

Неорганические материалы, 2020, T. 56, № 1, стр. 50-58
Синтез гидроксиапатита в присутствии оксиэтилидендифосфоновой кислоты и ионов Mg2+ в качестве ингибиторов кристаллизации
Н. В. Китикова 1, А. И. Иванец 1, *, И. Л. Шашкова 1
1 Институт общей и неорганической химии Национальной академии наук Беларуси
220072 Минск, ул. Сурганова, 9, корп. 1, Беларусь
* E-mail: Andreiivanets@yandex.ru
Поступила в редакцию 17.12.2018
После доработки 25.05.2019
Принята к публикации 19.06.2019
Аннотация
Осуществлен синтез гидроксиапатита (ГА) в присутствии оксиэтилидендифосфоновой кислоты и ионов Mg2+. Установлено влияние природы и концентрации ингибиторов кристаллизации, а также концентрации исходных реагентов на его физико-химические характеристики. Показана возможность регулирования кристаллической структуры и текстуры ГА путем направленного варьирования условий синтеза в присутствии малых концентраций ингибиторов кристаллизации (0.01–1.00 мол. %), что может быть использовано при получении материалов различного функционального назначения с улучшенными характеристиками.
ВВЕДЕНИЕ
Фосфаты кальция (ФК) широко применяются как материалы медицинского назначения, в качестве пищевых добавок, при решении экологических задач и др. Благодаря особенностям кристаллохимического строения и важной роли в различных биологических процессах наиболее изученным среди ФК является гидроксиапатит (ГА) [1]. Функциональные свойства ГА существенно зависят от способа и условий получения, определяющих его химический состав, кристаллическую структуру и характеристики пор, форму и размер частиц, морфологию поверхности. Наиболее активны в различных биологических и химических процессах ГА, обладающие низкой степенью кристалличности [2] и имеющие в составе замещенные ионы, что приводит к дестабилизации кристаллической структуры [3].
Наиболее распространенным способом получения ГА является метод осаждения из водных растворов, при котором физико-химические свойства ГА можно контролировать как варьированием условий его осаждения (температурой синтеза, рН реакционной среды, концентрацией реагентов, скоростью смешивания реагентов), так и введением в исходные растворы ингибиторов кристаллизации (ИКр). Кристаллизация ГА является чрезвычайно сложным процессом, состоящим из пяти стадий: 1) образование ионных кластеров и аморфного фосфата кальция (АФК); 2) стабилизация АФК; 3) переход от АФК к ГА через растворение и кристаллизацию; 4) рост кристаллов; 5) старение ГА в условиях, близких к равновесному состоянию [4, 5]. Применение ИКр оказывает различное влияние на протекание каждой из пяти стадий синтеза ГА [4, 6].
В качестве ингибиторов кристаллизации фосфатов кальция среди органических соединений ранее были изучены различные органические лиганды [7], полимеры [8]. Хорошо зарекомендовали себя также фосфонаты, характеризующиеся мостиком P–C–P и отличающиеся составом алкильного радикала, а среди неорганических – катионы некоторых металлов. При этом в большинстве работ приводятся результаты исследования кинетики подавления роста кристаллов в присутствии малых количеств фосфонатов [9, 10] либо функционализации поверхности ГА для придания новых свойств [11, 12]. Особый научный и практический интерес представляют комплексоны фосфонового ряда, содержащие фрагмент СН2РО3Н2. Такие соединения отличаются большей, чем у карбоксильных групп, электроотрицательностью и, соответственно, большей дентатностью, благодаря чему они образуют достаточно устойчивые комплексы с катионами кальция [13]. В литературе отсутствуют данные о применении фосфонатов для снижения степени кристалличности ГА с целью повышения его реакционной способности.
Действие катионов металлов можно рассматривать как модифицирование фосфатов кальция путем снижения степени кристаллизации ГА. Как показывают предыдущие исследования, этому способствуют ионы Zn2+ [14], Sr2+ [15], в то время как присутствие ионов Mn2+ приводит к формированию аморфной фазы [15]. Особенно детально изучено влияние ионов Mg2+, являющихся одними из основных заместителей ионов Ca2+ в составе костных тканей и играющих важную роль в биологических процессах [3, 4, 14–18].
Механизм действия данных ИКр, как правило, связывают со специфической адсорбцией вводимых реагентов на поверхности образующихся зародышей АФК, что препятствует их дальнейшей кристаллизации и тем самым повышает их реакционную способность [4, 10]. При этом изучены процессы образования ГА в широком интервале концентраций вводимых модификаторов, приводящих к изоморфным замещениям [3] или соосаждению различных фаз, как это обобщено Gozalian с соавторами [18] или получено Fadeev [16].
Учитывая многофакторность процесса кристаллизации и комплексное влияние условий синтеза на физико-химические свойства ГА, одной из актуальных задач при получении материалов различного функционального назначения на основе ГА является поиск оптимальной концентрации исходных реагентов в сочетании с минимальной концентрацией ИКр, обеспечивающих их высокую реакционную способность в различных процессах благодаря частичной аморфизации.
Целью настоящей работы является сравнительное исследование влияния малых концентраций (до 1.0 мол. %) органического – оксиэтилидендифосфоновой кислоты (ОЭДФ) – и неорганического (Mg2+) ИКр в зависимости от концентрации исходных реагентов на фазовый и химический состав ГА и его морфологию. Выбор веществ в качестве ИКр обусловлен их нетоксичностью и высокой ингибирующей способностью.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез ГА осуществляли химическим осаждением из растворов Ca(NO3)2 и H3PO4, доведенных раствором NH4OH до рН 10. Концентрации ионов Ca2+ (ССа = 0.083, 0.20 и 0.42 моль/л) и ${\text{РО}}_{4}^{{3 - }}$-ионов (СР = 0.05. 0.12 и 0.25 моль/л) в реакционных растворах после смешения соответствовали молярному соотношению ГА стехиометрического состава Са/Р = 1.67. Для изучения влияния ИКр на процесс осаждения ГА добавки Mg(NO3)2 вводили в Са-содержащий раствор из расчета 0.01, 0.1, 0.5 и 1.0 мол. % по отношению к ионам Са2+. Добавки ОЭДФ вводили в Р-содержащий раствор в таком же количестве, но по отношению к ${\text{РО}}_{4}^{{3 - }}$-ионам. Для получения образцов ГА P-содержащий раствор по каплям вводили в Са-содержащий раствор при постоянном перемешивании. Далее полученную суспензию оставляли на созревание в течение 24 ч без перемешивания с последующими отделением осадка на воронке Бюхнера и промывкой горячей водой и этанолом. Осадок ГА сушили на воздухе при комнатной температуре, затем при 100°С в течение 24 ч.
Рентгенофазовый анализ (РФА) и расчет параметров элементарной ячейки а и с, а также среднего размера кристаллитов τ были выполнены на рентгеновском дифрактометре D8 Advanced (Bruker, Германия) с использованием CuKα-излучения. Расчет τ по формуле Шеррера проводили по главному дифракционному пику 2θ = 31.7°–32.0°. Идентификацию фаз проводили по базе рентгенографических порошковых стандартов JCPDS PDF2. ИК-спектры снимали на ИК-Фурье-спектрометре Tenzor-27 в диапазоне 400–4000 см–1 (таблетки с KBr 800 мг, навеска 2 мг). Определение химического состава твердой фазы подробно описано в статье [19]. Элементный анализ выполнен с помощью JSM-5610 LV с системой энергодисперсионной рентгеновской спектроскопии (ЭДРС) JED-2201 JEOL (Japan).
Адсорбционные свойства и текстуру образцов оценивали из изотерм низкотемпературной (‒196°С) адсорбции–десорбции азота, измеренных объемным методом на анализаторе ASAP 2020 МР (Micromeritics, США). Удельную поверхность (Ауд) рассчитывали методом БЭТ (AБЭT) и одноточечным методом БЭТ (Asp), объем пор (Vпор) рассчитывали одноточечным методом по адсорбционной (Vsp. ads) и десорбционной (Vsp. des) ветвям изотермы. Средний диаметр пор для адсорбционной (Dsp. ads) и десорбционной (Dsp. des) ветвей изотермы рассчитывали по уравнению 4V/A. Перед анализом образцы вакуумировали в течение 1 ч при температуре 150°С и остаточном давлении 133.3 × 10–3 Па. Относительная ошибка определения Vпор составляла ±1%, Ауд и размера пор – ±15%.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно данным химического анализа, образцы ГА, синтезированные без ИКр, характеризуются молярным соотношением Са/Р в интервале 1.63–1.67, что свидетельствует об образовании стехиометрического и Са-дефицитного ГА (Са-дГА) (табл. 1). В случае введения ионов Mg2+ молярное соотношение Са/Р практически не зависит от концентрации исходных растворов. Содержание ионов Mg2+ не определяли, поскольку, согласно данным [16], можно считать, что при их концентрации в растворе до 10 ат. % сохраняется соотношение Ca/Mg в твердой фазе. ОЭДФ оказывает существенное влияние на химический состав. По мере снижения концентрации исходных растворов и увеличения количества вводимой ОЭДФ молярное соотношение Са/Р уменьшается до 1.56.
Таблица 1.
Химический состав по данным химического анализа и кристаллографические характеристики синтезированных образцов ГА
| Образец | СР в исходных растворах, моль/л | Количество ингибитора, мол. % |
Содержание элементов, ммоль/г | Молярное соотношение Са/Р | Параметры кристаллической структуры | τ, нм | ||
|---|---|---|---|---|---|---|---|---|
| Ca (Ca + Mg)* | Р | а, нм | с, нм | |||||
| Образцы без ингибитора | ||||||||
| ГА** | 9.96 | 5.98 | 1.67 | 0.942 | 0.688 | |||
| 1 | 0.05 | 0 | 9.14 | 5.62 | 1.63 | 0.944 | 0.687 | 8.86 |
| 6 | 0.12 | 0 | 9.32 | 5.59 | 1.67 | 0.947 | 0.689 | 9.09 |
| 11 | 0.25 | 0 | 9.12 | 5.48 | 1.66 | 0.948 | 0.689 | 8.44 |
| Ингибитор – Mg2+ | ||||||||
| 2 | 0.05 | 0.10 | 9.39 | 5.71 | 1.64 | 0.947 | 0.689 | 8.14 |
| 3 | 0.05 | 1.00 | 9.29 | 5.71 | 1.63 | 0.948 | 0.688 | 8.14 |
| 7 | 0.12 | 0.10 | 9.26 | 5.43 | 1.64 | 0.949 | 0.689 | 8.40 |
| 8 | 0.12 | 1.00 | 9.24 | 5.34 | 1.64 | 0.947 | 0.688 | 8.14 |
| 12 | 0.25 | 0.10 | 9.12 | 5.57 | 1.64 | 0.947 | 0.688 | 8.61 |
| 13 | 0.25 | 1.00 | 9.07 | 5.56 | 1.64 | 0.948 | 0.688 | 8.13 |
| Ингибитор – ОЭДФ | ||||||||
| 4 | 0.05 | 0.10 | 9.09 | 5.77 | 1.57 | 0.945 | 0.688 | 8.69 |
| 5 | 0.05 | 1.00 | 8.92 | 5.76 | 1.56 | – | – | – |
| 9 | 0.12 | 0.10 | 9.34 | 5.61 | 1.66 | 0.948 | 0.690 | 9.26 |
| 10 | 0.12 | 1.00 | 9.06 | 5.73 | 1.58 | 0.951 | 0.688 | 8.14 |
| 14 | 0.25 | 0.10 | 9.19 | 5.49 | 1.67 | 0.947 | 0.689 | 8.68 |
| 15 | 0.25 | 1.00 | 8.93 | 5.31 | 1.68 | 0.947 | 0.688 | 8.34 |
Результаты химического анализа хорошо согласуются с данными РФА. Так, образцы, синтезированные без ИКр, представляют собой ГА различной степени кристалличности, снижающейся с уменьшение концентрации исходных растворов (рис. 1), о чем свидетельствуют уменьшение интенсивности рефлексов и их разрешения, а также слияние рефлексов 211, 112 и 300. Введение в исходную реакционную смесь ионов Mg2+ в изученном диапазоне концентраций не отражается на фазовом составе продуктов при сохранении общей тенденции снижения степени кристалличности с уменьшением концентрации исходных растворов. В отличие от неорганического ИКр в случае ОЭДФ при синтезе из разбавленных растворов наблюдается аморфизация ГА. При максимальном содержании ОЭДФ полученный продукт представляет собой рентгеноаморфное соединение (рис. 1а, дифрактограмма 5), характеризующееся молярным соотношением Са/Р = 1.56, что наиболее близко по составу к трикальцийфосфату (ТКФ).
Рис. 1.
Рентгенограммы образцов, синтезированных без введения ингибиторов кристаллизации и в их присутствии из исходных растворов с концентрацией по фосфору 0.05 (a), 0.12 (б) и 0.25 моль/л (в) (нумерация образцов по табл. 1).
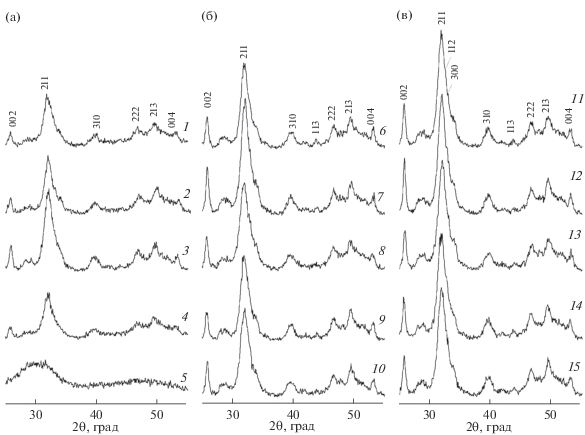
Изменение химического состава ГА отражается и на параметрах элементарной ячейки, особенно на величине а, существенно отличающихся от справочных данных для стехиометрического ГА (табл. 1). Это свидетельствует о различной степени разупорядоченности структуры. Причем при синтезе без ИКр отклонение значений параметров а и с увеличивается с ростом концентрации исходных растворов. В случае применения ИКр их влияние немного усиливается при уменьшении концентрации, а в исходных растворах наибольшей концентрации практически не проявляется.
Наиболее заметно различие влияния ионов Mg2+ и ОЭДФ на размеры кристаллитов. При использовании ионов Mg2+ независимо от концентрации исходных растворов происходит значительное снижение τ по сравнению с образцами, полученными как в присутствии ОЭДФ, так и без ИКр.
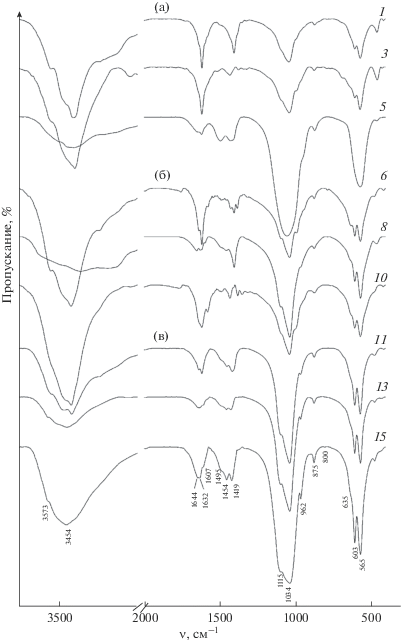
ИК-спектры позволяют уточнить состав синтезированных ФК (рис. 2). B спектрах всех образцов присутствуют полосы, характеризующие колебания ${\text{РО}}_{4}^{{3 - }}$-групп – деформационные в области 600–500 см–1 и валентные (в области 1100–1000 см–1) [19]. Характерные для ГА либрационные (около 630 см–1) и валентные колебания (около 3547 см‑1) структурных ОН-групп отсутствуют либо проявляются в виде плеча, что может быть обусловлено низкой степенью кристалличности [5]. Слабые полосы при 875 см–1 в сочетании с колебаниями в области 1620 см–1 отражают присутствие гидрофосфатных групп ${\text{НРО}}_{4}^{{2 - }},$ характерных для Са-дГА [7, 20]. Причем увеличение интенсивности полосы при 875 см–1 с ростом количества вводимых ИКр говорит об усилении отклонения состава ГА от стехиометрического. Полосы в области 1400–1430 см–1 обычно относят к ${\text{CO}}_{3}^{{2 - }}$-группам, которых трудно избежать при проведении синтеза на воздухе при высоком значении рН. Характерное для ГА расщепление полосы в области 600–500 см–1 сокращается при введении ОЭДФ и практически отсутствует для образца, полученного из разбавленного раствора с добавлением 1.0 мол. % ОЭДФ (рис. 2а, спектр 5), что соответствует рентгеноаморфному состоянию ТКФ. При увеличении исходных концентраций и введении 1% ОЭДФ на ИК-спектре (рис. 2в, спектр 15) заметна слабая полоса около 800 см–1, характеризующая колебания связи Р–С в структуре ОЭДФ [19].
Рис. 2.
ИК-спектры образцов, синтезированных без введения ингибиторов кристаллизации и в их присутствии из исходных растворов с CP = 0.05 (a), 0.12 (б) и 0.25 моль/л (в) (нумерация образцов по табл. 1).
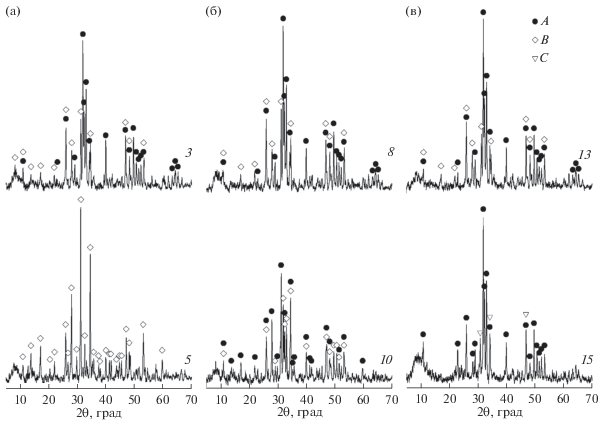
Данные ИК-спектроскопии подтверждаются результатами РФА этих образцов после их термообработки (ТО) при 1000°С (рис. 3). В большинстве случаев рентгенограммы свидетельствуют об образовании смеси ГА и ТКФ, что типично для Са-дГА при его ТО [20]. При этом рентгенограммы образцов ГА, синтезированных с добавлением ионов Mg2+, показывают, что с увеличением концентрации исходных растворов в присутствии данного ИКр происходит уменьшение доли ТКФ с 12 до 8%, что согласуется с результатами РФА исходных образцов (рис. 1) и литературными данными, согласно которым при увеличении содержания ионов Mg2+ до 1% доля ТКФ возрастает до 16% [17]. Добавление ОЭДФ в реакционный раствор приводит к кардинальным изменениям образующихся продуктов, зависящим от концентрации исходных растворов: в наиболее концентрированных растворах происходит формирование ГА с наименьшим содержанием примесной фазы (рис. 3, дифрактограмма 15), а в наиболее разбавленных растворах наблюдается формирование однофазного ТКФ (рис. 3, дифрактограмма 5).
Рис. 3.
Рентгенограммы термообработанных при 1000°С образцов ГА, синтезированных в присутствии добавок ионов Mg2+ и ОЭДФ из исходных растворов с CP = 0.05 (a), 0.12 (б) и 0.25 моль/л (в): A – ГА [76-694], B – Са3(РО4)2 (ромбоэдрическая сингония) [70-2065], C – Са3(РО4)2 (моноклинная сингония) [29-359] (нумерация образцов по табл. 1).
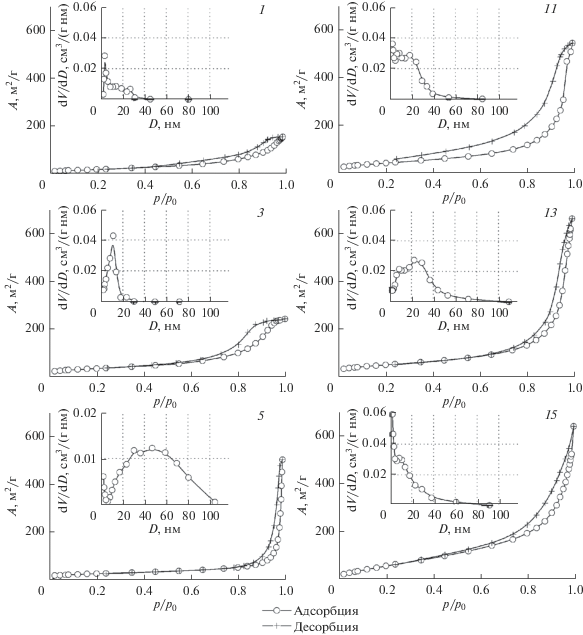
Изменения условий синтеза в присутствии ИКр приводят к варьированию адсорбционно-структурных характеристик ГА (табл. 2). Изотермы адсорбции–десорбции азота образцов ФК (рис. 4) относятся к IV типу по классификации ИЮПАК, характеризующему мезопористые тела [21]. Образцы ГА, полученные без ИКр и в присутствии ионов Mg2+ из разбавленных растворов, имеют схожий вид изотерм с петлей гистерезиса типа Н2, характеризующий сорбенты, поры которых образуют неоднородные по размеру и форме частицы (рис. 4, кривые 1, 3, 11). Для изотерм образцов ГА, полученных из наиболее концентрированных исходных растворов в присутствии и Mg2+, и ОЭДФ (рис. 4, кривые 13, 15), а также образца, синтезированного из разбавленного раствора в присутствии ОЭДФ (рис. 4, кривая 5), при высоких относительных давлениях характерна узкая петля гистерезиса, относящаяся к типу Н1 и указывающая на однородность частиц и пор. Увеличение крутизны средней части изотермы при больших относительных давлениях (p/p0 = 0.4–0.9), связанное с капиллярной конденсацией в мезопорах, говорит об увеличении поверхности мезопор. Расчеты адсорбционных характеристик, произведенные по полученным изотермам двумя способами (табл. 2), близки между собой и показывают, что в отсутствие ИКр с увеличением концентрации исходных растворов наблюдается рост Aуд и Vпор при среднем размере пор 13–21 нм. Использование ионов Mg2+ в качестве ИКр приводит к увеличению как Aуд, так и Vпор по сравнению с исходными образцами, полученными без ИКр. При этом эффект применения ионов Mg2+ снижается по мере увеличения концентрации исходных растворов. Введение ОЭДФ в разбавленных исходных растворах способствует значительному увеличению Vпор и их размера, а в наиболее концентрированных растворах – росту Aуд при одновременном уменьшении размера пор, что может быть обусловлено увеличением размера структурообразующих частиц.
Таблица 2.
Адсорбционные и текстурные свойства образцов, полученных без добавок ингибиторов и с введением 1 мол. % Mg2+ и ОЭДФ
| Ингибитор | Удельная поверхность, м2/г | Объем пор, см3/г | Размер пор, нм | |||
|---|---|---|---|---|---|---|
| Asp | AБЭT | Vsp. ads | Vsp. des | Dsp. ads | Dsp. des | |
| СР = 0.05 М | ||||||
| – | 72 | 75 | 0.233 | 0.242 | 13 | 13 |
| Mg | 123 | 127 | 0.375 | 0.367 | 12 | 12 |
| ОЭДФ | 75 | 79 | 0.516 | 0.735 | 28 | 39 |
| СР = 0.12 М | ||||||
| – | 119 | 125 | 0.373 | 0.366 | 13 | 12 |
| Mg | 176 | 184 | 0.743 | 0.766 | 17 | 18 |
| ОЭДФ | 81 | 90 | 0.361 | 0.346 | 18 | 17 |
| СР = 0.25 М | ||||||
| – | 155 | 162 | 0.791 | 0.834 | 20 | 21 |
| Mg | 172 | 179 | 0.925 | 0.983 | 22 | 23 |
| ОЭДФ | 207 | 251 | 0.779 | 0.854 | 15 | 17 |
Рис. 4.
Изотермы адсорбции–десорбции азота (А–р/рo) и кривые дифференциального распределения пор по размерам (dV/dD–D) образцов, полученных в отсутствие ингибиторов и при максимальном их содержании в исходных растворах с CP = 0.05 (1, 3, 5) и 0.25 моль/л (11, 13, 15) (нумерация образцов по табл. 1).
Наблюдаемое различие влияния ИКр различной природы можно объяснить их воздействием на процесс кристаллизации и взаимодействием с исходными компонентами. Так, ионы Mg2+ адсорбируются на образующихся первичных частицах ФК и препятствуют их дальнейшему росту, что приводит к снижению степени их кристалличности и размера кристаллитов (табл. 1), а также росту Aуд (табл. 2). В отличие от Mg2+ молекулы ОЭДФ способны образовывать сложные комплексы, захватывая несколько ионов Са2+, в результате чего происходят рост первичных частиц [13] и увеличение размера пор. Полученные данные подтверждают, что такие комплексы наиболее активно образуются при синтезе из разбавленных исходных растворов. Согласно данным РФА, в этом случае образуется аморфный продукт. С ростом концентрации исходных растворов происходит адсорбция молекул ОЭДФ, препятствующих дальнейшему росту первичных частиц, в результате чего увеличивается Ауд, снижается размер пор и возрастает их общий объем.
Результаты элементного анализа по данным ЭДРС подтверждают присутствие ИКр в составе образующихся продуктов. Наличие ионов Mg2+ детектируется в образцах, полученных в более концентрированных растворах, в количестве 0.06 ммоль/г при добавлении ИКр 0.1 мол. % и 0.15–0.29 ммоль/г при добавлении 1.0 мол. %. На присутствие ОЭДФ указывает наличие углерода в составе образцов, содержание которого увеличивается с ростом концентрации исходных растворов от 1.11 до 5.86 ммоль/г при введении 0.1 мол. % ИКр и от 5.15 до 8.74 ммоль/г при введении 1 мол. %. Часть углерода может относиться к ${\text{СО}}_{3}^{{2 - }}$-группам, о чем свидетельствуют ИК-спектры (рис. 2), однако даже при направленном синтезе ГА c содержанием 7.8 мас. % ${\text{СО}}_{3}^{{2 - }}$ количество углерода составляет 1.3 ммоль/г по [22].
ЗАКЛЮЧЕНИЕ
Изучены особенности фазового и химического состава ГА и его морфологии при проведении синтеза в интервале концентраций исходных растворов 0.05–0.25 моль/л по ${\text{РО}}_{4}^{{3 - }}$-ионам при молярном соотношении Са/Р = 1.67 и присутствии ионов Mg2+ и ОЭДФ как ИКр в количестве 0.01–1.0 мол. %. Установлено, что ингибирующий эффект ионов Mg2+ заключается в ограничении роста кристаллитов, способствующего формированию развитой поверхности, и усиливается с ростом концентрации исходных растворов и количества вводимой добавки. Введение ОЭДФ – сильного комплексообразователя – в разбавленные исходные растворы вызывает рост агломератов, что приводит к снижению степени кристалличности и в конечном итоге при максимальной дозе – аморфизации продукта и образованию ТКФ с развитой мезопористой структурой. При увеличении концентрации исходных растворов ОЭДФ препятствует росту кристаллов, что обеспечивает рост Ауд и Vпор.
Таким образом, применение ионов Mg2+ и ОЭДФ в качестве ИКр способствует формированию высокопористых ФК в широком диапазоне концентраций исходных растворов. Полученные данные могут быть использованы для регулирования кристаллической структуры и текстуры ГА при получении материалов различного функционального назначения с улучшенными характеристиками.
Список литературы
Haider A., Haider S., Han S.S., Kang I.-K. Recent Advances in the Synthesis, Functionalization and Biomedical Applications of Hydroxyapatite: a Review // RSC Adv. 2017. V. 7. P. 7442–7458.
Stötzel C., Müller F.A., Reinert F. et al. Ion Adsorption Behaviour of Hydroxyapatite with Different Crystallinities // Colloids Surf., B. 2009. V. 74. P. 91–95.
Bigi A., Boanini E., Gazzano M. Ion substitution in biological and synthetic apatites. Biomineralization and Biomaterials / Eds Aparicio C., Gineba M.-P. Amsterdam: Elsevier, 2016. Chapter 7. P. 235–266.
Ding H., Pan H., Xu X., Tang R. Towards a Detailed Understanding of Magnesium ions on Hydroxyapatite Crystallization Inhibition // Cryst. Growth Des. 2014. V. 14. P. 763–769.
Китикова Н.В., Шашкова Н.В., Зонов Ю.Г. и др. Влияние фазовых превращений в процессе синтеза кальцийдефицитного гидроксиапатита на его химический состав и структуру // Неорган. материалы. 2007. Т. 43. № 10. С. 1246–1255.
Bertran O., del Valle L.J., Revilla-Lopez G. et al. Synergistic Approach to Elucidate the Incorporation of Magnesium Ions into Hydroxyapatite // Chem. Eur. J. 2015. V. 21. P. 2537–2546.
Van der Houwen J.A.M., Cressey G., Cressey B.A., Valsami-Jones E. The effect of organic ligands on the crystallinity of calcium phosphate // J. Cryst. Growth. 2003. V. 249. P. 572–583.
Shimabayashi S., Uno T. Crystal Growth of Calcium Phosphates in the Presence of Polymeric Inhibitors. Calcium Phosphate in Biological and Industrial Systems / Ed. Amjad Z. N.Y.: Kluwer Academic Publishers, 1998. Chapter 9. P. 195–215.
Zieba A., Sethuraman G., Perez F. et al. Influence of Organic Phosphonates on Hydroxyapatite Crystal Growth Kinetics // Langmuir. 1996. V. 12. P. 2853–2858.
Amjad Z. The Influence of Polyphosphates, Phosphonates, and Poly(carboxylic acids) on the Crystal Growth of Hydroxyapatite // Langmuir. 1987. V. 3. P. 1063–1069.
Agougui H., Aissa A., Maggi S., Debbabi M. Phosphonate-Hydroxyapatite Hybrid Compounds Prepared by Hydrothermal Method // Appl. Surf. Sci. 2010. V. 257. P. 1377–1382.
Daniels Y., Lyczko N., Nzihou A., Alexandratos S.D. Modification of Hydroxyapatite with Ion-Selective Complexants: 1-hydroxyethane-1,1-Diphosphonic Acid // Ind. Eng. Chem. Res. 2015. V. 54. № 2. P. 585–596.
Дятлова Н.М., Темкина В.Я., Попов К.И. Комплексоны и комплексонаты металлов. М.: Химия, 1988. 544 с.
Kanzaki N., Onuma K., Treboux G. et al. Inhibitory Effect of Magnesium and Zinc on Crystallization Kinetics of Hydroxyapatite (0001) Face // J. Phys. Chem. B. 2000. V. 104. P. 4189–4194.
Bracci B., Torricelli P., Panzavolta S. et al. Effect of Mg2+, Sr2+, and Mn2+ on the Chemico-Physical and in vitro Biological Properties of Calcium Phosphate Biomimetic Coatings // J. Inorg. Biochem. 2009. V. 103. P. 1666–1674.
Фадеева И.В., Шворнева Л.И., Баринов С.М., Орловский В.П. Синтез и структура магнийсодержащих гидроксиапатитов // Неорган. материалы. 2003. Т. 39. № 9. С. 1102–1105.
Кубарев О.Л., Баринов С.М., Комлев В.С. Распределение магния при синтезе бифазных фосфатов кальция // Докл. АН. 2008. Т. 418. № 4. С. 497–499.
Gozalian A., Behnamghader A., Daliri M., Moshkforoush A. Synthesis and Thermal Behavior of Mg-Doped Calcium Phosphate Nanopowders via the Sol Gel Method // Sci. Iran. F. 2011. V. 18. P. 1614–1622.
Иванец А.И., Китикова Н.В., Шашкова И.Л., Кульбицкая Л.В. Стабильность кальциевых и магниевых фосфатных сорбентов в растворах хлорида кальция и комплексообразующих реагентов // Журн. прикл. химии. 2015. Т. 88. № 2. С. 227–233.
Dorozhkina E.I., Dorozhkin S.V. Mechanism of the Solid-State Transformation of a Calcium-Deficient Hydroxyapatite (CDHA) into Biphasic Calcium Phosphate (BCP) at Elevated Temperatures // Chem. Mater. 2002. V. 14. P. 4267–4272.
Thommes M., Kaneko K., Neimark A.V. et al. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Thechnical Report) // Pure Appl. Chem. 2015. V. 87. P. 1051–1069.
Barralet J.E., Best S.M., Bonfield W. Effect of Sintering Parameters on the Density and Microstructure of Carbonate Hydroxyapatite // J. Mater. Sci.–Mater. Med. 2000. V. 11. P. 719−724.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы