

Неорганические материалы, 2021, T. 57, № 1, стр. 47-56
Интеркалирование гептамолибдата аммония в межслоевое пространство природных алюмосиликатов
В. Д. Кошевар 1, *, В. Г. Шкадрецова 1
1 Институт общей и неорганической химии Национальной академии наук Беларуси
220072 Минск, ул. Сурганова, 9, корп. 1, Беларусь
* E-mail: koshevar@igic.bas-net.by
Поступила в редакцию 02.04.2020
После доработки 27.08.2020
Принята к публикации 31.08.2020
Аннотация
Осуществлено интеркалирование бентонита как путем его одностадийной обработки раствором гептамолибдата аммония ((NH4)6Mo7O24 ⋅ 4Н2O) с добавками лимонной кислоты, так и в двухстадийном процессе – при воздействии на первой стадии парами диметилсульфоксида. В первом случае межплоскостное расстояние (d001) бентонита было увеличено от 16.2 до 21.8 Å, а во втором – до 23 Å. При этом степень внедрения гептамолибдата аммония составила 36–40 мас. %. Интеркалаты на основе каолина с d001 = 10.98 Å при 2θ = 7.52° ввиду жесткости сочленения его структурных пакетов получены только в двухстадийном процессе. Степень внедрения прекурсоров в структуру каолина – 17 мас. %, а температурная стойкость соединения внедрения – до 220°С.
ВВЕДЕНИЕ
В последнее время количество работ, посвященных исследованию локализации соединений металлов с переходной валентностью в микро- и мезопорах (“нанореакторах”) и их фото- и термохимического превращения, сопровождающихся синтезом наночастиц, постоянно растет. Основная задача при использовании таких “нанореакторов” – предотвратить слияние и рост твердых частиц, образующихся при фото- или термолизе этих соединений, и способствовать проявлению уникальных эффектов, связанных с этим: сдвиг полос спектров поглощения, люминесценции, кардинальное изменение электрических, магнитных, адсорбционных, каталитических других свойств [1–5]. Ряд публикаций, касающихся этого направления исследований, посвящены получению и преобразованию соединений, внедренных не только в микрополости, но и в мицеллярные или микроэмульсионные среды [6–9]. Таким путем были получены нанокластеры оксидов меди, сульфида кадмия, золота и т.д. [8, 9]. Оригинальные статьи, посвященные исследованию механизма внедрения соединений d-элементов VI группы в межслоевое пространство слоистых природных минералов, почти не встречаются, хотя применение таких матриц для синтеза наночастиц актуально благодаря широкой возможности и доступности внедрения в их структуру различных соединений.
Целью настоящей работы является исследование механизма интеркалирования гептамолибдата аммония (ГМА) и сопутствующих прекурсоров в межплоскостные области природных алюмосиликатов для последующего твердофазного синтеза полиоксометаллатных комплексов молибдена с нетипичной химией, обусловленной многочисленными валентными состояниями, и получения путем их фото- и термического превращения наночастиц оксидов этого металла, имеющих высокие адсорбционные и каталитические свойства, в частности, при селективном окислении низших олефинов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При исследовании использовали каолин обогащенный марки ПР-2 (ГОСТ 21285-75) и бентонит ЕВ (Бельгия), содержащий 90% монтморрилонита, со средними размерами частиц 5.0 и 0.5 мкм соответственно. В качестве интеркалантов использовали раствор гептамолибдата аммония ((NH4)6Mo7O24 ⋅ 4Н2O) с добавками лимонной кислоты (электродонорного и комплексообразующего агента), а также диметилсульфоксид (ДМСО) квалификации “ч. д. а”. Для интеркалирования алюмосиликатов путем пропитки в водной среде была использована композиция следующего состава (г/л): гептамолибдат аммония – 200, лимонная кислота – 200, вода – остальное (ГМА-Л). Выбор такого состава был обусловлен данными [10, 11], которые свидетельствуют о том, что в смешанных водных растворах такого состава, а также в пропитанных ими образцах целлюлозных бумаг и пленок при УФ-облучении формировались полиоксометаллатные комплексы Мо (молибденовые “сини”) с максимумом поглощения в области 700–710 нм.
Навеску алюмосиликатов массой 100 г, предварительно прогретую в течение 2 ч при температуре при 200°С с целью удалении физически адсорбированной воды, обрабатывали при комнатной температуре ГМА-Л объемом 140 мл при периодическом перемешивании в течение 2 ч. Осадок отделяли от маточного раствора фильтрованием на воронке Бюхнера и сушили в затемненном помещении при 25°С до постоянной массы. При двухстадийном процессе интеркалирования, по аналогии с [6], образцы алюмосиликатов на первой стадии выдерживали в течение суток в специальной камере, насыщенной парами ДМСО, а затем сразу же подвергали жидкофазной обработке раствором ГМА-Л.
Контроль за изменениями, происходящими в обработанных таким образом алюмосиликатах, осуществляли методами рентгенографического анализа, термического анализа и ИК-Фурье-спектроскопии (ИК-спектроскопия).
Съемку порошковых дифрактограмм выполняли на дифрактометре ДРОН-3 с использованием CuKα-излучения при скорости съемки 1 град/мин при напряжении 30 кВ и анодном токе 15 мА. Фазовую идентификацию образцов проводили с использованием программного пакета WinXpow (Version 1.04, Yan.-1999) и базы рентгенографических порошковых стандартов JCPDS PDF2 (Version 1.21, may-1999 (пополнена в 2005)). Количественное содержание идентифицированных фаз в образцах рентгенографическим методом определяли с помощью программы бесстандартного количественного анализа многофазных порошковых образцов Quan, входящей в состав программного пакета WinXpow. В используемой программе реализован метод корундовых чисел, в котором оценку количественного состава смеси проводят с использованием корундового числа RIR (Reference Intensity Ratio) по методу Чанга.
Кривые термического анализа снимали на дериватографе Q 1500D (фирма MOM, система Паулик–Паулик–Эрдей) в температурном интервале 20–900°С на воздухе. Масса навески составляла 200 мг, скорость подъема температуры – 5°С/мин. В качестве эталона использовали прокаленный оксид алюминия “х. ч.”.
ИК-спектроскопические исследования проводили на ИК-спектрометре с Фурье-преобразованием М 2000 Series фирмы MTDAC (США) в области 400–4000 см–1 с разрешением 4 см–1. Зарегистрированные спектры обрабатывали с помощью программы Grams/32 фирмы Galactic (США).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
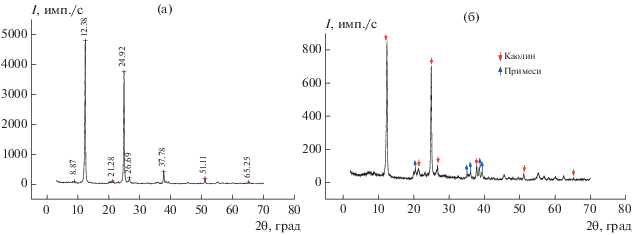
На дифрактограммах каолина, термообработанного при 200°С (рис. 1а), наблюдаются рефлексы по базальным отражениям со значениями межплоскостных расстояний d в Å: 7.13 [001] (2θ = 12.38°), 3.56 [002] (2θ = 24.92), 1.784 [004] (2θ = 37.8). Воздействие на данный глинистый минерал раствором ГМА-Л в указанных выше условиях не приводит к изменению положения основных рефлексов, а только к снижению их интенсивностей, что может свидетельствовать об отсутствии проникновения данного прекурсора в межпакетную область и образования интеркалатов внедрения (рис. 1б).
Рис. 1.
Дифрактограммы термообработанного каолина (а) и каолина после 2-часовой пропитки в ванне, содержащей ГМА-Л (б) (примеси: кварц, гидрослюда, полевой шпат).
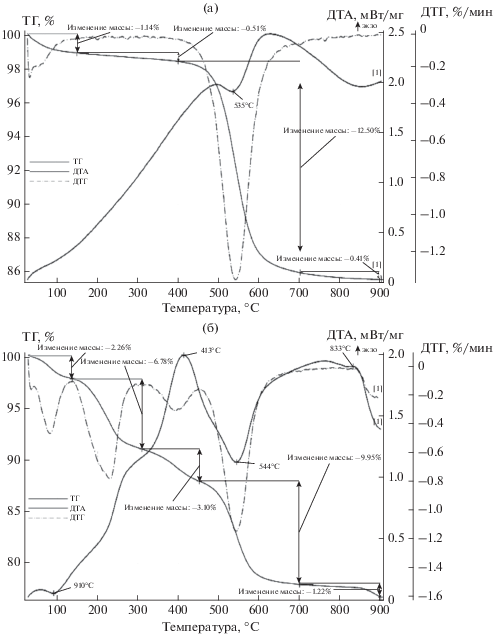
Результаты термического анализа образца каолина после обработки составом ГМА-Л (рис. 2б) существенно отличаются от данных контрольного образца (рис. 2а). Так, появляется эндоэффект при 91°С, вызванный, по-видимому, удалением кристаллогидратной воды ГМА-Л. Наблюдали смещение экзоэффекта, связанного с выделением аммиака при разложении индивидуального ГМА (352°С), в более высокотемпературную область (413°С), а также смещение эндоэффекта, вызванного потерей структурной воды каолина, от 535 к 544°С, что свидетельствует об адсорбции ГМА-Л поверхностью каолина с образование прочного комплекса.
Рис. 2.
Результаты термического анализа исходного каолина (а) и обработанного жидкофазным ГМА-Л (б).
Формирование такого комплекса подтверждается данными ИК-спектроскопии. В частности, понижаются интенсивности пиков в области 3620–3694 см–1 и при 1670 см–1, соответствующих валентным и деформационным колебаниям структурных ОН-групп каолина. Наблюдаются полосы при 3143 и 3001 см–1, принадлежащие первичной и связанной группам N–H и NH2 ГМА (но с некоторым смещением). Обнаружено поглощение в области 1721 см–1, которое можно отнести к валентным колебаниям кислотных групп (–СООН) лимонной кислоты.
Значительные различия в исследуемых образцах наблюдаются в случае применения двухстадийного интеркалирования каолина с обработкой его на 1-й стадии парами ДМСО. Так, приведенная на рис. 3а дифрактограмма свидетельствует о внедрении ДМСО в межплоскостное пространство каолина с образованием интеркалата каолин–ДМСО, что подтверждается появлением пика с d = 10.98 Å [001] при 2θ = 7.52°. Рефлекс же, соответствующий базальному отражению исходного каолина с d = 7.13 Å [001] при 2θ = 12.38°, заметно ослабевает, а с увеличением длительности интеркалирования полностью исчезает. Из результатов термического анализа каолина, интеркалированного ДМСО (не приводятся по причине ограничения объема статьи), следует, что в результате воздействия его паров образуется достаточно устойчивый интеркалат с содержанием внедренного ДМСО до 17%, который начинает распадаться при 220°С.
Рис. 3.
Дифрактограммы каолина, подвергнутого двухстадийному интеркалированию сначала ДМСО (а), а затем жидкофазным ГМА-Л (б).
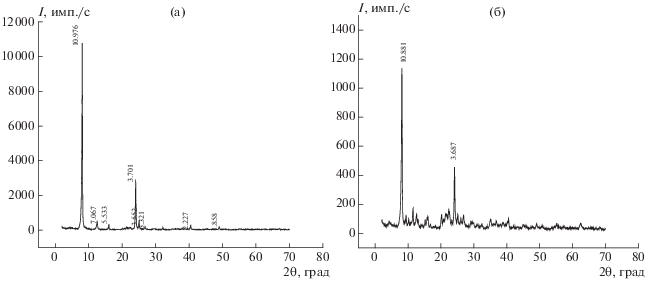
Проведение второй стадии интеркалирования каолина с использованием ГМА-Л (рис. 3б) не влияет на положение основного рефлекса интеркалата каолин–ДМСО, а только несколько снижает его интенсивность, что может говорить о частичном вытеснении ДМСО с образованием более сложного по составу интеркалата каолин–ДМСО-ГМА-Л.
ИК-спектры каолина до и после его обработки в двухстадийном процессе также подтверждают образование интеркалатов. В частности, зарегистрировано понижение интенсивности полос структурных и поверхностных ОН-групп и их смещение в область 3502 см–1, а также уменьшение интенсивности полосы при 1632 см–1, принадлежащей деформационным колебаниям ОН-групп. Кроме того, появляются пики в области 2852–2920 см–1, обусловленные валентными колебаниями СН3-групп ДМСО. Возникает также новая полоса при 3573 см–1, интенсивность которой возрастает с увеличением длительности интеркалирования.
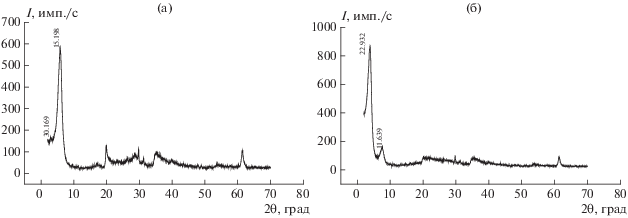
Характер интеркалирования другого алюмосиликата (бентонита) заметно отличается от аналогичного процесса с участием каолина. На рис. 4 представлены дифрактограммы исходного бентонита в сухом состоянии и обработанного жидким ГМА-Л. Из рис. 4а следует, что для данного минерала в исходном состоянии наиболее сильный рефлекс 001 соответствует межплоскостному расстоянию d = 16.2 Å при 2θ = 4.96°. После воздействия прекурсоров в течение 1 ч при 20°С d основного базального отражения увеличивается до 18.9 Å, что свидетельствует об образовании интеркалата бентонит–ГМА-Л. Увеличение времени пропитки при комнатной температуре до 2 ч приводит к образованию интеркалата с d = 21.8 Å (рис. 4б). Однако следует заметить, что процесс интеркалирования за это время сопровождается заметной аморфизацией минерала, о чем свидетельствует появление на дифрактограммах выраженного галло.
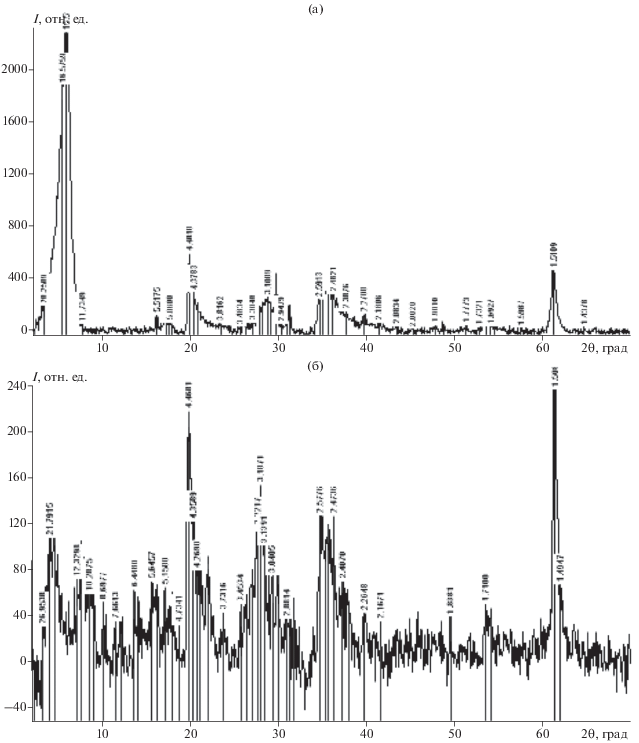
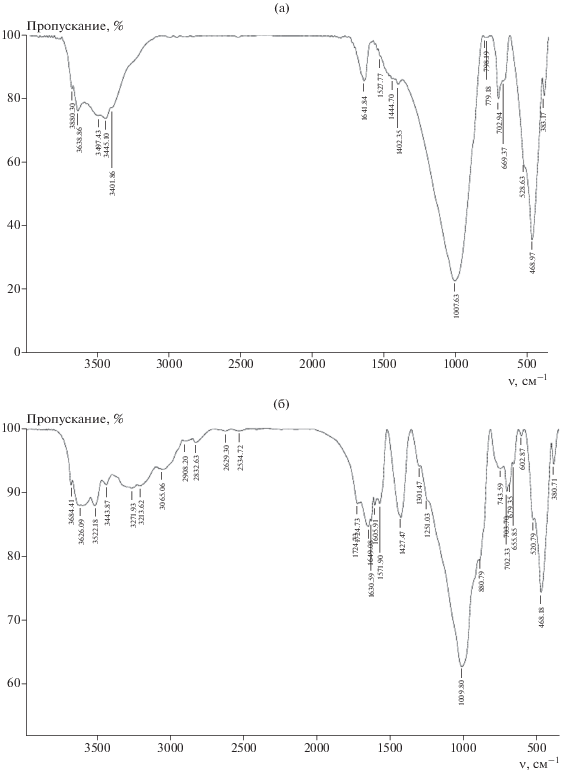
Механизмы взаимодействия ГМА-Л с бентонитом исследовали с применением ИК-спектроскопии (рис. 5). Для сухого бентонита, термообработанного в течение 2 ч при температуре 200°С, наблюдаются следующие полосы поглощения (рис. 5а): в области 3638–3680 см–1 (валентные колебания структурных ОН-групп, перпендикулярных силикатной плоскости); в области 3445–3497 см–1 (пики, принадлежащие валентным колебаниям структурных ОН-групп, направленных к октаэдрическим вакансиям); при 3402 см–1 (валентные колебания ОН-групп остатков адсорбированной воды); при 1645 см–1 (деформационные колебаниям ОН-групп); в области 1370–1481 см–1 (валентные колебания Si–O в слоистых силикатах); интенсивный пик при 1010 см–1 (валентные Si–O и деформационные колебания Al–O–H); пик слабой интенсивности при 795 см–1 (симметричные колебания мостиковой связи Si–O–Si, характерной для тетраэдра SiO4); пики в области 657–703 см–1 (валентные колебания связей Аl–O и Si–O–Аl; поглощение большой интенсивности при 468 см–1 (деформационные колебания Si–O).
Уже в течение первого часа воздействия на данный минерал интеркалантом ГМА-Л (рис. 5б) наблюдаются существенные изменения в ИК-спектрах по сравнению с исходным бентонитом (рис. 5а). Так, зафиксировано смещение полосы при 3636 см–1, принадлежащей валентным колебаниям ОН-групп, направленных к октаэдрическим вакансиям, а также при 3497 см–1. Появляются полосы в диапазоне частот 3213–3271 см–1, присущие симметричным валентным колебаниям катиона ${\text{NH}}_{4}^{ + },$ а также полосы низкой интенсивности при 2832 и 2908 см–1, являющиеся комбинацией полос аммонийной структуры. Обнаружено поглощение при 1427 см–1, принадлежащее валентным колебания связи Мо–О, и слабые пики при 2354 и 2620 см–1, наличие которых можно отнести к колебаниям СН- и СН2-групп, а при 1724 см–1 – СООН-групп лимонной кислоты. С увеличением времени интеркалирования интенсивности полос, фиксируемых на рис. 5б, значительно возрастают и положение их продолжает изменяться. Все это свидетельствует о том, что процесс интеркалирования раствора ГМА-Л в межплоскостное пространство бентонита сопровождается сложным механизмом межмолекулярных взаимодействий, включающих, по-видимому, ионный обмен, образование ковалентных и донорно-акцепторных связей. При этом какие-либо существенные изменения в поглощениях функциональных групп алюмосиликатного скелета (500–100 см–1) при таких временах интеркалирования не обнаружены.
Результаты термического анализа этих образцов показали, что для исходного бентонита характерно наличие эндоэффекта с максимумом при 117°С, обусловленного удалением адсорбированной воды, присутствие которой связано с высокой удельной поверхностью частиц минерала, ее проникновением в межплоскостное пространство данного алюмосиликата и высокой набухаемостью последнего (рис. 6а). Этот эффект позволяет установить состав и характер гидратированных обменных катионов щелочных металлов, так как они влияют на интенсивность и форму термических эффектов. Потеря структурной воды бентонитом происходит в диапазоне температур 500–600°С. При этих же температурах возможно выделение конституционной воды у примесных минералов группы гидрослюды, которые в небольших количествах присутствуют в составе бентонита. На кривой ДТА наблюдается также экзотермический максимум при 768°С, обусловленный удалением оставшейся гидроксильной воды и разрушением структуры бентонита.
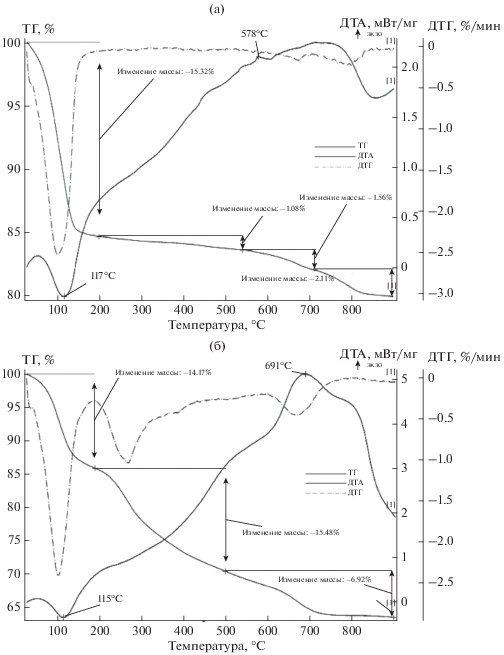
После обработки исходного бентонита составом ГМА-Л при 20°С в течение 2 ч (рис. 6б) наблюдали эндоэффект с максимумом при 115°С, при этом потеря массы образца составляла 14.17%. В интервале температур 200–500°С происходит разложение катиона аммония с выделением аммиака и потерей массы образца 15.5%. Интеркалирование сопровождается некоторым снижением термостабильности бентонита, на что указывает смещение экзоэффекта с максимумом при 768°С (исходный бентонит), связанного с его структурными изменениями, в более низкотемпературную область (691°С). При увеличении времени обработки наблюдается дальнейшее смещение эндо- и экзоэффектов в более низкотемпературные области. Из анализа представленных результатов следует, что проникновение данного интеркаланта в межслоевое пространство бентонита составляет около 36 мас. %.
На рис. 7 представлены дифрактограммы бентонита в сухом состоянии и обработанного парами ДМСО на первой стадии двухстадийного процесса интеркалирования. Из рис. 7а следует, что для изучаемого исходного бентонита наиболее сильный рефлекс соответствует межплоскостному расстоянию 15.2 Å. После воздействия паров ДМСО d основного базального отражения 001 увеличивается до 23 Å (рис. 6б), что свидетельствует об образовании интеркалата бентонит–ДМСО.
Обработка бентонита парами ДМСО приводит также к существенным изменениям ИК-спектров (рис. 8). Особенно это касается области проявления валентных колебаний ОН-групп этого минерала (структурных, свободных поверхностных, вступающих в водородные связи), а именно, при 3410–3680 см–1. В частности, наблюдаются исчезновение пика при 3638 см–1, характерного для исходного бентонита, ввиду смещения его в область 3542 см–1, а также увеличение интенсивностей пиков при 3680 и 3410 см–1. Кроме того, зафиксированы поглощения в виде трехплечевого пика с полосами при 3001, 2922 и 2854 см–1, связанные с присутствием в структуре бентонита ДМСО, которые принадлежат валентным колебаниям связей S=O, C–H и H–C–H последнего, а также поглощение при 1400 см–1, характерное для деформационного колебания S=O-групп (рис. 8б).
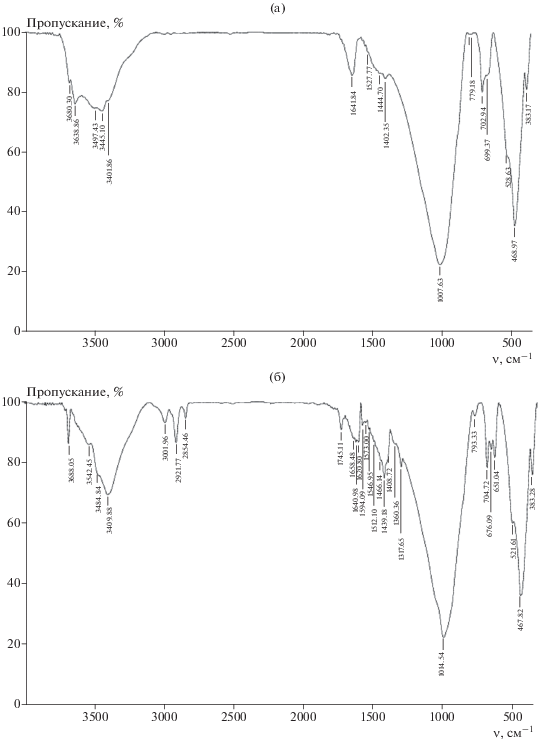
ДТА образцов показал, что для исходного бентонита характерно наличие эндоэффекта с максимумом при 127°С, обусловленного удалением адсорбированной воды. После обработки в парах ДМСО этот эндоэффект смещается в более низкотемпературную область по причине вытеснения некоторого объема адсорбированной бентонитом воды молекулами ДМСО при его внедрении в межпакетное пространство. Интеркалирование сопровождается некоторой потерей термостабильности бентонита, на что указывает смещение экзоэффекта при 768°С, связанного с его структурным переустройством, в более низкотемпературную область. Степень внедрения ДМСО в межплоскостное пространство бентонита составляет 27%, что превышает, как и ожидалось, степень внедрения этого интеркаланта в межслоевое пространство каолина. Обработка данного интеркалата жидкофазной смесью ГМА-Л на 2-й стадии процесса уменьшает d до 21 Å, что может быть вызвано вытеснением ДМСО в результате внедрения в межслоевое пространство ГМА-Л.
ЗАКЛЮЧЕНИЕ
Методами рентгенографического анализа, термического анализа и ИК-спектроскопии показано, что интеркалирование ГМА с добавкой лимонной кислоты в слоистые алюмосиликаты существенно зависит от структурного устройства минералов и фазового состояния прекурсоров. Так, низкотемпературная обработка каолина жидкофазным составом на основе ГМА-Л не позволяет осуществить его внедрение в межплоскостные области этого минерала, что обусловлено как жестким сочленением его структурных пакетов, так и стерическим затруднением, связанным с размерами интеркаланта. Формирование соединения каолин–ГМА-Л с d = 10.98 Å [001] при 2θ = 7.52° и степенью внедрения прекурсоров 17 мас. % оказалось возможным только в результате двухстадийного процесса интеркалирования с воздействием на 1-й стадии на данный глинистый минерал парами ДМСО.
Благодаря более высокой подвижности структурных пакетов бентонита его интеркалаты получены с использованием как одностадийного (обработка жидкофазным ГМА-Л с расширением межплоскостного расстояния до 21.8 Å), так и 2‑стадийного процессов с образованием интеркалата бентонит–ДМСО с d основного базального отражения 001 23 Å, однако во втором случае зафиксировано более эффективное проникновение прекурсоров в межплоскостную область (до 40 мас. %) и с меньшими энергозатратами.
Процесс интеркалирования ГМА в межплоскостное пространство данных природных алюмосиликатов сопровождается сложным механизмом межмолекулярных взаимодействий, включающих ионный обмен, образование ковалентных и донорно-акцепторных связей. При этом каких-либо существенных изменений в структуре алюмосиликатных скелетов при используемых режимах интеркалирования не обнаружено.
В дальнейшем наши исследования будут направлены на изучение механизма фотохимического превращения полученных интеркалатов с образованием полиоксометаллатных соединений молибдена и их глубокого термолиза с целью синтеза наночастиц и нанокластеров различных оксидов этого металла.
Список литературы
Tomas J.K. Photophysical and Photochemical Processes on Clay Surfaces // Acc. Chem. Res. 1988. V. 21. P. 275–280.
Yamase T., Kurozumi J. Isopolyanions of Molybdenum and Tungsten as Photocatalysts for Hydration of Acetylene // Inorg. Chim. Acta. 1984. № 83. P. 25–32.
Bechinger C., Oefinger G., Herminghaus S., Leiderer P. On the Fundamental Role of Oxygen for the Photochromic Effect of WO3 // J. Appl. Phys. 1993. V. 74. № 7. P. 4527–4532.
Стахеев А.Ю. Электронные и каталитические свойства наночастиц металлов и полупроводников на оксидных и цеолитных носителях: Дис. ... докт. хим. наук. М. 2004. 268 с.
Вячеславов А.С. Синтез магнитных нанокомпозитов на основе микро- и мезопористых алюмосиликатов: Дис. ... канд. хим. наук. М. 2008. 145 с.
Кошевар В.Д., Будейко Н.Л. Получение наноструктур диоксида титана в межслоевом пространстве глинистых минералов // Коллоид. журн. 2009. Т. 71. № 5. С. 632–637.
Ozin G.A., Oskar S., Procopowich R.A. Smart: New Forms of Tungsten and Molybdenum Oxides // Acc. Chem. Res. 1992. V. 25. № 12. P. 553–560.
Берданова Е.И., Ларин А.М., Шахновская О.Л., Романовский Б.В. Получение и свойства высокодисперсного оксида меди в цеолитной матрице // Изв. Академии наук. Сер. хим. 1997. № 10. С. 1761–1765.
Смоленцева Е.В. Формирование активных состояний золота в модифицированных цеолитах: Автореф. дис. ... канд. хим. наук. Томск. 28 с.
Шпилевская Л.Е., Кошевар В.Д. Получение, свойства и применение светочувствительных систем на основе парамолибдата аммония // ЖПХ. 1998. Т. 71. № 8. С. 1263–1269.
Андреев В.Н., Никитин С.Е., Климов В.А., Козыре С.В. и др. Исследование фотохромных кластерных систем на основе оксидов молибдена методом ЭПР-спектроскопии // ФТТ. 2001. Т. 43. № 4. С. 755–758.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы