, что говорит о значительном падении резервно-адаптационного потенциала клеток мозга и свидетельствует как о снижении устойчивости системы к действию свободных радикалов, так и о нарушении процессов метаболизма белков. Подобные изменения отмечались также в скелетной, сердечной и гладкомышечной тканях при умеренной ГГц у взрослых животных [54, 55].
Карбонилирование аминокислотных остатков способствует агрегации и фрагментации белковых молекул и, как следствие, резкому повышению их чувствительности к деградации протеазами [56]. Дестабилизация лизосомальной мембраны в результате окислительного стресса вызывает высвобождение и активацию лизосомальных цистеиновых протеаз – катепсинов, что вызывает повреждение клеточных структур [57]. В наших экспериментах в ткани мозга крыс с пренатальной ГГц возрастала общая протеолитическая активность при pH 5.5, что указывает на активацию лизосомальных протеаз. Кроме того, мы наблюдали усиление активности цитоплазматических протеаз, что согласуется с данными об увеличении активности внелизосомальной фракции протеаз в клетках гладкой мускулатуры, печени и почек при ГГц [55]. Полученные данные свидетельствуют об активации системы протеолиза в первую неделю развития мозга крыс с пренатальной ГГц.
Триггером ОМБ является окислительный стресс, характерный для ГГц и показанный в нашем исследовании у животных первой недели после рождения, у которых наблюдалось высокое содержание H2O2 и МДА с одновременным снижением активности антиоксидантных ферментов – SOD, CAT, GPx и GR.
В условиях ГГц снижение активности SOD и CAT может быть обусловлено как инактивацией ферментов вследствие окисления тирозиновых аминокислотных остатков пероксинитритами и другими АФК, так и с угнетением их экспрессии за счет изменения метилирования ДНК [1, 19, 23, 30, 45, 53]. В условиях окислительного стресса из-за быстрого окисления глутатиона соотношение восстановленного/окисленного глутатиона падает [53]. Снижение активности как глутатионпероксидазы, так глутатионредуктазы, вызванное ГГц, может приводить к нарушению метаболизма глутатиона и уменьшению его уровня [8, 53, 58]. В результате полного или частичного истощения запасов глутатиона в клетках мозга крыс будет усиливаться карбонилирование белков и активность сериновых протеаз [60–62], что и наблюдалось в головном мозге крыс с пренатальной ГГц.
ЗАКЛЮЧЕНИЕ
Результаты нашей работы показывают значительное усиление необратимого процесса ОМБ в головном мозге новорожденных крыс с пренатальной ГГц вследствие развития окислительного стресса. Принимая во внимание описанные здесь эффекты и литературные данные, можно предложить, что в условиях ГГц матери в тканях мозга новорожденных крыс происходит ослабление антиоксидантной защиты, преобладание генерации АФК над их деградацией, накопление супероксидных и гидроксильных радикалов, что стимулирует перекисное окисление липидов и активирует ОМБ, которая, в свою очередь, может служить источником генерации новых свободных радикалов и дальнейшей инактивации антиоксидантных систем, приводя к клеточной гибели и нарушению развития нервной ткани развивающегося организма.
Список литературы
Арутюнян А.В., Керкешко Г.О., Милютина Ю.П., Щербицкая А.Д., Залозняя И.В. // Биохимия. 2021. Т. 86. № 6. С. 871–884.
Marseglia L., D’Angelo G., Manti S., Arrigo T., Barberi I., Reiter R.J., Gitto E. // Oxid. Med. Cell. Longev. 2014. № 358375.
Мальцева Н.В., Волчегорский И.А., Шемяков С.Е. // Нейрохимия. 2017. Т. 34. № 1. С. 54–61.
Baydas G., Koz S.T., Tuzcu M., Nedzvetsky V.S., Etem E. // Int. J. Dev. Neurosci. 2007. V. 25. P. 133–139.
Koz S.T., Gouwy N.T., Demir N., Nedzvetsky V.S., Etem E., Baydas G. // Int. J. Dev. Neurosci. 2010. V. 28. P. 325–329.
Troen, A.M. // Prog. Neuropsychopharm. Biol. Psychiatry. 2005. V. 29. P. 1140–1151.
Арутюнян А.В., Пустыгина А.В., Милютина Ю.П., Залозняя И.В., Козина Л.С. // Молекулярная медицина. 2015. № 5. С. 41–46.
Yakovleva O., Bogatova K., Mukhtarova R., Yakovlev A., Shakhmatova V., Gerasimova E., Ziyatdinova G., Hermann A., Sitdikova G. // Biomolecules. 2020. V. 10. № 7. P. 995.
Yakovleva O.V., Ziganshina A.R., Dmitrieva S.A., Arslanova A.N., Yakovlev A.V., Minibayeva F.V., Khaertdinov N.N., Ziyatdinova G.K., Giniatullin R.A., Sitdikova G.F. // Oxid. Med. Cell. Longev. 2018. V. 2018. № 2. P. 2746837–2746837.
Sharma M., Tiwari M., Tiwari R.K. // Basic Clin. Pharmacol. Toxicol. 2015. V. 117. P. 287–296.
Herrmann W., Obeid R. // Clin Chem Lab Med. 2011 V. 49. № 3. P .435–441.
Gerasimova E., Burkhanova G., Chernova K., Zakharov A., Enikeev D., Khaertdinov N., Giniatullin R., Sitdikova G. // Behavioural Brain Resh. 2021. V. 409. P. 1–8.
Beard R.S., Reynolds J.J., Bearden S.E. // Blood. 2011. V. 118. № 7. P. 2007–2014.
Милютина Ю.П., Щербицкая А.Д., Салтыкова Е.Д., Козина Л.С., Журавин И.А., Наливаева Н.Н., Ару-тюнян А.В. // Росс. физ. журнал. им. И.М. Сеченова. 2017. Т. 103. № 11. С. 1280–1291.
Dai C., Fei Y., Li J., Shi Y., Yang X. // BioMed. Res. International. 2021. V. 2021. № 6652231.
Bolton A.D., Phillips M.A., Constantine-Paton M. // J. Neurophysiol. 2013. V. 16. № 110. P. 1567–1582.
Abushik P.A., Niittykoski M., Giniatullina R., Shakirzyanova A., Bart G., Fayuk D. // J. Neurochem. 2014. V. 11. № 129. P. 264–274.
Lipton S.A., Kim W.K., Choi Y.B., Kumar S., D’Emilia D.M., Rayuda P.V., Arnelle D.R., Stampler J.S. // Proc. Natl. Acad. Sci. U.S.A. 1997. V. 94. P. 5923–5928.
Sibarov D.A., Abushik P.A., Giniatullin R., Antonov S.M. // Front. Cell. Neurosci. 2016. V. 10. № 246.
Курмашова Е.Д., Гатаулина Э.Д., Зефиров А.Л., Ситдткова Г.Ф., Яковлев А.В. // Росс. физ. журнал. им. И.М. Сеченова. 2019. Т. 105. № 10. С. 1236–1246.
Арутюнян А.В., Милютина Ю.П., Щербицкая А.Д., Керкешко Г.О., Залозняя И.В., Михель А.В. // Биохимия. 2020. Т. 85. № 2. С. 248–259.
Арутюнян А.В., Козина Л.С., Арутюнов В.А. // Журнал акушерства и женских болезней. 2010. № 59. С. 16–23.
Pustygina A.V., Milyutina Y.P., Zaloznyaya I.V., Arutyunyan A.V. // Neurochem. J. 2015. V. 9. P. 60–65.
Longoni A., Bellaver B., Bobermin L.D., Santos C.L., Nonose Y., Kolling J., Dos Santos T.M., de Assis A.M., Quincozes-Santos A., Wyse A.T.S // Mol. Neurobiol. 2018. V. 55. № 3. P. 1966–1976.
Shcherbitskaia A.D., Vasilev D.S., Milyutina Yu.P., Tumanova N.L., Mikhel A.V., Zalozniaia I.V., Arutjunyan A.V. // Cells. 2021. V. 10. № 6. P. 1536.
Вьюшина А.В., Притворова А.В., Флеров М.А. // Нейрохимия. 2012. Т. 29. № 3. С. 240–246.
Cindrova-Davie T., Herrera E.A., Niu Y., Kingdom J., Giussani D.A., Burton G.J. // Am. J. Pathol. 2013. V. 182. P. 1448–1458.
Дубинина Е.Е. Физиологические и клинико-биохимические аспекты. СПб.: Медицинская пресса, 2006. 400 с.
Sibrian-Vazquez V, Escobedo J.O., Lim S., Samoei G.K., Strongin R.M. // Proc. National Academy of Sciences. 2010. V. 107. № 2. P. 551–554.
Perla-Kajan J., Twardowski T., Jakubowski H. // Amino Acids. 2007. V. 12. № 32. P. 561–572.
Stadtman E.R. // Free Radical Res. 2006. V. 40. P. 1250−1258.
Reinheckel T., Noack H., Lorenz S., Wiswedel I., Augustin W. // Free Radical Res. 1998. V. 29. P. 297–305.
Телушкин П.К. // Проблемы эндокринологии. 1998. Т. 44. № 3. С. 35–37.
Nyström T. // EMBO J. 2005. V. 24. № 7. P. 1311–1317.
Gerasimova E.E., Yakovleva O.V., Burkhanova G., Ziyatdinova G., Khaertdinov N., G. Sitdikova G. // BioNanoScience. 2017. V. 7. № 1. P. 55–158.
Parasuraman S., Raveendran R., Kesavan R. // J. Pharmacology & Pharmacotherapeutics. 2010. V. 1. № 2. P. 87–93.
Teixera de Oliveira D, Souza-Silva E., Tonussi C.R. // Scand. J. Lab. Anim. Sci. 2009. V. 36. № 2. P. 109–113.
Śliwa-Jóźwik A., Jóźwik1 A., Fronczyk W., Guszkiewicz1 A., Kołątaj A. // Animal Science Papers and Reports. 2004. V. 22 № 2. P. 237–245.
Fulle S., Di Donna S., Puglielli C., Pietrangelo T., Beccafico S., Bellomo R., Protasi F., Fanò G. // Exp. Gerontol. 2005. V. 40. P. 189–197.
Weydert C.J., Cullen J.J. // Nat Protoc. 2010. V. 5. P. 51–66.
Brunk, U.T. Neuzil J., Eaton J.W. // Redox Rep. 2001. V. 6. № 2. P. 91–97.
Aruoma O., Halliwell B., Laughton M.J. // Biochem. J. 1989. V. 258. № 2. P. 617–620.
Болдырев А.А. // Соросовский образовательный журнал 2001. № 4. С. 21–28.
Hoffman K.B., Murray B.A., Lynch G., Munirathinam S., Bahr B.A. // Neuroscience Res. 2001. V. 39. № 2. P. 167–173
Blaise S.A., Nédélec E., Schroeder H., Alberto J.M., Bossenmeyer-Pourié C., Guéant J.L., Daval J.L. // The American Journal of Pathology. 2007. V. 170. № 2. P. 667–679.
Yakovlev A.V., Kurmasheva E.D., Giniatullin R., Khalilov I., Sitdikova G.F. // Neurosci. 2017. V. 340. P. 153–165.
Губский Ю.И., Беленичев И.Ф., Павлов С.В., Левицкий Е.Л., Бухтиярова Н.В. // Современные проблемы токсикологии. 2006 Т. 2. С. 37–43.
Bizzozero O.A. // Handbook of Neurochemistry and Molecular Neurobiology / Ed. Lajtha A., Banik N., Ray S.K. Boston: Springer, 2009. P. 543–562.
Wehr N.B., Levine R.L. // Methods Mol. Biol. 2013. V. 965. P. 265–281.
Streck E.L., Matte C., Vieira P.S., Rombaldi F., Wannmacher C.M.D., Wajner M., Wyse A.T.S. // Neurochem. Res. 2002. V. 27. № 12. P. 1593–1598.
Matte C., Mackedanz, V., Stefanello, F.M., Schererna E.B.S. Andreazza A.C., Zanotto C., Moro A.M., Garcia S.C., Gonçalves C.A., Erdtmann B., Salvador M., Wyse A.T.S. // Neurochem. Int. 2009. V. 54. P. 7–13.
Durmaz A., Dikmen N. // J Enzyme Inhib. Med. Chem. 2007. V. 22. № 6. P. 733–738.
Lubos E., Loscalzo J., Handy D.E. // Antioxid. Redox Signal. 2007. V. 9. P. 1923–1940.
Ильичева А.С., Фомина М.А. // Российский медико-биологический вестник имени академика И.П. Павлова. 2015. Т. 1. С. 45–51.
Фомина М.А., Терентьев А.А. // Российский медико-биологический вестник имени академика И.П. Павлова. 2018. Т. 26. № 2. С. 195–212.
Shringarpure R., Davies K.J.A. // Free Radic. Biol. Med. 2002. V. 32. № 11. P. 1084–1089.
Tiwari S.C., Soni R.M. // J. Alzheimer’s Disease & Parkinsonism. 2014. № 4. P. 5–9.
Durmaz A., Dikmen N. // J. Enzyme Inhib. Med. Chem. 2007. V. 22. № 6. P. 733–738.
Dasgupta A., Zheng J., Bizzozero O.A. / ASN NEURO. 2012. V. 4. № 3. art: e00084.
Zheng J., Hu Ch.-L, Shanley K.L., Bizzozero O.A. // Neurochem. Res. 2018. V. 43. P. 609–618.
Mandaviya P.R., Stolk L., Heil S.G. // Mol. Genet. Metab. 2014. V. 113. P. 243–252.
de Moreira S.D., Figueiró P.W., Siebert C., Prezzi C.A., Rohden. F, Guma F.C.R., Manfredini V., Wyse A.T.S. // Neurotox. Res. 2018. V. 33. № 3. P. 580–592.
Дополнительные материалы отсутствуют.