

Неорганические материалы, 2019, T. 55, № 10, стр. 1116-1122
Структура и нанотвердость компактной керамики на основе гидроксиапатита
В. М. Иевлев 1, 2, *, А. В. Костюченко 3, Г. С. Кочлар 1, В. И. Путляев 1
1 Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
2 Воронежский государственный университет
394006 Воронеж, Университетская пл., 1, Россия
3 Воронежский государственный технический университет
394026 Воронеж, Россия
* E-mail: rnileme@mail.ru
Поступила в редакцию 18.01.2019
После доработки 19.03.2019
Принята к публикации 21.03.2019
Аннотация
Методом просвечивающей электронной микроскопии исследована субструктура, а методом наноиндентирования твердость и модуль упругости керамики гидроксиапатита, полученной холодным прессованием нанокристаллического порошка гидроксиапатита с последующим отжигом в течение 1–12 ч при 1150°С. Установлено, что в процессе компактирования реализуется процесс срастания наночастиц исходного порошка, обеспечивающий минимальную твердость компакта при его 50%-ной относительной плотности. С увеличением времени отжига твердость и модуль упругости керамики увеличиваются вследствие совершенствования ее субструктуры: увеличения размера зерен, уменьшения размера и количества пор.
ВВЕДЕНИЕ
Синтез и свойства керамики из гидроксиапатита (ГА, Ca10(PO4)(OH)2) как материала, применяемого в инженерии костной ткани, экстенсивно исследуются. Синтез керамики проводят спеканием порошкового компакта, активируемым традиционной термической обработкой, микроволновым нагреванием, искровым плазменным спеканием. Для нанесения фосфат-кальциевых керамических покрытий на имплантаты применяют спрей-плазменный процесс, высокочастотное магнетронное распыление [1–3].
Кинетику процесса спекания ГА исследовали методами термомеханического анализа, дилатометрии, высокотемпературной микроскопии. Показано, что процесс спекания можно представить тремя последовательными стадиями [1–3]. Считается, что первая стадия начинается от 400°С и проявляется в уменьшении площади свободной поверхности порошкового компакта в результате срастания, начинающегося с образования шеек между частицами. Этот процесс происходит без уплотнения компакта до 700–800°С. Коалесценция частиц порошка происходит посредством поверхностной диффузии [4, 5]. В зависимости от метода исследования величину энергии активации поверхностной диффузии оценивали в 120 [5] или 190–290 кДж/моль [6]. Для стехиометрического ГA скорость уменьшения площади поверхности остается низкой, константа скорости k при 700°C составляет всего 9.2 × 10–3. Стадия уплотнения начинается от 750°С [6]. С увеличением количества шеек между частицами порошка объем пор уменьшается, образец уплотняется и формируются границы зерен. При более высоких температурах происходит уплотнение в результате выхода пор, а также возможна рекристаллизация. Механизм выхода пор пока не установлен. На третьем этапе спекания возможно частичное дегидроксилирование ГА с превращением его в оксигидроксиапатит. Спекание ГA без давления обычно проводят в интервале температур 1100–1250°С. В этом интервале уплотнение происходит без образования жидкой фазы. Исследования подтвердили, что вторичные фазы (кристаллические или аморфные) не образуются [7]. ГА остается стабильным до 1450°С в атмосфере воздуха [8–10].
Уплотнение и рост зерен посредством собирательной рекристаллизации – конкурирующие процессы, поскольку происходит расширение интервала размеров зерен, увеличивается неоднородность структуры. Анализ работ по спеканию показывает, что установлены общие закономерности уплотнения в процессе синтеза ГА-керамики, но остается открытым вопрос о микроскопических механизмах, реализуемых на разных стадиях процесса.
При некоторой разнице в количественной оценке твердости керамики из порошков разного происхождения общая закономерность – ее увеличение с повышением температуры спекания. В зависимости от среды, в которой реализуется спекание, увеличение температуры процесса выше 1100°С может привести к уменьшению твердости [11, 12]. Предварительное прессование заготовок из нанокристаллических порошков приводило к снижению температуры спекания до 670°С, и микротвердость образцов керамики достигала 5.8 ГПа [13]. Зависимость твердости керамики от размера порошков проявляется как следствие разной возможности компактирования и достигаемой пористости, размера зерен, достигаемого в процессе рекристаллизации.
Цель настоящей работы – количественная оценка твердости образцов керамики на основе ГА на последовательных стадиях спекания и ее корреляции с изменением плотности керамики.
Приведенные в табл. 1 данные о твердости образцов керамики, покрытий и микрокристаллов ГА указывают на широкий диапазон величины твердости (от 1.4 до 8.4 ГПа) как следствие различий субструктуры и плотности. Твердость, определяемая методом микроиндентирования, не может быть объективной фундаментальной характеристикой керамики, поскольку в зоне локализованной деформации возможно разрушение образца, а в случае керамики ГА возможны существенные структурные превращения (например, аморфизация [28]). Поэтому можно считать, что наноиндентирование при малой величине нагрузки на индентор дает более объективную оценку твердости.
Таблица 1.
Зависимость зеренной субструктуры, плотности и твердости керамики ГА от температуры спекания
| Температура спекания, °С | Относительная плотность, % | Средний размер зерен, мкм | Максимальная твердость, ГПа | Источник |
|---|---|---|---|---|
| 1100 | 97 | 0.3–0.5 | 5.6 | [11] |
| 1200 | – | 0.5-1 | 9.5 | [12] |
| 1300 | ~86 | ~1 | 1.42 ± 0.02 | [13] |
| – | 99.6 | 1 | 4.5 ± 0.6 | [14] |
| 1150–1250 | 88.5–95.9 | 0.78–2.32 | ~5.11 | [15] |
| 1200 | 97.1 | 1.25 ± 0.31 | 4.45 ± 0.18 | [16] |
| 1000 | 97.7 ± 0.5 | 0.17 ± 0.09 | 8.4 ± 0.4 | [17] |
| 750 | ~99 | <0.05 | 5.7 | [18] |
| 1100 | ~78 | 90 | 2.04 ± 0.02 | [19] |
| 1300 | 98.4 ± 0.2 | 0.16–17 | 4.87 ± 0.15 | [20] |
| 1250 | >99 | 2.03 | 6.08 ± 028 | [21] |
| 1300 | >87 | – | 2.40 | [22] |
| 1200 | 97–98 | 1–5 | 2.94–3.04 | [23] |
| 1200 | 81 | >0.13 | 4.85 | [24] |
| 1200 | 85 | 2–5 | 2.55 | [25] |
| 1100 | >99.5% | 2–6 | 7.04 | [26] |
| 950 | 97–99% | – | 5.5 | [27] |
Исследование методом просвечивающей электронной микроскопии (ПЭМ) показывает [28, 29], что высокая локальная пластичность образцов ГА позволяет избежать образования трещин: механизм пластической деформации в этом материале не дислокационный.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходный порошок ГА синтезировали из раствора по реакции
Раствор (NH4)2HPO4 (1 M) добавляли по каплям к раствору Ca(NO3)2 (0.6 М) со скоростью 3.5 мл/мин. Реакцию проводили при 60°С при интенсивном перемешивании. Раствор доводили до рН 9 добавлением 25%-ного водного раствора аммиака. После выдерживания в течение 30 мин осадок собирали на фильтровальной бумаге. Фильтрат сушили при комнатной температуре. Затем осадок дезагрегировали помолом в шаровой мельнице в среде ацетона при весовом соотношении порошок : ацетон : шары 1 : 1 : 3. Затем порошок сушили при комнатной температуре в течение 2 ч, пропускали через сито 200 мкм и прессовали при 250–300 МПа в таблетки диаметром 8 мм на гидравлическом прессе Carver PG-10.
Термообработку (ТО) таблеток ГА проводили на воздухе при 1150°С в течение 1, 3, 6 и 12 ч. Скорость выхода на заданную температуру составляла 10°С/мин. Образцы оставляли в печи для охлаждения до комнатной температуры.
Плотность образцов рассчитывали исходя из объема и массы таблетки, ее изменение по мере спекания и уплотнения оценивали по отношению к теоретической плотности ГА (3.16 г/см3 [30]).
Фазовый состав образцов исследовали методами рентгеновской дифрактометрии (XRD, ARL Х’TRA), субструктуру тонких срезов керамики – методом ПЭМ (Karl Zeis Libra 2D, FEI Titan 80-300). Наноиндентирование проводили на нанотвердомере CSM Instruments с алмазным индентором Берковича. Тонкие срезы образцов исходной керамики и компактного фрагмента для исследования методом ПЭМ готовили на микроскопе Quanta 3D.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Структура. На рис. 1 приведены рентгеновские дифрактограммы исходного компакта и образцов керамики, образовавшихся в результате отжига в течение 1, 3 и 12 ч при 1150°С. Из них следует, что исходный компакт имеет нанокристаллическую субструктуру. Перекрывающиеся разрешаемые широкие пики соответствуют ГА.
Рис. 1.
Дифрактограммы исходного компакта (1) и керамики после отжига при 1150°С в течение 1 (2), 3 (3) и 12 (4) ч.
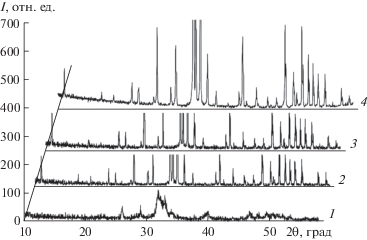
ПЭМ-изображение фрагментов, полученных ультразвуковым диспергированием исходного компакта (рис. 2), подтверждает его нанокристаллическую субструктуру с размером зерен до 20 нм, которые образуют компактные агрегаты размером до 200 нм, что позволяет сделать вывод о начале спекания в процессе компактирования порошка. ТО в течение 1 ч приводит к завершению формирования субмикрокристаллической зеренной структуры. Увеличение продолжительности ТО не приводит к заметным изменениям ширины пиков.

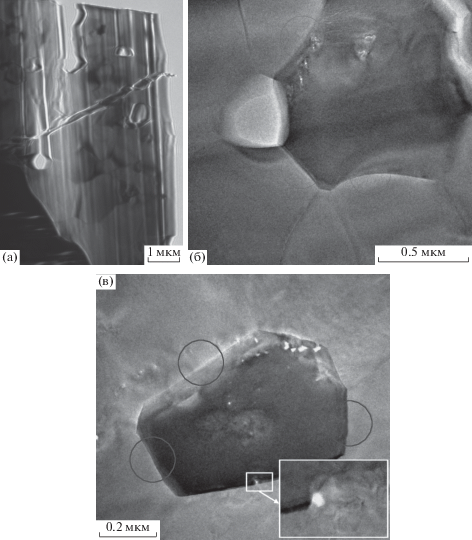
ПЭМ-изображение тонкого среза образца, синтезированного в процессе отжига в течение 1 ч (рис. 3а) показывает наличие пор субмикронного размера в количестве до 5 × 10–5 см–2. Субструктуру характеризует рис. 3б: размер зерен в пределах от 0.2 до 1.2 мкм есть следствие прошедшего процесса спекания, включая и начало стадии рекристаллизации.

Термообработка до 12 ч приводит к увеличению размера зерен вследствие продолжения собирательной рекристаллизации. Плотность пор субмикронного размера уменьшается в 2 раза (рис. 4а), они локализуются в зернограничных узлах (рис. 4б). Поры нанометрового размера (5–25 нм) – вблизи границ и на границах зерен (рис. 4б, 4в), что может быть следствием их движения к границам зерен.
Механические свойства. На рис. 5 приведены диаграммы нагрузка на индентор (Р)-глубина погружения индентора (h), полученные по результатам индентирования исходных компактов и образцов керамики. Из них следует упруго-пластический характер деформации и закономерное увеличение твердости с увеличением продолжительности отжига.
Рис. 5.
Диаграммы Р-h, полученные по результатам индентирования неотожженных и отожженных при 1150°С образцов керамики ГА при максимальной нагрузке на индентор Р = 5 (а) и 100 мН (б): 1 – исходный компакт; 2 – керамика, отожженная в течение 1 ч; 3 – 6 ч; 4 – 12 ч.
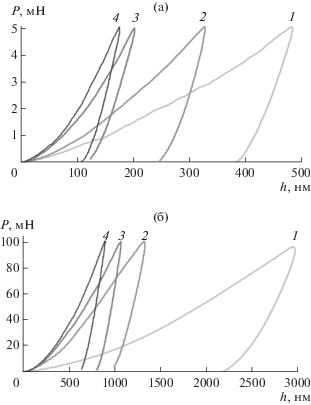
Характерная для всех образцов и для обеих нагрузок на индентор большая доля пластической деформации в работе индентирования согласуется с результатами наноиндентирования нанокристаллических покрытий, полученных методом магнетронного распыления [28, 29], и микрокристаллов ГА [31], положенными в обоснование механизма их пластической деформации в условиях сосредоточенной нагрузки (межкластерное проскальзывание как аналог межзеренного проскальзывания в нанокристаллических материалах). При невозможности дислокационного механизма деформации кристаллического ГА реализации межкластерного проскальзывания способствует особенность структурной организации кристаллов ГА: наличие характерной структурной единицы – кластера Познера Ca9(PO4)6 [32].
В табл. 2 сведены результаты измерения твердости (Н), модуля упругости (Е) и доли пластической деформации (δ = Wplast/Wtotal) в работе индентирования исходного компакта и керамики после спекания при разной продолжительности отжига для нагрузки на индентор 5 и 100 мН.
Таблица 2.
Механические свойства керамики ГА при различной нагрузке на индентор
| Время ТО, ч | Н, ГПa | Е, ГПa | δ, % | Н, ГПa | Е, ГПa | δ, % |
|---|---|---|---|---|---|---|
| 5 мН | 100 мН | |||||
| 0 | 0.8 ± 0.3 | 13.4 ± 2.9 | 75.8 ± 3.1 | 0.4 ± 0.2 | 7.7 ± 2.5 | 79.3 ± 2.7 |
| 1 | 2.7 ± 0.4 | 57.3 ± 6.4 | 74.0 ± 3.4 | 3.1 ± 0.4 | 55.5 ± 8.9 | 74.4 ± 3.3 |
| 6 | 7.0 ± 1.6 | 87.3 ± 10.0 | 60.7 ± 4.5 | 4.6 ± 0.9 | 90.7 ± 10.2 | 72.1 ± 4.1 |
| 12 | 9.2 ± 1.8 | 127.3 ± 18.1 | 55.4 ± 4.9 | 6.9 ± 0.4 | 117.2 ± 10.6 | 66.0 ± 4.3 |
Наблюдаемое снижение твердости образцов керамики, отожженных в течение 6 и 12 ч, при увеличении максимальной нагрузки на индентор указывает на наличие более компактного (менее пористого) по сравнению с объемом образцов приповерхностного слоя субмикронной толщины. Твердость приповерхностного слоя образца, отожженного в течение 12 ч, достигает в отдельных испытаниях 11 ГПа и соответствует твердости микрокристаллов ГА [31], что может быть следствием большей плотности приповерхностного слоя керамики, поскольку поверхности раздела (и в первую очередь свободная поверхность) служат стоком свободного объема (нанопор) в процессе уплотнения керамики.
Рисунок 6 характеризует зависимость Н и Е от продолжительности отжига образцов и изменение плотности, отражающее стадийность процесса спекания. Закономерное увеличение твердости и модуля упругости коррелирует с уплотнением образцов в процессе спекания.
Рис. 6.
Зависимости Н (1), Е (2) (полученных при нагрузке на индентор 5 мН) и относительной плотности ρ (3) керамики ГА от времени отжига (τ).
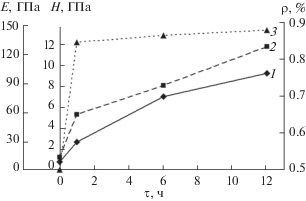
Доля пластической деформации в работе индентирования снижается по мере увеличения продолжительности отжига керамики и достигает после 12-часового отжига значений, характерных для нанокристаллических ГА-покрытий и компактной керамики [28].
Необходимо подчеркнуть, что оцененную твердость керамики с 50%-ной долей пор в объеме нельзя считать коррелирующей с зеренной субструктурой керамики, необходима ее коррекция с использованием статистики Вейбулла [33].
ЗАКЛЮЧЕНИЕ
В процессе компактирования реализуется процесс срастания наночастиц исходного порошка, обеспечивающий минимальную твердость компакта при его 50%-ной относительной плотности. Твердость и модуль упругости керамики ГА увеличиваются с увеличением времени отжига как следствие совершенствования ее субструктуры (увеличение размера зерен, уменьшение размера и количества пор). Относительно большая доля пластической деформации в работе индентировании плотной керамики подтверждает положение о ее высокой локальной пластичности и о кластерном механизме пластической деформации кристаллического ГА.
Список литературы
Rahaman M.N. Ceramic Processing and Sintering // Mater. Manufact. Proc. 1997. V. 12. № 3. P. 555–564. https://doi.org/10.1002/maco.19960470511
Barsoum M.W. Fundamentals of Ceramics. Boca Raton: CRC. 2003. 624 p.
Kang S.-J.L. Sintering: Densification, Grain Growth and Microstructure. Amsterdam: Elsevier, 2005. 265 p.
Raynaud S., Champion E. Bemache Assollant. Calcium Phosphate Apatites with Variable Ca/P Atomic Ratio II. Calcination and Sintering // Biomaterials. 2002. V. 23. № 4. P. 1073–1080.
Bemache-Assollant D., Ababou A., Champion E., Heughebaert M. Sintering of Calcium Phosphate Hydroxyapatite Ca10(PO4)6(OH)2 I. Calcination and Particle Growth // J. Eur. Ceram. Soc. 2003. V. 23. № 2. P. 229–241.
Putlayev V., Veresov A., Pulkin M., Soin A., Kuznetsov V. Silicon-substituted Hydroxyapatite Ceramics (Si-Hap): Densification and Grain Growth Through the Prism of Sintering Theories // Materialwissenschaft Werkstofftech. 2006. V. 37. № 6. P. 416–421. https://doi.org/10.1002/mawe.200600007
Kleebe H.J., Bres E.F., Bernache-Assolant D., Ziegler G. High-Resolution Electron Microscopy and Convergent-Bearn Electron Diffraction of Sintered Undoped Hydroxyapatite // J. Am. Ceram. Soc. 1997. V. 80. № 1. P. 37–44. https://doi.org/10.1111/j.1151-2916.1997.tb02788.x
Zhou J., Zhang X., Chen J., Zeng S., De Groot K. High Temperature Characteristics of Synthetic Hydroxyapatite // J. Mater. Sci.: Mater. Med. 1993. V. 4. № 1. P. 83–85. https://doi.org/10.1007/BF00122983
Raynaud S., Champion E., Bemache-Assollant D., Thomas P. Calcium Phosphate Apatites with Variable Ca/P Atomic Ratio I. Synthesis, Characterisation and Thermal Stability of Powders // Biomaterials. 2002. V. 23. № 4. P. 1065–1072. https://doi.org/10.1016/S0142-9612(01)00218-6
Liao C.J., Lin F.H., Chen K.S., Sun J.S. Thermal Decomposition and Reconstruction of Hydroxyapatite in Air Atmosphere // Biomaterials. 1999. V. 20. P. 1807–1813. https://doi.org/10.1016/S0142-9612(99)00076-9
Petrakova N.V., Lysenkov A.S., Ashmarin A.A., Egorov A.A., Fedotov A.Yu., Shvomeva L.I., Komlev V.S., Barinov S.M. Effect of Hot Pressing Temperature on the Microstructure and Strength of Hydroxyapatite Ceramic // Inorg. Mater.: Appl. Res. 2013. V. 4. № 4. P. 362–367. https://doi.org/10.1134/S2075113313040072
Pramanik S., Agarwal A.K., Rai K.N., Garg A. Development of High Strength Hydroxyapatite by Solid-State-Sintering Process // Ceram. Int. 2007. V. 33. № 3. P. 419–426. https://doi.org/10.1016/j.ceramint.2005.10.025
Goller G., Oktar F.N., Agathopoulos S., Tulyaganov D.U., Ferreira J.M.F., Kayali E.S., Peker I. Effect of Sintering Temperature on Mechanical and Micro Structural Properties of Bovine Hydroxyapatite (BHA) // J. Sol-Gel Sci. Technol. 2006. V. 37. № 3. P. 111–115. https://doi.org/10.1007/s10971-006-6428-9
Hervas I., Montagne A., Van Gorp A., Bentoumi M., Thuault A., Lost A. Fracture Toughness of Glasses and Hydroxyapatite: A Comparative Study of 7 Methods by Using Vickers Indenter // Ceram. Int. 2016. P. 42. P. 12 740–12 750. https://doi.org/10.1016/j.ceramint.2016.05.030
Song J., Liu Y., Zhang Y., Jiao L. Mechanical Properties of Hydroxyapatite Ceramics Sintered From Powders with Different Morphologies // Mater. Sci. Eng. A. 2011. V. 528. P. 5421–5427. https://doi.org/10.1016/j.msea.2011.03.078
Chen B., Zhang T., Zhang J., Lin Q., Jiang D. Microstructure and Mechanical Properties of Hydroxyapatite Obtained by Gel-Casting Process // Ceram. Int. 2008. V. 34. P. 359–364. https://doi.org/10.1016/j.ceramint.2006.10.021
Bose S., Dasgupta S., Tarafder S., Bandyopadhyay A. Microwave-Processed Nanocrystalline Hydroxyapatite: Simultaneous Enhancement of Mechanical and Biological Properties // Acta Biomater. 2010. V. 6. P. 3782–3790. https://doi.org/10.1016/j.actbio.2010.03.016
Фомин А.С., Баринов С.М., Иевлев В.М., Смирнов В.В., Михайлов Б.П., Кущев С.Б., Белоногов Е.К., Дроздова Н.А. Нанокристаллическая гидроксиапатитовая керамика // Неорган. материалы. 2009. Т. 45. № 10. С. 1271–1274. https://doi.org/10.1134/S0012500808010084
Wang P.E., Chaki T.K. Sintering Behaviour and Mechanical Properties of Hydroxyapatite and Dicalcium Phosphate // J. Mater. Sci.: Mater. Med. 1993. V. 4. № 2. P. 150–158. https://doi.org/10.1007/BF00120384
Pazarlioglu S., Salman S. Sintering Effect on the Micro Structural, Mechanical, and in Vitro Bioactivity Properties of a Commercially Synthetic Hydroxyapatite // J. Aust. Ceram. Soc. 2017. V. 53. P. 391–401. https://doi.org/10.1007/s41779-017-0048-4
Muralithran G., Ramesh S. The Effects of Sintering Temperature on the Properties of Hydroxyapatite // Ceram. Int. 2000. V. 26. P. 221–230. https://doi.org/10.1016/S0272-8842(99)00046-2
Herliansyah M.K., Hamdi M., Ide-Ektessabi A., Wildan M.W., Toque J.A. The Influence of Sintering Temperature on the Properties of Compacted Bovine Hydroxyapatite // Mater. Sci. Eng. C. 2009. V. 29. P. 1674–1680. https://doi.org/10.1016/j.msec.2009.01.007
Gibson R., Ke S., Best S.M., Bonfield W. Effect of Powder Characteristics on the Sinterability of Hydroxyapatite Powders // J. Mater. Sci.: Mater. Med. 2001. V. 12. P. 163–171. /https://doi.org/10.1023/A:1008930313194
Veljovic D., Zalite I., Palcevskis E., Smiciklas I., Petrovic R., Janac’kovic D. Microwave Sintering of Fine Grained HAP and HAP/TCP Bioceramics // Ceram. Int. 2010. V. 36. P. 595–603. https://doi.org/10.1016/j.ceramint.2009.09.038
Veljovic’ D., Palcevskis E., Dindune A., Putic’ S., Balac’ I., Petrovic’ R., Janac’kovic D. Microwave Sintering Improves the Mechanical Properties of Biphasic Calcium Phosphates from Hydroxyapatite Microspheres Produced from Hydrothermal Processing // J Mater. Sci. 2010. V. 45. P. 3175–3183. https://doi.org/10.1007/s10853-010-4324-8
Gua Y.W., Loha N.H., Khora K.A., Tora S.B., Cheang P. Spark Plasma Sintering of Hydroxyapatite Powders // Biomaterials. 2002. V. 23. P. 37–43.
Ramesh S., Tan C.Y., Tolouei R., Amiriyan M., Purbolaksono J., Sopyan I., Teng W.D. Sintering Behavior of Hydroxyapatite Prepared from Different Routes // Mater. Design. 2012. V. 34. P. 148–154.
Иевлев В.М., Костюченко А.В., Белоногов Е.К., Баринов С.М. Твердость и природа микропластичности гидроксиапатита // Неорган. материалы. 2013. Т. 49. № 4. С. 434–440. https://doi.org/10.1134/S0020168513040043
Иевлев В.М., Костюченко А.В., Даринский Б.М., Баринов С.М. Твердость и микропластичность нанокристаллических и аморфных фосфат-кальциевых покрытий // Физика твердого тела. 2014. Т. 56. № 2. С. 318–325. https://doi.org/10.1134/S1063783414020127
Peon E., Fuentes G., Delgado J.A., Morejon L., Almirall A., Garcia R. Preparation and Characterization of Porous Blocks of Synthetic Hydroxyapatite // Lat. Am. Appl. Res. 2004. V. 34. P. 225–228.
Viswanath B., Raghavan R., Ramamurty U., Ravishankar N. Mechanical Properties of Hydroxyapatite Single Crystals // Scr. Mater. 2007. V. 57. P. 361–364. https://doi.org/10.1016/j.scriptamat.2007.04.027
Posner A.S., Betts F. Synthetic Amorphous Calcium Phosphate and Its Relation to Bone Mineral Structure // Acc. Chem. Res. 1975. V. 8. P. 273–281. https://doi.org/10.1021/ar50092a003
Fan X., Case E.D., Ren F., Shu Y., Baumann M.J. Porosity Dependence of the Weibull Modulus for Hydroxyapatite and Other Brittle Materials // J. Mech. Behav. Biomed. Mater. 2012. V. 8. P. 21–36. https://doi.org/10.1016/j.jmbbm.2011.12.010
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы