

Неорганические материалы, 2019, T. 55, № 1, стр. 3-10
Электронное строение и температура ферромагнитного перехода Ga1 – xMnxAs в неэмпирическом методе локального обменаВ. Г. Яржемский, С. В. Мурашов, А. Д. Изотов
В. Г. Яржемский 1, *, С. В. Мурашов 1, А. Д. Изотов 1
1 Институт общей и неорганической химии им. Н.С. Курнакова Российской академии наук
119991 Москва, Ленинский пр., 31, Россия
* E-mail: vgyar@igic.ras.ru
Поступила в редакцию 25.05.2018
Аннотация
Методом функционала электронной плотности (ФЭП) рассчитаны зонная структура и плотности состояний разбавленного магнитного полупроводника Ga1 – xMnxAs. Получено, что часть Mn3d-состояний гибридизуется с валентной зоной на уровне Ферми. Расчеты магнитных свойств проводились многомасштабным методом, в котором использовались плотности состояний из метода ФЭП и значения обменных интегралов, рассчитанные методом Хартри-Фока для атома Mn. Теоретическая температура ферромагнитного перехода TC Ga0.9375Mn0.0625As согласуется с экспериментальными данными.
ВВЕДЕНИЕ
Магнитные полупроводники являются перспективными материалами для спинтроники, т.е. электроники, в которой для управления токами кроме заряда электрона используется его спин [1, 2]. Основным способом получения этих материалов является добавление 3d-элементов в известные полупроводники, например GaAs, InSb, CdGeAs2 [1–5]. Поскольку параметры кристаллических структур магнитных материалов отличаются от параметров полупроводников, растворимость магнитных атомов в этих полупроводниках обычно составляет всего несколько процентов. Такие материалы часто называют разбавленными магнитными полупроводниками (РМП). Наибольший коэффициент замещения Ga на Mn в GaAs составляет около 0.053, а температура Кюри TC – 110 К [1]. Недавние исследования методом стоячих волн в жестком рентгеновском диапазоне (SW-HARPES) показали, что Mn находится в узлах решетки на месте Ga [6]. Несмотря на то что состав, свойства и электронное строение РМП известны, до сих пор нет единого мнения о типе обменного взаимодействия, определяющего переход в ферромагнитное состояние. Подходы к расчетам ферромагнитных свойств РМП включают неэмпирические расчеты электронного строения в различных модификациях метода функционала электронной плотности (ФЭП) и параметрические модельные подходы к расчету магнитных свойств [7–9]. В частности, используется параметризация Гейзенберга в виде [8]
(1)
${{T}_{C}} = \frac{2}{3}{{k}_{B}}\left( {x\sum\limits_j {{{z}_{j}}{{J}_{{0j}}}{{S}^{2}}} } \right),$В модели RKKY [10] взаимодействие между спинами S1 и S2, находящимися в точках r1 и r2, описывается Гамильтонианом
(2)
${{H}_{{int}}} = {{J}^{2}}{{S}_{1}}{{S}_{2}}\frac{{2m{{k}_{F}}\cos (2{{k}_{F}}\left| {{{r}_{1}} - {{r}_{2}}} \right|)}}{{{{{\left( {2\pi } \right)}}^{3}}{{{\left| {{{r}_{1}} - {{r}_{2}}} \right|}}^{3}}}}.$Модели магнетизма типа Зинера (см., например, [7]) тесно связаны с относительными энергетическими положениями уровня Ферми (EF), валентной зоны и т.н. примесной зоны, т.e. зоны, образованной 3d-электронами магнитной примеси. В модели двойного обмена d-уровень переходного металла находится в запрещенной зоне а в модели p-d-обмена d-уровень лежит ниже края валентной р-зоны [7]. На основании экспериментальных данных был сделан вывод о том, что уровень Ферми находится в примесной зоне и что, изменяя его положение, можно менять TC [11]. Однако исследования методом HARPES показали, что Mn 3d-электроны гибридизованы с валентной зоной при энергиях связи от 0 до 4 эВ [12]. Согласно данным резонансной фотоэмиссии и теоретическим расчетам, Mn3d-состояния находятся в валентной зоне GaAs в широком диапазоне энергий [13]. Экспериментальный спектр имеет основной пик 3d-электронов при энергии связи 3.2 эВ [13] и небольшой пик вблизи уровня Ферми. Методом фотоэлектронной спектроскопии с угловым разрешением (ARPES) получено, что энергия уровня Ферми равна –0.3 эВ в GaMnAs с TC около 100 K, что соответствует pd-модели Зинера, а также существенное уменьшение плотности состояний вблизи EF [14]. Другие исследования методом ARPES показали, что вблизи точки Γ максимум валентной зоны исходного полупроводника находится вблизи EF и гибридизован с примесными Mn3d-состояниями [15]. Инфракрасные оптические данные доказали, что уровень Ферми находится в примесной зоне Mn, и авторы не исключают его перекрытия с валентной зоной [16]. Отмечалось также [17], что некоторые качественные различия экспериментальных данных связаны с условиями синтеза и что в оптимально синтезированных образцах примесная зона и зона исходного полупроводника всегда смешиваются и перекрываются. Поэтому термины валентная зона и примесная зона являются условными.
На основании анализа спектров магнитного кругового дихроизма был сделан вывод о pd-обменном взаимодействии в Ga1 – xMnxAs и о том, что носители тока в примесной зоне играют важную роль в формировании ферромагнетизма в этом материале [18].
Согласно спектрам магнитного кругового дихроизма spd-обменное взаимодействие в ферромагнитном Ga0.97Mn0.03As локализовано в точке Γ, где зеемановское расщепление равно 120 мэВ, а в точке L оно составляет всего 8 мэВ [19]. Резонансная туннельная спектроскопия GaMnAs показала, что валентная зона GaAs не сливается с примесной зоной и что валентная зона практически не магнитная даже в GaMnAs с высоким значением TC (154 K) [20].
Недавние исследования методом AEPES показали, что Mn существенно меняет зону GaAs в широком диапазоне энергий [21]. Дисперсная зона, индуцированная Mn, обнаружена также выше валентной зоны исходного полупроводника. Однако плотность состояний в этой зоне недостаточна для обеспечения высокой плотности носителей тока, обнаруженной экспериментально [21]. Резонансные туннельные эксперименты с GaMnAs показали, что при низких концентрациях Mn примесная зона лежит в запрещенной зоне полупроводника [22]. Когда концентрация Mn близка к концентрации, соответствующей ферромагнитному переходу, зоны сливаются, но при более высоких концентрациях Mn начальное положение примесной зоны в запрещенной зоне исходного полупроводника восстанавливается [22].
Результаты рассмотренных экспериментальных работ и теоретических расчетов [6, 8, 13, 23–26] указывают на гибридизацию части 3d-электронов c валентной зоной полупроводника. Теоретические расчеты описывают магнетизм в РМП с учетом параметров взаимодействия локализованных 3d-электронов с электронами зоны проводимости.
Модель Андерсона учитывает кулоновское и обменное взаимодействие, локализованное на центре Mn [27]. Гамильтониан Андерсона взаимодействия локализованных d-электронов с s‑электронами зоны проводимости может быть записан в виде:
(3)
${{H}_{{sd}}} = \sum\limits_{k,{\sigma }} {{{V}_{{dk}}}\left( {c_{{k{\sigma }}}^{ * }{{c}_{{d{\sigma }}}} + c_{{d{\sigma }}}^{ * }{{c}_{{k{\sigma }}}}} \right)} .$Этот тип sd-взаимодействия является чисто одноэлектронным и отличается от sd-обменного взаимодействия, которое входит в теории типа Зинера. Вследствие этого величина взаимодействия не зависит от энергии “примесной” d-зоны относительно зоны проводимости исходного полупроводника.
В наших предыдущих работах [25, 26] рассмотрено локальное обменное взаимодействие s-электронов валентной зоны с d-электронами Mn, причем обменный интеграл рассчитывался с использованием точных атомных волновых функциях. Однако вследствие малых плотностей состояний s-электронов на атомных центрах Mn результирующие значения TС оказались заниженными. В настоящей работе учитываются не только одноцентровые обменные sd- и pd-интегралы, но и кулоновские dd-интегралы. Отметим, что в то время как одноцентровые d-d-интегралы мультипольности 2- и 4-го порядка называются кулоновскими, их роль такая же, как и у обменных одноцентровых sd- и pd-интегралов: они минимизируют энергию состояния с наибольшим спином.
МЕТОД РАСЧЕТА И РЕЗУЛЬТАТЫ
Современные программы расчетов электронного строения кристаллов [28–30] используют потенциалы типа “ультрасофт” [31], которые соответствуют волновым функциям усредненного терма, поэтому в дальнейшем рассматривается вклад обменных интегралов в полную энергию относительно среднего терма. В настоящей работе расчеты электронной структуры выполнялись в методом ФЭП в приближении GGA (обобщенное градиентное приближение) [29].
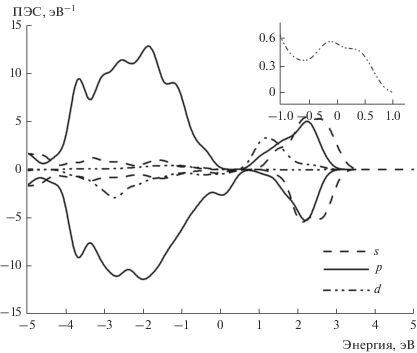
На рис. 1 приведена теоретическая зонная структура исходного полупроводника GaAs. Характерной особенностью этого расчета, как и многих других расчетов полупроводников методом ФЭП [32], является заниженная ширина запрещенной зоны. Теоретическая ширина запрещенной зоны 0.52 эВ существенно меньше экспериментального значения 1.519 эВ [33]. Теоретическая ширина запрещенной зоны GaAs 1.36 эВ была получена методом функций Грина G0W0, требующим существенно больших вычислительных ресурсов [32]. Для расчетов разбавленных систем необходимо существенное увеличение элементарной ячейки, поэтому они требуют пропорционального увеличения вычислительных ресурсов. В настоящей работе ширина зоны не является параметром теории, поэтому мы ограничились одноэлектронным подходом. Теоретические плотности электронных состояний (ПЭС) GaAs представлены на рис. 2, из которого видно, что валентная зона полупроводника состоит в основном из p-орбиталей Ga и As с небольшой примесью s-орбиталей, в то время как в зоне проводимости ПЭС s- и p-орбиталей примерно совпадают.
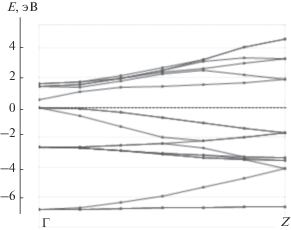
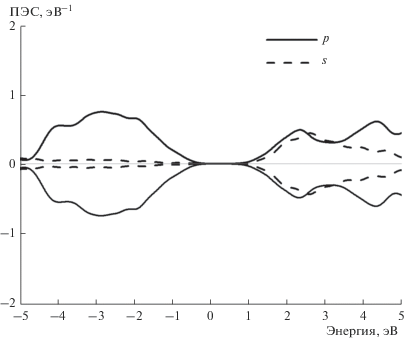
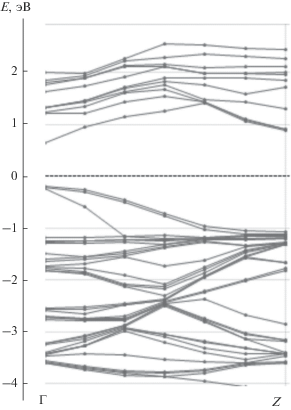
Зонные структуры состояний спин-вверх и спин-вниз для случая, когда 1/16 часть атомов Ga заменяется на Mn, приведены на рис. 3 и 4 соответственно. Зонная структура для состояний спин-вниз соответствует полупроводнику (см. рис. 4), в то время, в случае состояний спин-вверх наблюдается переcечение валентной зоны с уровнем Ферми (см. рис. 3). В замещенном полупроводнике появляются также почти плоские зоны с энергией около –1 эВ, которые соответствуют d-состояниям. Характерным отличием замещенного материала от чистого GaAs является небольшой вклад Mn3d-орбиталей в валентную зону, который виден на рис. 5, где приведены ПЭС. Наибольшая плотность состояний спин-вниз имеет энергию –3эВ. Также имеется отличная от нуля плотность состояний спин-вниз чуть ниже уровня Ферми, показанная на вставке к рис. 5. Выше уровня Ферми основной вклад в ПЭС дают состояния спин-вверх.
Рис. 3.
Зонная структура Ga0.9375Mn0.0625As в направлении ΓZ (спин-вверх-состояния); ноль шкалы энергий соответствует EF.
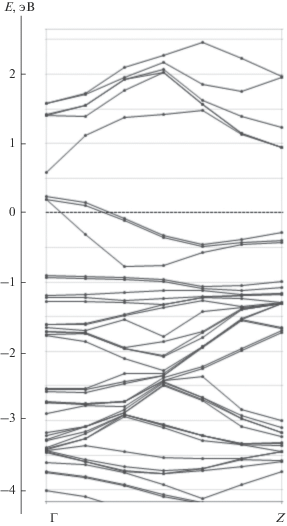
Рис. 4.
Зонная структура Ga0.9375Mn0.0625As в направлении ΓZ (спин-вниз-состояния); ноль шкалы энергий соответствует EF .
Рассмотрим применяемую в настоящей работе модель локального обмена на примере основного терма атома Mn. В случае заряда +2 число Mn3d-электронов не изменяется и основной терм – 3d 5(6S). В рассматриваемых соединениях атомы 3d-элемента занимают положение трехвалентного иона Ga, и Mn имеет конфигурацию 3d 4, основной терм которой 5D. Вычисления в этих двух случаях аналогичны, поэтому в дальнейшем будем рассматривать только терм 3d 5(6S). Вблизи атома Mn энергия s-электронов валентной зоны со спином, параллельным спинам 3d 5-оболочки (это состояние обозначается как 3d 5(6S)4s(7S)), ниже энергии состояния, в котором спин s-электронов антипараллелен спинам 3d 5-оболочки, т.е. состояния 3d 5(6S)4s(5S). Это утверждение соответствует расширению правила Хунда на случай двух оболочек, которое подтверждается расчетами. В отсутствие магнитного упорядочения вероятности состояний спин-вверх и спин-вниз пропорциональны их статистическим весам, и это среднее состояние обозначим 3d 5(6S)4s(av). В рассматриваемой модели разность энергий между состояниями 3d 5(6S)4s(av) и 3d 5(6S)4s(7S), умноженная на вклад s-электронов Mn в валентную зону, равна вкладу s-электронов в энергию ферромагнитного упорядочения. Это соответствует тому, что в формуле (3) сумма по k заменяется плотностью s-состояний, а матричный элемент Vdk, заменяется матричным элементом Vds, который рассчитывается на точных атомных волновых функциях. В случае заполненной валентной зоны полный спин равен нулю и уменьшение полной энергии может достигаться за счет разного заполнения подзон спин-вверх и спин-вниз p- и s-электронами. При этом спин-вверх поляризация s-электронов на Mn сопровождается спин-вниз-поляризацией p-электронов на Mn. Понижение энергии на магнитном центре пропорционально разности обменных интегралов между Mn3d-электронами и s- и p-электронами валентной зоны. При ферромагном фазовом переходе такое уменьшение энергии происходит на всех центрах. Отметим, что здесь термины спин-вверх и спин-вниз относятся к ориентации спина электрона валентной зоны по отношению к спину основного терма атома Mn. В то же время, в одноэлектронных расчетах, результаты которых приведены на рис. 1–5, термины спин-вверх и спин-вниз относятся к ориентации спина по отношению к моменту одного электрона.
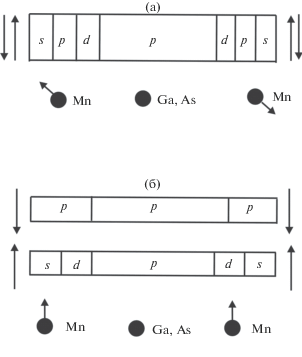
Если атом Mn замещает трехвалентный Ga, то один из его 3d-электронов участвует в химической связи. Это подтверждается результатами расчетов [7]. В этом случае часть плотности d-состояний находится вблизи уровня Ферми, а основным состоянием иона Mn будет 3d 4(5D). При этом надо учитывать изменение кулоновского взаимодействия в d-оболочке в зависимости от ориентации спина d-электрона, гибридизованного с валентной зоной. Перераспределение d- и p-электронов валентной зоны на Mn между состояниями спин-вверх и спин-вниз приводит к некоторому уменьшению полной энергии. Модель проиллюстрирована на рис. 6. В парамагнитном случае s-, p- и d-электроны на Mn одинаково заполняют подзоны спин-вверх и спин-вниз. В ферромагнетике заселенность спин-вверх подзоны Mn3d-электронами превосходит заселенность Mn3d-электронами спин-вниз зоны. При этом Mn4s- и Mn4p-электроны преимущественно заселяют спин-вниз-зону. Уменьшение полной энергии обусловлено различием обменных интегралов между локализованными 3d-электронами и s-, p- и d-электронами валентной зоны. Таким образом, важной особенностью настоящей модели является то, что заполненная валентная зона остается неполяризованной по спину, а уменьшение полной энергии достигается за счет перераспределения s-, p- и d-ПЭС между подзонами спин-вверх и спин-вниз. Низкая спин-поляризация валентной зоны согласуется с экспериментальными данными [19, 20].
Рис. 6.
Уменьшение полной энергии за счет изменения парциальных плотностей состояний спин-вверх и спин-вниз заполненных подзон: а – плотности состояний спин-вверх и спин-вниз (по отношению к спину атома Mn) для каждого типа орбиталей совпадают; б – плотность d-состояний спин-вверх превышает плотность d-состояний спин-вниз, для s- и p-состояний обратное соотношение.
Применяемая многомасштабная модель требует точного вычисления кулоновских матричных элементов и соответствующих коэффициентов для отдельного атома и расчета электронного строения материала. Атомные волновые функции были рассчитаны с использованием атомного комплекса программ методом Хартри–Фока [34].
Энергия атомного nl-электрона, связанного с атомным термом 3dN(LS) в полный терм $L{\text{'}}S{\text{'}}$ может быть представлена в виде:
(4)
${{U}_{l}} = \sum\limits_\kappa {(3d,nl\left| {{{R}^{\kappa }}} \right|nl,3d)\bar {C}_{{LS,L{\text{'}}S{\text{'}}}}^{\kappa }(3d,nl)} .$Первый сомножитель – это обменный интеграл, рассчитываемый с использованием атомных волновых функций [34], второй – коэффициент, рассчитываемый с использованием теории углового момента [35, 36]. Верхняя черта означает, что этот коэффициент рассчитывается относительно средней энергии. Суммирование идет по всем мультипольностям κ кулоновского взаимодействия, в частности: для s-электронов κ = 2, для p-электронов κ = 1, 3.
Плотность Mn3d-электронов на уровне Ферми, гибридизованных с валентной зоной, существенно меньше плотностей состояний на 2–3 эВ ниже уровня Ферми (см. рис. 5). Спин электрона на уровне Ферми, участвующего в химической связи, может иметь спин-поляризацию, параллельную спинам остовных 3d-электронов, а может не иметь спин-поляризации. В первом случае полная энергия уменьшается. Энергия спин-вверх-поляризации одного 3d-электрона относительно остовных 3d-электронов, связанных в основной терм, рассчитывается по формуле
(5)
${{U}_{d}} = \frac{1}{n}\sum\limits_\kappa {(3d,3d\left| {{{R}^{\kappa }}} \right|3d,3d)\bar {C}_{{LS}}^{\kappa }(3d,3d)} ,$Для вычисления температуры Кюри TC использовалась приближенная формула [7]
где Δε – понижение полной энергии за счет спин-поляризации в расчете на одну формульную единицу.В табл. 1 приведены кулоновские интегралы, коэффициенты и рассчитанные на их основе энергии и температуры переходов.
Таблица 1.
Теоретические атомные параметры и рассчитанные на их основе вклады в температуры Кюри TC для Ga0.9375Mn0.0625As
| Тип интеграла | Интеграл, эВ | Коэффициент | Энергия, эВ | TC, К |
|---|---|---|---|---|
| ${{R}^{2}}(3d3d\left| {{1 \mathord{\left/ {\vphantom {1 {{{r}_{{12}}}}}} \right. \kern-0em} {{{r}_{{12}}}}}} \right|3d3d)$ | 10.31 | –0.3968 | –1.327* | 145 |
| ${{R}^{4}}(3d3d\left| {{1 \mathord{\left/ {\vphantom {1 {{{r}_{{12}}}}}} \right. \kern-0em} {{{r}_{{12}}}}}} \right|3d3d)$ | 6.41 | –0.3968 | ||
| ${{R}^{2}}(3d3s\left| {{1 \mathord{\left/ {\vphantom {1 {{{r}_{{12}}}}}} \right. \kern-0em} {{{r}_{{12}}}}}} \right|3s3d)$ | 0.889 | –0.5 | –0.444 | 21 |
| ${{R}^{1}}(3d4p\left| {{1 \mathord{\left/ {\vphantom {1 {{{r}_{{12}}}}}} \right. \kern-0em} {{{r}_{{12}}}}}} \right|4p3d)$ | 0.391 | –0.3333 | –0.196 | |
| ${{R}^{3}}(3d4p\left| {{1 \mathord{\left/ {\vphantom {1 {{{r}_{{12}}}}}} \right. \kern-0em} {{{r}_{{12}}}}}} \right|4p3d)$ | 0.313 | –0.2143 |
Энергии спин-поляризации для 4s-электронов рассчитывалась путем задания в программе ФЭП параметра дополнительной энергии на центре, иногда называемого параметром Хаббарда. Величина этого параметра равнялась теоретической разности обменных интегралов для s- и p-электронов (см. табл. 1). Из данных табл. 1 видно, что спин-поляризации s-электронов на Mn соответствует TC = 21 K, что значительно меньше экспериментальной величины TС = 110 K [1]. Такой подход неприменим для d-электронов, поскольку здесь имеет место поляризация остовными d-электронами небольшой части d-электронов валентной зоны. Учитывалась поляризация d-электронов, ПЭС которых находятся между минимумом с энергией –0.5 эВ и EF (см. вставку к рис. 5). Интеграл ПЭС в этой области составляет 0.265. Теоретическое значение TС = 145 K, полученное с использованием этой величины и атомных интегралов табл. 1, находится между двумя экспериментальными значениями: 110 K [1] и 188 K [17], полученными для таких же концентраций Mn.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ И ЗАКЛЮЧЕНИЕ
Согласно результатам расчетов ПЭС и зонной структуры РМП Ga0.9375Mn0.0625As, Mn3d-орбитали гибридизованы с валентной зоной в области энергий от EF до –4 эВ, что согласуется с экспериментальными данными фотоэлектронной спектроскопии [12]. Детали теоретической электронной структуры также согласуются с экспериментальными данным. В частности, на теоретической кривой ПЭС d-электронов имеются большой максимум с энергией –2.9 эВ и малый максимум с энергией –0.8 эВ (см. рис. 5), которым соответствуют похожие максимумы в спектрах резонансной фотоэмисси [13]. Согласно теоретическим расчетам, при замещении 1/16 атомов Ga на Mn появляются плоские зоны с энергией –1 эВ и дисперсные зоны с энергией связи –3 эВ (см. рис. 4), причем полная плотность состояний растет в направлении от EF до –2.5 эВ (см. рис. 5), что качественно согласуется с фотоэлектронными спектрами [14, 15].
Температуры Кюри, рассчитанные многомасштабным методом, аналогичным методу [27], в котором кулоновские и обменные интегралы на магнитном центре вычисляются точно с использованием атомных волновых функций Хартри–Фока, а ПЭС рассчитываются с использованием пакетов ФЭП, находятся в согласии с экспериментальными данными. Указанное согласие подтверждает предложенную модель ферромагнетизма в РМП, согласно которой магнитное упорядочение спинов на атомах Mn может быть достигнуто за счет перераспределения s-, p- и d-орбиталей между подзонами спин-вверх и спин-вниз, т.е. без спин-поляризации валентной зоны.
Список литературы
Ohno H. Making Nonmagnetic Semiconductors Ferromagnetic // Science. 1998. V. 281. P. 951–956.
Dietl T. A Ten-Year Perspective on Dilute Magnetic Semiconductors and Oxides // Nature Materials. 2010. V. 9. P. 965–974.
Маренкин С.Ф., Трухан В.М., Федорченко И.В., Труханов С.В., Шелковая Т.В. Магнитные и электрические свойства композита Cd3As2 + MnAs // Журн. неорган. химии. 2014. Т. 59. № 4. С. 511–516.
Маренкин С.Ф., Изотов А.Д., Федорченко И.В., Новоторцев В.М. Синтез магнитогранулированных структур в системах полупроводник-ферромагнетик // Журн. неорган. химии. 2015. Т. 60. № 3. С. 343–348.
Novotortsev V.M., Kochura A.V., Marenkin S.F. New Ferromagnetics Based on Manganese-Chalcopyrites AIIBIVCV2 // Inorg. Mater. 2010. V. 46. № 13. P. 1421–1436.
Nemšák S., Gehlmann M., Kuo C.-T.,2, Lin S.-C., Schlueter C., Mlynczak E., Lee T.-L., Plucinski L., Ebert H., Di Marco I., Minár J., Schneider C.M., Fadley C.S. Element- and Momentum-Resolved Electronic Structure of the Dilute Magnetic Semiconductor Ga1 – xMnxAs // arxiv 1801.06587.
Sato K., Bergqvist L., Kudrnovský J., Dederichs P.H., Eriksson O., Turek I., Sanyal B., Bouzerar G., Katayama-Yoshida H., Dinh V.A., Fukushima T., Kizaki H., Zeller R. First-Principles Theory of Dilute Magnetic Semiconductors // Rev. Mod. Phys. 2010. V. 82. P. 1633–1690.
Szwacki G.N., Majewski J.A., Dietl T. (Ga, Mn)As under pressure: A First-Principles Investigation // Phys. Rev. B. 2015. V. 91. P. 184409.
Kudrnovsky J., Drchal V., Turek I. Exchange and SpinOorbit Induced Phenomena in Diluted (Ga, Mn)As from First Principles // Phys. Rev. 2016. V. 94. P. 054428.
Ruderman M.A., Kittel C. Inderect Exchange Coupling of Nuclear Moments by Conduction Electrons // Phys. Rev. 1954. V. 96. P. 99–102.
Dobrowolska M., Tivakornsasithorn K., Liu X., Furdyna J.K., Berciu M., Yu K.M., Walukiewicz W. Controlling the Curie Temperature in (Ga, Mn)As through Location of the Fermi Level within the Impurity Band // Nature Materials. 2012. V. 11. P. 444–449.
Gray A.X., Minar J., Ueda S., Stone P.R., Yamashita Y., Fujii, J., Braun J., Plucinski L., Schneider C.M., Panaccione G., Ebert H., Dubon O.D., Kobayashi K., Fadley C.S. Bulk Electronic Structure of the Dilute Magnetic Semiconductor Ga1 – xMnxAs through Hard X-ray Angle-Resolved Photoemission // Nature Mater. 2012. V. 11. P. 957–962.
Di Marco I., Thunstrom P., Katsnelson M.I., Sadowski J., Karlsson K., Lebegue S., Kanski J., Eriksson O. Electron Correlations in MnxGa1 – xAs as Seen by Resonant Electron Spectroscopy and Dynamical Mean Field Theory // Nat. Commun. 2013. V. 4. P. 2645.
Souma S., Chen L., Oszwałdowski R., Sato T., Matsukura F., Dietl T., Ohno H., Takahashi T. Fermi Level Position, Coulomb Gap, and Dresselhaus Splitting in (Ga, Mn)As // Sci. Rep. 2016. V. 6. P. 27266.
Kobayashi M., Muneta I., Takeda Y., Harada Y., Fujimori A., Krempaský J., Schmitt T., Ohya S., Tanaka M., Oshima M., Strocov V.N. Unveiling the Impurity Band Induced Ferromagnetism in the Magnetic Semiconductor (Ga, Mn)As // Phys. Rev. B. 2014. V. 89. P. 205204.
Burch K.S., Shrekenhamer D.B., Singley E. J., Stephens J., Sheu B.L., Kawakami R.K., Shiffer P., Samarth N., Awschalom D.D., Basov D.N. Impurity Bands in a High Temperature Ferromagnetic Semiconductor // Phys. Rev. Lett. 2006. V. 97. P. 087208.
Nemec P., Nova V., Tesarova N., Rozkotova E., Reichlova H., Butkovicova D., Trojanek F., Olejn K., Maly P., Campion R.P., Gallagher B.L., Sinova J., Jungwirth T. The Essential Role of Carefully Optimized Synthesis for Elucidating Intrinsic Material Properties of (Ga, Mn)As // Nat. Commun. 2013. V. 4. P. 2426.
Ando K., Saito H., Agarwal K.C., Debnath M.C., Zayets V. Origin of the Anomalous Magnetic Circular Dichroism Spectral Shape in Ferromagnetic Ga1 – xMnxAs: Impurity Bands Inside the Band Hap // Phys. Rev. Lett. 2008. V. 100. P. 067204.
Tanaka H., Jadwisienczak W.M., Saito H., Zayets V., Yuase S., Ando K. Localized sp–d Exchange Interaction in Ferromagnetic Ga1 − xMnxAs Observed by Magnetic Circular Dichroism Spectroscopy of L Critical Points // J. Phys. D.: Appl. Phys. 2014. V. 47. P. 355001.
Ohya S., Takata K., Tanaka M. Nearly Non-Magnetic Valence Band of the Ferromagnetic Semiconductor GaMnAs // Nature Phys. 2011. V. 7. P. 342–347.
Kanski J., Ilver L., Karlsson K., Ulfat I., Leandersson M., Sadowski J., Di Marco I. Electronic Structure of (Ga, Mn)As Revisited // New J. Phys. 2017. V. 19. P. 023006.
Muneta I., Ohya S., Terada H., Tanaka M. Sudden Restoration of the Band Ordering Associated with the Ferromagnetic Phase Transition in a Semiconductor // Nat. Commun. 2016. V. 7. P. 12013.
Мурашов С.В., Яржемский В.Г., Нефедов В.И., Муравьев Э.Н. Электронное строение магнитных полупроводников Cd1 – xMnxGeAs2 и Cu1 – xMnxGaTe // Журн. неорган. химии. 2007. T. 52. № 8. С. 1327–1331.
Яржемский В.Г., Мурашов С.В., Нефедов В.И., Муравьев Э.Н. Электронное строение и химическая связь в магнитных полупроводниках MnxCd1 – xGeAs2 и MnxZn1 – xGeAs2 // Неорган. материалы. 2008. Т. 44. № 11. С. 1300–1306.
Яржемский В.Г., Мурашов С.В., Изотов А.Д. Электронное строение и обменное взаимодействие в магнитных полупроводниках Ga1 – xMnxAs и In1 – xMnxAs // Неорган. материалы. 2016. Т. 52. № 2. С. 119–123.
Яржемский В.Г., Мурашов С.В., Изотов А.Д. Расчет электронного строения и обменного взаимодействия в полупроводниках InSb и GaAs при солегировании Mn и Ni // Неорган. материалы. 2017. Т. 53. № 11. С. 1158–1162.
Anderson P.W. Localized Magnetic States in Metals // Phys. Rev. 1961. V. 124. P. 41–53.
Gonze X., Jollet F., Araujo F.A., Adams D., Amadon B. et al. Recent developments in the ABINIT software Package // Comput. Phys. Commun. 2016. V. 205. P. 106–131.
Clark S.J., Segall M.D., Pickard C.J., Hasnip P.J., Probert M.J., Refson K., Payne M.C. First |Principles Methods Using CASTEP // Z. Kristallogr. 2005. V. 220. № 5–6. P. 567–570.
Kresse G., Hafner J. Norm-Conserving and Ultrasoft Pseudopotentials for First-Row and Transition-Elements // J. Phys.: Condens. Matter. 1994. V. 6. P. 8245–8257.
Vanderbilt D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism // Phys. Rev. B. 1990. V. 41. P. 7892–7895(R).
Pela R.R., Marques M.,Teles L.K. Comparing LDA-1/2, HSE03, HSE06 and G0W0 Approaches for Band Gap Calculations of Alloys // J. Phys.: Condens. Matter. 2015. V. 27. № 50. P. 505502.
Vurgaftman I., Meyer J.R., Ram-Mohan I.R. Band Parameters for III–V Compound Semiconductors and Their Alloys // J. Appl. Phys. 2001. V. 89. P. 5815.
Амусья М.Я., Семенов С.К., Чернышева Л.В. АТОМ-М алгоритмы и программы исследований атомных и молекулярных процессов. C.-Петербург: Наука, 2016. 553 с.
Amusia M.Ya., Chernysheva L.V., Yarzhemsky V.G. Handbook of Theoretical Atomic Physics. Springer, 2012. 799 p.
Собельман И.И. Введение в теорию атомных спектров. М.: Наука, 1977. 319 с.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы