

Журнал неорганической химии, 2019, T. 64, № 1, стр. 38-50
Катионно-анионные комплексы PdII с катионом адамантилимидазолия: синтез, структурные исследования и МАО-ингибирующая активностьМ. С. Денисов, М. В. Дмитриев, Д. В. Ерошенко, П. А. Слепухин, С. П. Шавкунов, В. А. Глушков
М. С. Денисов 1, *, М. В. Дмитриев 2, Д. В. Ерошенко 1, П. А. Слепухин 3, 4, С. П. Шавкунов 2, В. А. Глушков 1, 2
1 Институт технической химии УрО РАН
614013 Пермь, ул. Академика Королева, 3, Россия
2 Пермский государственный национальный исследовательский университет
614990 Пермь, ул. Букирева, 15, Россия
3 Уральский федеральный университет им. первого Президента России Б.Н. Ельцина
620002 Екатеринбург, ул. Мира, 19, Россия
4 Институт органического синтеза им. И.Я. Постовского УрО РАН
620990 Екатеринбург, ул. С. Ковалевской, 22/20, Россия
* E-mail: denisov.m@itcras.ru
Поступила в редакцию 28.02.2018
После доработки 04.07.2018
Принята к публикации 19.04.2018
Аннотация
Получены новые катионно-анионные комплексы палладия(II) из адамантилзамещенных солей имидазолия и исследовано влияние структуры адамантилзамещенных солей и условий синтеза на строение этих комплексов с анионами Pd(ДМСО)${\text{Hal}}_{3}^{ - }$ (1–4), ${\text{P}}{{{\text{d}}}_{2}}{\text{Br}}_{6}^{{2 - }}$ (5, 6) или ${\text{PdCl}}_{4}^{{2 - }}$ (7). Получен ряд комплексов палладия(II), активных против моноаминоксидазы Б, установлены закономерности влияния состава и структуры на их биологическую активность. Строение комплексов подтверждено методами рентгеноструктурного анализа, проведено кондуктометрическое исследование комплекса 1. Установлено, что МАО-ингибирующая активность синтезированных комплексов находится на уровне препаратов сравнения: 17.6% остаточной активности фермента при ингибировании комплексом 3 по сравнению с 16.9% у препарата сравнения (селегелин). Комплексы с бромом в качестве лиганда обладают большей активностью, чем с хлором. Результаты исследования могут быть использованы в области металлоорганической и бионеорганической химии.
ВВЕДЕНИЕ
Комплексы палладия(II) c P-, S- и N-лигандами широко используются как катализаторы реакций кросс-сочетания [1, 2]. В последние годы большой интерес вызывают комплексы Pd(II) и Pd(0) с N-гетероциклическими карбеновыми лигандами (Pd–NHC-комплексы) [3–5]. Прямой метод синтеза Pd–NHC-комплексов, предложенный Херрманном [6], заключается во взаимодействии ацетата палладия(II) с солями имидазолия в ДМСО при нагревании в интервале температур 50–120°С [7–11]. Этим же методом можно получать комплексы палладия(II) из солей 1,2,4-триазолия [12] и для так называемых “аномальных” (“abnormal”) карбенов [13, 14]. Нами установлено, что применение данной методики к солям 1-адамантил-3-бензилимидазолия неожиданно привело к получению анионных комплексов палладия(II) с ДМСО, в которых в качестве катиона выступает имидазолий. Вероятно, причиной такого поведения является стерический фактор. В литературе известны примеры, когда из-за стерической перегруженности имидазольного лиганда карбеновые комплексы не образуются, а вместо них получаются катионно-анионные комплексы с катионом имидазолия [15–17]. Известны аналогичные катионно-анионные комплексы палладия, в том числе с ДМСО и катионами различных типов [18–23], но противоионы имидазолия встречаются редко [24–26]. В литературе есть также примеры, когда ДМСО включается в комплекс Pd–NHC с координацией кислорода на палладий [27]. Есть примеры формального комплекса палладия с NHC и ДМСО, где сера координирована палладием [28, 29], однако правильнее его рассматривать как сокристаллизацию комплексов PdII–NHC и PdII–ДМСО. Стоит отметить, что в работах 2017 г. именно анион [Pd(ДМСО)${\text{Cl}}_{3}^{ - }$] признан активным катализатором реакций Хека [29] и Сузуки [30]. Описаны подобные (c остатком соли имидазолия) комплексы с ДМСО для Pt(II) [31–33], Rh(I) [34] и Ru(II) [35–38], причем последние обладают противораковым действием. Существуют работы, в которых продемонстрировано использование катионно-анионных комплексов палладия как альтернативы противораковому препарату цисплатину [39–46].
Болезни Паркинсона и Альцгеймера, а также депрессивные состояния и ряд других нейродегенеративных заболеваний связаны с нарушением работы фермента моноаминоксидазы Б (МАО-Б), который локализован в нейронах головного мозга. Существует целый ряд лекарственных средств МАО-ингибиторов, применяемых в терапии депрессивных состояний и болезней Альцгеймера и Паркинсона. На данный момент на рынке присутствуют препараты моклобемид и разагилин (с обратимым и необратимым ингибирующим механизмом действия на МАО соответственно), но только последний рекомендован к лечению болезни Паркинсона [47]. Другие лекарственные препараты против нейродегенеративых заболеваний задействуют иные механизмы действия. Очевидно, необходим принципиально новый подход к получению МАО-ингибиторов, обладающих высокой активностью и меньшей токсичностью.
Целью работы является синтез катионно-анионных тригалогенид-сульфоксидных, мостиковых комплексов и тетрагалогенидных комплексов палладия(II) с адамантилзамещенными катионами имидазолия, изучение их строения и МАО-ингибирующей активности, установление закономерностей анти-МАО-активности в зависимости от состава комплексов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ИК-спектры регистрировали на Фурье-спектрометре VERTEX 80v в тонкой пленке, полученной испарением раствора вещества в хлороформе непосредственно на стекле NaCl или в суспензии вазелинового масла. Спектры ЯМР 1Н и 13С записывали в CDCl3 на приборе Bruker 400 при 400 и 100 МГц соответственно, внутренний стандарт – ГМДС (гексаметилдисилоксан); при записи спектров ЯМР 13С внутренним стандартом служили остаточные сигналы растворителя. Температуру плавления определяли на приборе ПТП-2. Элементный анализ (C, H, N, S) проводили на приборе Vario EL Cube (Elementar Analysensysteme GmbH, Германия). Для колоночной хроматографии применяли Silicagel 60 (Alfa Aesar, 0.032–0.070 мм) или Celite Standard Super (Alfa Aesar). Элюировали высушенным хлористым метиленом. В работе использовали PdCl2 и Pd(OAc)2 (Alfa Aesar, Великобритания); коммерческие препараты кинурамин, разагилин и селегелин фирмы Sigma-Aldrich, реактивы ДМСО (х. ч.), KBr (ч. д. а.) и K2CO3 (х. ч.) отечественного производства использовали без дополнительной очистки. Метилен хлористый (х. ч.) сушили перегонкой над оксидом фосфора(V). Исходные соли имидазолия синтезировали по методу, опубликованному нами ранее в работе [48].
Рентгеноструктурный анализ (РСА) соединений 1 и 3 выполнен на оборудовании ЦКП “Спектроскопия и анализ органических соединений” ИОС УрО РАН, соединений 4–7 – в Пермском государственном национальном исследовательском университете. Эксперименты проведены на автоматических дифрактометрах Xcalibur 3 (соединения 1 и 3) и Xcalibur Ruby (соединения 4–7) по стандартной процедуре (MoKα-излучение, графитовый монохроматор, ω-сканирование с шагом 1°). Введена эмпирическая поправка на поглощение. Решение и уточнение структур выполнено в программной оболочке Olex2 [49]. Структуры решены с помощью программы ShelXS [50] и уточнены с использованием программы ShelXL [51] в анизотропном для неводородных атомов приближении. Рисунки структур созданы в программе Mercury 3.3 (Build RC5) [52]. Часть атомов водорода добавлена в рассчитанные положения и включена в уточнение в модели “наездника” с зависимыми тепловыми параметрами, часть уточнена независимо в изотропном приближении. Координаты разупорядоченных атомов аниона соединения 6 уточнены с использованием мягких ограничений SAME. Основные кристаллографические параметры и результаты уточнения структуры представлены в табл. 1.
Таблица 1.
Основные кристаллографические параметры и результаты уточнения структуры
| Соединение | 1 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| Брутто-формула | C25H37Cl3N2OPdS | C25H37Br3N2OPdS | С22H31Br3N2OPdS | C24H33Br3N2Pd | C24H29Br3N2OPd | C46H64Cl4N4OPd |
| М | 626.38 | 759.76 | 717.68 | 695.65 | 707.62 | 937.21 |
| T, K | 295(2) | 140.00(10) | 295(2) | 295(2) | 295(2) | 295(2) |
| Сингония | Моноклинная | Моноклинная | Триклинная | Моноклинная | Триклинная | Триклинная |
| Пр. гр. | P21/c | P21/c | P1 | P21/n | P1 | P1 |
| a, Å | 12.6101(10) | 12.5697(2) | 11.158(3) | 15.208(3) | 9.0489(13) | 12.1207(13) |
| b, Å | 22.6182(11) | 22.7347(4) | 11.207(2) | 9.6043(19) | 11.712(2) | 13.8340(14) |
| c, Å | 9.6095(7) | 9.60871(16) | 11.3123(15) | 18.695(3) | 12.965(2) | 14.3768(13) |
| α, град | 90.00 | 90.00 | 101.159(14) | 90.00 | 109.401(16) | 89.226(8) |
| β, град | 97.508(6) | 96.5381(17) | 102.275(17) | 109.590(19) | 101.512(14) | 85.153(8) |
| γ, град | 90.00 | 90.00 | 108.48(2) | 90.00 | 91.251(13) | 68.885(10) |
| V, Å3 | 2717.3(3) | 2728.00(8) | 1257.9(5) | 2572.5(9) | 1264.2(4) | 2240.4(4) |
| Z | 4 | 4 | 2 | 4 | 2 | 2 |
| ρрасч, г/см3 | 1.531 | 1.850 | 1.895 | 1.796 | 1.859 | 1.389 |
| μ, мм–1 | 1.076 | 5.172 | 5.603 | 5.395 | 5.494 | 0.692 |
| F(000) | 1288 | 1504.0 | 704 | 1368 | 692 | 980 |
| Размеры кристалла, мм | 0.45 × 0.29 × 0.16 | 0.19 × 0.12 × 0.07 | 0.56 × 0.21 × 0.04 | 0.48 × 0.28 × 0.11 | 0.57 × 0.35 × 0.12 | 0.53 × 0.27 × 0.19 |
| 2θ, град | 5.34 < 2θ < 52.76 | 4.84 < 2θ < 61.56 | 5.87 < 2θ < 58.97 | 5.99 < 2θ < 58.69 | 4.61 < 2θ < 58.66 | 4.29 < 2θ < 58.84 |
| Индексы отражений | –11 < h < 15, –28 < k < 26, –12 < l < 11 |
–15 ≤ h ≤ 17, –16 ≤ k ≤ 31, –11 ≤ l ≤ 13 |
–14 ≤ h ≤ 10, –15 ≤ k ≤ 13, –14 ≤ l ≤ 15 |
–14 ≤ h ≤ 19, –12 ≤ k ≤ 9, –23 ≤ l ≤ 25 |
–11 ≤ h ≤ 12, –15 ≤ k ≤ 13, –15 ≤ l ≤ 17 |
–11 ≤ h ≤ 15, –19 ≤ k ≤ 17, –18 ≤ l ≤ 17 |
| Собрано отражений | 20 226 | 15 077 | 10 881 | 13 194 | 10 415 | 19 180 |
| Независимых отражений | 5487 (Rint = 0.0504) | 7478 (Rint = 0.0187) | 5848 (Rint = 0.0576) | 6048 (Rint = 0.0419) | 5856 (Rint = 0.0531) | 10 440 (Rint = 0.0355) |
| Данные/ограниченных /параметров | 5487/0/299 | 7478/0/303 | 5848/0/273 | 6048/0/275 | 5856/8/323 | 10 440/0/522 |
| Goodness-of-fit on F2 | 1.004 | 0.998 | 1.034 | 1.011 | 1.035 | 1.042 |
| R-факторы [I ≥ 2σ(I)] | R1 = 0.0425, wR2 = 0.0848 |
R1 = 0.0592, wR2 = 0.1756 |
R1 = 0.0636, wR2 = 0.1380 |
R1 = 0.0475, wR2 = 0.0812 |
R1 = 0.0757, wR2 = 0.1528 |
R1 = 0.0449, wR2 = 0.1056 |
| R-факторы [все отр.] | R1 = 0.1109, wR2 = 0.0915 |
R1 = 0.0741, wR2 = 0.1905 |
R1 = 0.1283, wR2 = 0.1825 |
R1 = 0.1029, wR2 = 0.1016 |
R1 = 0.1628, wR2 = 0.2076 |
R1 = 0.0622, wR2 = 0.1201 |
| Δρē/ē Å–3 | 0.859/–0.310 | 2.75/–4.23 | 1.144/–1.278 | 0.677/–0.973 | 0.751/–1.260 | 1.466/–0.839 |
Результаты РСА зарегистрированы в Кембриджской базе структурных данных в виде cif-файла под номерами ССDC: 1541626 (комплекс 1), 1541627 (комплекс 3), 1824095 (комплекс 4), 1541628 (комплекс 5), 1541629 (комплекс 6), 1541630 (комплекс 7). Эти данные находятся в свободном доступе и могут быть запрошены по адресу: www.ccdc.cam.ac.uk.
1-Адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолий диметилсульфоксидтрихлорпалладат (1). В 5 мл ДМСО растворяли хлорид 1-адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолия (153 мг, 0.412 ммоль) и хлорид палладия(II) (73 мг, 0.412 ммоль), перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Перекристаллизовывали из смеси хлористый метилен-этилацетат. Получили коричнево-красные кристаллы. Выход 85% (220 мг), tпл = 159–164°С.
| C | H | N | S | |
| Найдено, %: | 47.47; | 6.14; | 4.41; | 4.69. |
| Для С25H37Cl3N2OPdS | ||||
| вычислено, %: | 47.93; | 5.59; | 4.47; | 5.12. |
ИК-спектр (тонкая пленка), ν, cм–l: 3124, 3087, 3051, 2986, 2916 (CH2 Ad), 2856, 1613, 1546, 1452, 1416, 1381, 1362, 1346, 1309, 1295, 1253, 1238, 1215, 1152 (NCHN), 1142 (S–O), 1105, 1025, 855, 827, 816, 798, 752 (Ar–H), 693, 663, 637, 620, 426.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.82 (к, 6 H, Ad, J = 9.0 Гц); 2.29–2.36 (м, 18 Н, Ad + MeAr); 3.31 (c, 6 H, ДМСО); 5.73 (с, 2 Н, СН2Ar); 6.79 (с, 1 Н, Im); 6.93 (с, 2 Н, Ar); 7.38 (с, 1 Н, Im); 9.77 (с, 1 Н, 2-Im).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 19.54 (MeAr); 20.56 (MeAr); 29.01 (CH Ad); 34.84 (CH2 Ad); 42.40 (CH2 Ad); 44.26 (ДМСО); 48.10 (CH2Ar); 60.34 (C Ad); 117.67 (Im); 119.92 (Im); 125.25 (C Ar); 129.33 (CH Ar); 134.50 (2-Im); 137.88 (C Ar); 139.14 (C Ar).
1-Адамантил-3-[(2,3,5,6-тетраметилфенил)метил]-1Н-имидазолий диметилсульфоксидтрихлорпалладат тригидрат (2). В 5 мл ДМСО растворяли хлорид 1-адамантил-3-[(2,3,5,6-тетраметилфенил)метил]-1Н-имидазолия (159 мг, 0.412 ммоль) и хлорид палладия(II) (73 мг, 0.412 ммоль), перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Растворяли в этилацетате, высаживали петролейным эфиром (40–70°С); к декантированной нижней фазе (масло) добавляли каплю ацетонитрила и оставляли испаряться при комнатной температуре. Получили желтый порошок. Выход 83% (237 мг), tпл(СН3СN) = 52–55°С.
| C | H | N | S | |
| Найдено, %: | 44.54; | 7.17; | 3.98; | 4.23. |
| Для С26H39Cl3N2OPdS · 3Н2О | ||||
| вычислено, %: | 44.97; | 6.53; | 4.03; | 4.62. |
ИК-спектр (вазелиновое масло), ν, cм–l: 3163, 3123 (Ar–H), 3081, 3058, 3007, 1552, 1416, 1346, 1311, 1297, 1256, 1235, 1213, 1152 (NCHN), 1104 (Im–H), 1092 (S–O), 1065, 1027, 986, 946, 919, 901, 878, 866, 760 (Ar–H), 688, 663, 640, 621, 425.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.84 (к, 6 H, Ad, J = 8.9 Гц); 2.01 (с, 3 H, Ad); 2.24 (c, 6 H, MeAr); 2.27 (c, 6 H, MeAr); 2.33 (c, 6 H, Ad); 2.36 (с, 6 H, ДМСО); 3.33 (c, 6 H, H2O); 5.82 (с, 2 Н, СН2Ar); 6.77 (с, 1 Н, Im); 7.06 (с, 2 Н, Ar); 7.31 (с, 1 Н, Im); 9.81 (с, 1 Н, 2-Im).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 15.43 (MeAr); 19.89 (MeAr); 29.04 (CH Ad); 34.87 (CH2 Ad); 42.46 (CH2 Ad); 44.30 (ДМСО); 48.89 (CH2Ar); 60.37 (C Ad); 117.28 (Im); 120.04 (Im); 128.01 (C Ar); 132.82 (C Ar); 133.88 (CH Ar); 134.30 (C Ar); 134.62 (2-Im).
1-Адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолий диметилсульфоксидтрибромпалладат (3). В 5 мл ДМСО растворяли тетрафторборат 1-адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолия (174 мг, 0.412 ммоль) и ацетат палладия(II) (92 мг, 0.412 ммоль), добавляли бромид калия (294 мг, 2.429 ммоль) и перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Хроматографировали на силикагеле. Перекристаллизовывали из ацетонитрила. Получили красные кристаллы. Выход 48% (150 мг), tпл = 143–145°С.
| C | H | N | S | |
| Найдено, %: | 37.94; | 5.87; | 2.57; | 3.08. |
| Для С25H37Br3N2OPdS | ||||
| вычислено, %: | 40.30; | 5.20; | 3.62; | 4.14. |
ИК-спектр (пленка), ν, cм–l: 3125, 3088, 3052, 2984, 2915 (CH2 Ad), 2856, 1613, 1546, 1452, 1416, 1381, 1362, 1346, 1309, 1295, 1253, 1236, 1216, 1152 (NCHN), 1142 (S–O), 1104, 1024, 855, 827, 816, 798, 752 (Ar–H), 693, 662, 637, 620, 424.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.82 (д, 6 H, Ad, J = 9.0 Гц); 2.27–2.36 (м, 18 Н, Ad + MeAr); 2.64 (c, 6 H, ДМСО); 5.76 (с, 2 Н, СН2Ar); 6.85 (с, 1 Н, Im); 6.92 (с, 2 Н, Ar); 7.34 (с, 1 Н, Im); 9.67 (с, 1 Н, 2-Im).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 20.29 (MeAr); 21.01 (MeAr); 29.46 (CH Ad); 35.34 (CH2 Ad); 43.07 (CH2 Ad); 44.25 (ДМСО); 49.08 (CH2Ar); 60.98 (C Ad); 118.73 (Im); 120.94 (Im); 125.25 (C Ar); 129.82 (CH Ar); 134.50 (2-Im); 138.33 (C Ar); 139.65 (C Ar).
1-Адамантил-3-бензил-1Н-имидазолий диметилсульфоксидтрибромпалладат (4). В 5 мл ДМСО растворяли бромид 1-адамантил-3-бензил-1Н-имидазолия (166 мг, 0.443 ммоль) и ацетат палладия(II) (100 мг, 0.443 ммоль), добавляли бромид калия (158 мг, 1.330 ммоль) и перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Хроматографировали на силикагеле. Перекристаллизовывали из ацетонитрила. Получили красные призмы. Выход 25% (80 мг), tпл(СН3СN) = 147–149°С.
| C | H | N | S | |
| Найдено, %: | 36.87; | 4.23; | 3.93; | 4.37. |
| Для С22H31Br3N2OPdS | ||||
| вычислено, %: | 36.87; | 4.23; | 3.93; | 4.34. |
ИК-спектр (пленка), ν, cм–l: 3141, 3123, 3081, 2915 (CH2 Ad), 2849, 1538, 1447, 1342, 1307, 1254, 1148 (H+), 1113, 1104 (S–O), 1026, 840, 823, 816, 736, 708 (Ar–H), 681, 656, 637, 626, 620.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.84 (к, 6 H, Ad, J = 14.0 Гц); 2.37 (с, 6 Н, Ad) 2.64 (c, 6 H, ДМСО); 3.42 (т, 3 Н, Ad, J = 3.6 Гц); 5.79 (с, 2 Н, СН2Ar); 7.32 (т, 1 Н, Im, J = 0.4 Гц); 7.41–7.47 (м, 4Н, м-Ph, п-Ph, Im); 7.55 (дд, 2 Н, o-Ph, J = 7.6, J = = 1.2 Гц); 9.60 (с, 1 Н, 2-Im).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 29.03 (CH Ad); 34.87 (CH2 Ad); 40.63 (CH2 Ad); 42.53 (ДМСО); 53.90 (CH2Ar); 60.68 (C Ad); 118.24 (Im); 121.60 (Im); 128.90 (CH Ar); 128.93 (CH Ar); 128.96 (CH Ar); 129.13 (C Ar); 132.51 (2-Im).
Бис-{1-адамантил-3-[(2,3,5,6-тетраметилфенил)метил]-1Н-имидазолий} гексабромдипалладат полугидрат (5). В 5 мл ДМСО растворяли (0.180 г) тетрафторборат 1-адамантил-3-[(2,3,5,6-тетраметилфенил)метил]-1Н-имидазолия (180 мг, 0.412 ммоль) и ацетат палладия(II) (92 мг, 0.412 ммоль), добавляли бромид калия (294 мг, 2.429 ммоль) и перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Хроматографировали на силикагеле. Перекристаллизовывали из ацетонитрила. Получили кристаллы темно-бордового цвета. Выход 36% (601 мг), tпл(СН3СN) = 255–256°С.
ИК-спектр (вазелиновое масло), ν, cм–l: 3158, 3126, 3088, 2734, 1558, 1544, 1414, 1344, 1320, 1307, 1294, 1283, 1269, 1251, 1187, 1145, 1111, 1101, 1063, 1044, 1017, 994, 984, 904, 880, 833, 817, 775, 752 (Ar–H), 737, 700, 686, 660, 637, 615.
Спектр ЯМР 1Н (ДМСО-d6, δ, м.д.): 1.73 (т, 6 H, Ad, J = 2.8 Гц); 2.14 (т, 6 H, Ad, J = 3.8 Гц); 2.30 (т, 3 Н, Ad, J = 1.6 Гц); 5.48 (с, 2 Н, СН2Ar); 7.09 (с, 1 H, Ar); 7.30 (т, 1 Н, Im, J = 2.0 Гц); 7.98 (т, 1 Н, Im, J = 2.0 Гц); 9.26 (т, 1 Н, 2-Im, J = 1.8 Гц).
Спектр ЯМР 13C (ДМСО-d6, δ, м.д.): 15.31 (MeAr); 20.05 (MeAr); 28.90 (CH Ad); 34.82 (CH2 Ad); 41.55 (CH2 Ad); 47.69 (CH2Ar); 59.40 (C Ad); 119.71 (Im); 121.55 (Im); 129.20 (C Ar); 132.60 (CH Ar); 133.64 (2-Im); 134.00 (C Ar); 134.06 (C Ar).
Бис-{1-адамантил-3-[(1-нафтил)метил]-1Н-имидазолий} гексабромдипалладат моногидрат (6). В 5 мл ДМСО растворяли тетрафторборат 1-адамантил-3-[(1-нафтил)метил]-1Н-имидазолия (177 мг, 0.412 ммоль) и ацетат палладия(II) (92 мг, 0.412 ммоль), добавляли бромид калия (294 мг, 2.429 ммоль) и перемешивали 2 ч при 50°С, затем 3 ч при 110°С. Отгоняли ДМСО под вакуумом, остаток растворяли в хлористом метилене и фильтровали через целит. Остаток хроматографировали на силикагеле. Перекристаллизовали из ацетонитрила. Получили крупные темно-бордовые, почти черные призмы. Выход 39% (119 мг), tпл(СН3СN) = 121–124°С.
ИК-спектр (вазелиновое масло), ν, cм–l: 3163, 3120, 3057, 1598, 1542, 1414, 1357, 1347, 1306, 1306, 1253, 1235, 1218, 1194, 1166, 1144 (NCHN), 1105, 1097, 1063, 1021, 968, 878, 862, 843, 827, 815, 804, 780 (Ar–H), 687, 665, 637, 623, 537, 512, 412.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.78 (дд, 6 H, Ad, J = 12.2, 1.2 Гц); 1.88 (дд, 6 H, Ad, J = 12.2, 1.2 Гц); 2.38 (с, 3 H, Ad); 2.47 (д, 6 Н, Ad, J = 2.4 Гц); 6.39 (с, 2 Н, СН2Ar); 7.49 (т, 1 Н, Im, J = 1.8 Гц); 7.50–7.57 (м, 3 H, 2,3-нафтил+Im), 7.67 (тт, 1 Н, 4-нафтил, J = 7.7, 1.4 Гц); 7.82 (дд, 1 Н, 5-нафтил, J = 7.2, 0.8 Гц); 7.92 (тт, 2 Н, 7,6-нафтил, J = 7.4, 0.4 Гц); 8.15 (дд, 1 Н, 8-нафтил, J = 8.4, 0.8 Гц); 9.42 (т, 1 Н, 2-Im, J = 3.8 Гц).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 29.06 (CH Ad); 34.87 (CH2 Ad); 42.68 (CH2 Ad); 52.08 (CH2Ar); 60.97 (C Ad); 118.81 (Im); 122.41 (Im); 122.53 (CH Ar); 125.20 (CH Ar); 126.08 (CH Ar); 127.47 (CH Ar); 127.86 (CH Ar); 128.58 (CH Ar); 129.03 (C Ar); 130.08 (CH Ar); 130.65 (C Ar); 132.73 (C Ar); 133.49 (2-Im).
Бис(1-адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолий) тетрахлорпалладат полугидрат (7). В 5 мл ацетонитрила растворяли хлорид 1-адамантил-3-[(2,4,6-триметилфенил)метил]-1Н-имидазолия (306 мг, 0.824 ммоль), хлорид палладия(II) (73 мг, 0.327 ммоль) и прокаленный карбонат калия (336 мг, 2,429 ммоль), перемешивали 3 ч при 80°С. Реакционную массу охлаждали, фильтровали и оставляли испаряться при комнатной температуре. Получили красные кристаллы. Выход 26% (102 мг), tпл(СН3СN) = 222–224°С.
ИК-спектр (пленка), ν, cм–l: 3114, 3080, 3040, 2974, 2913 (CH2 Ad), 2855, 1613, 1546, 1452, 1379, 1308, 1252, 1215, 1153 (NCHN), 1104, 855, 749 (Ar–H), 693, 678, 661, 638.
Спектр ЯМР 1Н (СDСl3, δ, м.д.): 1.73 (т, 6 H, Ad, J = 3.0); 2.14 (д, 6 Н, Ad, J = 3.2 Гц); 2.22 (с, 3Н, Ad); 2.26 (c, 9 H, MeAr); 3.27 (1/2Н, Н2О); 5.42 (с, 2 Н, СН2Ar); 6.97 (с, 2 Н, Ar); 7.38 (т, 1 Н, Im, J = = 1.8 Гц); 8.01 (т, 1 Н, Im, J = 2.0 Гц); 9.38 (т, 1 Н, 2-Im, J = 1.6 Гц).
Спектр ЯМР 13C (СDСl3, δ, м.д.): 19.33 (MeAr); 20.58 (MeAr); 28.87 (CH Ad); 34.79 (CH2 Ad); 41.50 (CH2 Ad); 46.97 (CH2Ar); 59.33 (C Ad); 119.79 (Im); 121.9268 (Im); 126.79 (C Ar); 129.37 (CH Ar); 133.91 (2-Im); 137.99 (C Ar); 138.44 (C Ar).
Кондуктометрия. Электропроводность растворов соли 1 в ацетонитриле определяли в кондуктометрической ячейке с гладкими платиновыми электродами (постоянная сосуда составляла 0.366 см–1), для измерений использовали автоматизированный прибор Solartron-1280C. Амплитуда переменного сигнала составляла 20 мВ, диапазон частот от 20 000 до 1000 Гц. Ячейку термостатировали при 295 K с точностью ±1°С. Полученные данные приведены в табл. 2.
Таблица 2.
Электропроводность растворов комплекса 1
| m, ммоль/л СH3CN | κ × 106, Cм/см |
|---|---|
| 0 | 2.13 |
| 0.2 | 17.4 |
| 0.3 | 20.9 |
| 0.4 | 37.3 |
Определение МАО-ингибирующей активности. Ферментативную активность МАО в гомогенате головного мозга мышей определяли по методу [53] с модификациями [54], основанному на флуориметрическом измерении 4-гидроксихинолина, образующегося при ферментативном окислении кинурамина. Гомогенат готовили из ткани головного мозга мышей в соотношении 1 г ткани мозга на 8 мл буфера (0.1 М фосфатно-солевой буфер, рН 7.4). Гомогенат центрифугировали при 1000 g в течение 30 мин, затем надосадочную жидкость отбирали и центрифугировали при 10 000 g в течение 30 мин. Осадок разводили в 0.1 М фосфатно-солевом буфере при рН 7.4 до содержания белка 125 мкг/мл. Концентрацию белка определяли методом Лоури [55]. Для определения активности МАО в 96-луночный черный планшет (SPL) вносили суспензию митохондрий по 100 мкл/лунка и инкубировали при 37°С в течение 30 мин. Исследуемые соединения растворяли в ДМСО, разводили в фосфатно-солевом буфере до конечной концентрации 100 мкМ и добавляли к суспензии митохондрий по 100 мкл/лунка. Через 30 мин инкубации ферментативную реакцию инициировали добавлением неселективного субстрата кинурамина (Sigma-Aldrich) с концентрацией 0.2 мг/мл по 50 мкл/лунка. Затем планшеты инкубировали при 37°С в течение 30 мин, после чего реакцию останавливали добавлением 50 мкл 10%-ной трихлоруксусной кислоты и 50 мкл 1 M NaOH. Интенсивность флуоресценции 4-гидроксихинолина, который был образован из кинурамина, измеряли при длине волны излучения 380 нм и длине волны возбуждения 320 нм с помощью планшетного спектрофотометра FLUOstar Optima (BMG Labtech, GmbH). В качестве положительного контроля использовали суспензию митохондрий с добавлением ДМСО до конечной концентрации 0.1%. Активность фермента в контрольном эксперименте принимали за 100%. В качестве препаратов сравнения использовали специфичные ингибиторы МАО-Б разагилин (Sigma-Aldrich) и селегелин (Sigma-Aldrich), которые также вносили в концентрации 100 мкМ. Все эксперименты проводили трижды.
МТТ-тест. Цитотоксическую активность полученных соединений (1, 3) изучали с помощью МТТ-теста в отношении культур клеток человека: карциномы толстой кишки (HCT116) и рабдомиосаркомы (RD TE 32). Культуры клеток выращивали в среде DMEM с добавлением 10% эмбриональной телячьей сыворотки, 2мM L-глутамина и 1% гентамицина при 37°C и 5% CO2 во влажной атмосфере. Вещества вносили в монослой клеток в концентрациях от 10–4 до 10–6 M. Через 72 ч инкубации в обработанные культуры клеток вносили МТТ-краситель. Кристаллы формазана растворяли в 100 мкл ДМСО. Оценивали оптическую плотность с помощью планшетного спектрофотометра FLUOstar Optima (BMG Labtech, GmbH), рассчитывая IC50 – концентрацию вещества, токсичную для 50% клеток [56].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В качестве солей имидазолия мы использовали хлориды, бромиды или тетрафторбораты 1-адамантил-3-бензилимидазолия, замещенные в арильном остатке, синтезированные нами ранее [48]. Эти соли были взяты также из тех соображений, что адамантан – хорошая фармакофорная группа [57]. Комплексы 1 и 2 были получены нагреванием соответствующих солей адамантил-имидазолия с PdCl2 в ДМСО (схема 1). Строго говоря, в оригинальной методике Херрманна [6] в качестве источника палладия был использован не хлорид палладия(II), а ацетат палладия(II). Нами показано, что использование ацетата палладия(II) вместо хлорида палладия(II) приводит также к веществу 1 (подтверждено РСА), но с меньшим выходом по палладию (74 и 85% соответственно). Вероятно, первоначально образуется комплекс с ацетильными лигандами, а недостающие атомы хлора берутся из дихлорметана, который был использован в процессе выделения. Херрманн в работе [6] описал пример сохранения ацетатного лиганда на палладии при комплексобразовании с солями имидазолия с последующей его заменой на другие лиганды.
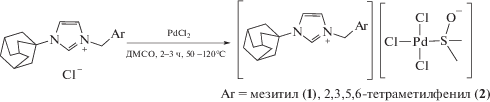
Соответствующие трибромидные комплексы 3, 4 были получены из ацетата палладия(II) (тример) и тетрафторбората 1-адамантил-3-мезитилметилимидазолия или бромида бензилимидазолия при добавлении в реакционную массу трех эквивалентов KBr (схема 2).
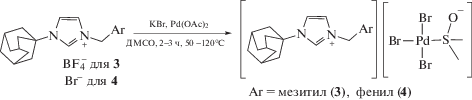
Схема 2.
Нами были предприняты попытки получить аналогичные комплексы с иными ароматическими фрагментами. Интересно, что в случае 2,3,5,6-тетраметилфенильного (дурильного) или 1-нафтильного заместителя исходного тетрафторбората (с трехкратным избытком KBr) с последующей очисткой продуктов на колонке с силикагелем были выделены комплексы Pd(II) 5 и 6 соответственно с анионом [Pd2Br6]2– и мостиковыми атомами брома (схема 3). Эти комплексы новые, но аналогичные известным структурам [58].
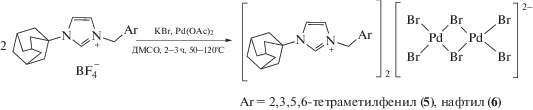
Схема 3.
Нами была также предпринята попытка провести реакцию в отсутствие ДМСО. Проведение реакции в ацетонитриле в присутствии K2CO3 в качестве основания привело к образованию тетрахлоридного комплекса палладия(II) 7, хотя и с небольшим выходом (26%) по причине частичного восстановления Pd(II) с образованием палладиевого зеркала (схема 4). Комплекс 7 новый, но органические комплексы с анионом ${\text{Pd(Hal)}}_{{\text{4}}}^{{2 - }}$ хорошо известны [59–63]. Возможно, что первоначально образуются комплексы именно такого типа, но затем один из атомов галогена заменяется на ДМСО [64]. В любом случае возможное взаимопревращение комплексов 5, 6 → 3 или 7 → 1, 2 требует дополнительных исследований.
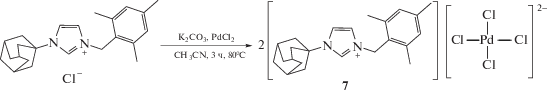
Схема 4.
Превращение, показанное на схеме 4, можно осуществить в кипящей воде вместо ацетонитрила, что позволит сэкономить время реакции (сокращается до 5 мин), при этом не происходит образования палладиевого зеркала, но выход практически не меняется (25 и 26% соответственно).
В ИК-спектрах комплексов 1–7 наблюдается интенсивная полоса в области 1144–1153 см–1, характерная для катиона адамантилимидазолия. Эта полоса отсутствует в PdII–NHC-комплексах адамантилимидазолия, описанных нами ранее [48]. Кроме того, в ИК-спектрах соединений 1–4 присутствуют интенсивные полосы валентных колебаний S–O в области 1092–1142 см–1, характерные для комплексов транс-PdCl2(ДМСО)2 [65].
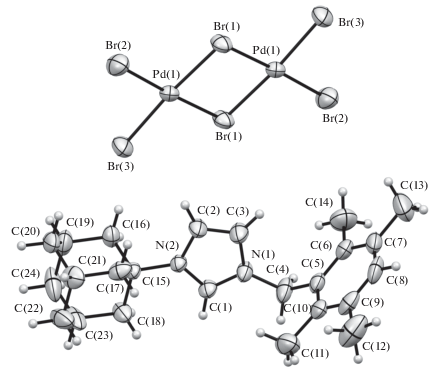
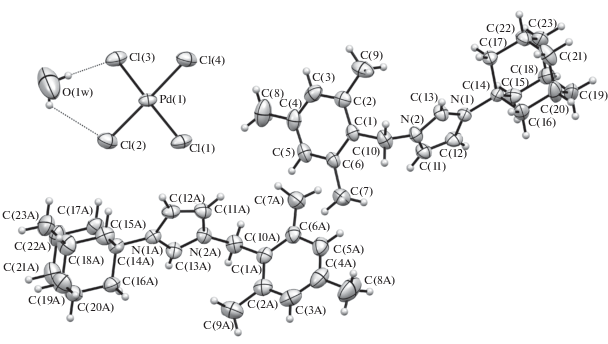
Структура комплексов 1, 3–7 подтверждена рентгеноструктурным анализом (рис. 1–6).
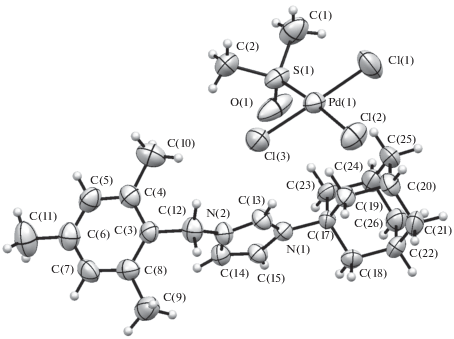
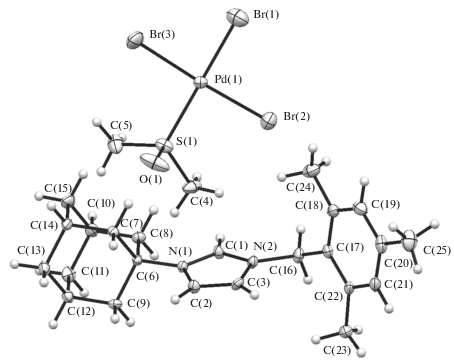
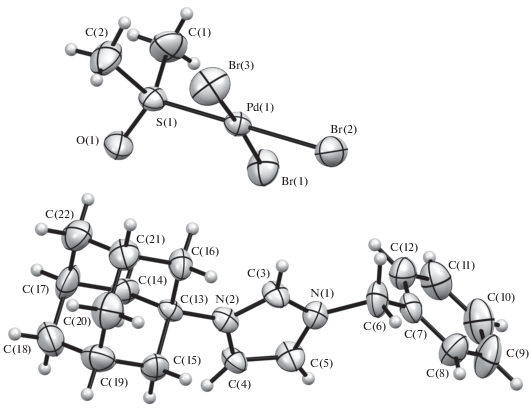
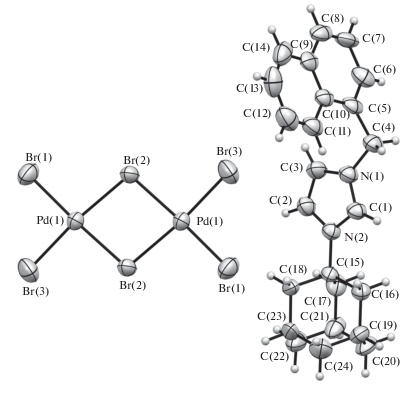
По данным РСА, комплексы 1 и 3 изоструктурны, кристаллизуются в центросимметричной пространственной группе моноклинной системы. Очевидные различия связаны с природой комплексных анионов, геометрические параметры которых соответствуют найденным ранее [19–26]. Комплекс 4 кристаллизуется в центросимметричной пр.гр. P1 триклинной сингонии. Геометрия аниона в комплексе 4 близка к геометрии аниона в комплексе 3. Распределение длин связей в имидазольном цикле катиона имидазолия в комплексах 1, 3 и 4 указывает на ожидаемо сильную делокализацию заряда. Отличительной особенностью кристаллической упаковки комплексов является наличие укороченных (на 0.15–0.30 Å меньше суммы ван-дер-ваальсовых радиусов) контактов С–H…Hal между катионом имидазолия и комплексным анионом. Подобные контакты, вероятно, указывают на сильную поляризацию С–Н-связей и проявление С–Н-кислотности катиона имидазолия.
Комплекс 5 кристаллизуется в центросимметричной пр. гр. моноклинной сингонии. Центросимметричный двухъядерный анион плоский в пределах 0.08 Å с характерной для структур подобного типа геометрией [58].
Комплекс 6 кристаллизуется в центросимметричной пр. гр. P1 триклинной сингонии в виде сольвата с молекулой воды (на рисунке не показана). Анион, разупорядоченный по двум позициям с заселенностью минорной компоненты 0.411(17), по структуре аналогичен аниону в комплексе 5. Значимые специфические межмолекулярные взаимодействия в кристаллах комплексов 5, 6 отсутствуют.
Два кристаллографически независимых катиона и анион комплекса 7 кристаллизуются в центросимметричной пр. гр. P1 триклинной сингонии в виде сольвата с молекулой воды. Геометрия катионов в комплексах 5–7 близка к таковой в комплексах 1, 3, 4. Атом палладия в анионе имеет обычное плоскоквадратное окружение. Помимо водородных связей между молекулой воды и атомами хлора в стабилизации кристаллической упаковки задействовано несколько укороченных контактов C–H⋅⋅⋅O и C–H⋅⋅⋅Cl, в том числе с участием связей С–Н катиона имидазолия.
Известно, что солеобразный характер комплексов можно подтвердить наличием у них электропроводности в растворе [66]. Ионный характер наших комплексов был подтвержден изучением их электропроводности на примере комплекса 1, когда с ростом концентрации соли в ацетонитриле наблюдается закономерный рост этого параметра раствора (табл. 2).
Вещества 1–3, 5–7 испытаны нами как ингибиторы МАО на митохондриях мозга мышей. МАО-ингибирующая активность установлена для всех соединений, и она на уровне препаратов сравнения разагалина и селегелина, хотя и не превзошла их (табл. 3). Показано, что бромидные комплексы 3, 5, 6 превосходят хлоридные 1, 2, 7 по активности. Из них наибольшую активность проявил комплекс 3. Исходная для него соль имидазолия также проверена, МАО-ингибирующая активность для нее не выявлена.
Таблица 3.
Влияние соединений 1–6 (100 мкМ)* на активность моноаминоксидазы
| Соединение | Активность фермента МАО, % |
|---|---|
| Контроль | 97.0 ± 14.3 |
| Разагалин | 17.2 ± 3.3 |
| Селегелин | 16.9 ± 2.6 |
| 1 | 20.1 ± 2.2 |
| 2 | 19.2 ± 4.6 |
| 3 | 17.6 ± 4.3 |
| 5 | 18.5 ± 2.4 |
| 6 | 17.9 ± 2.2 |
| 7 | 20.8 ± 2.5 |
Согласно литературным данным, для комплексов металлов при испытании биологической активности имеют значение растворитель [67] и катионы [68]. В частности, при испытании на МАО-ингибирующую активность используется фосфатно-солевой буфер, что может привести к образованию труднорастворимого фосфата палладия, как утверждается в работе [69]. Контрольными опытами нами установлено, что за время испытания МАО-ингибирующей активности (1.5 ч при 37°С, солефосфатный буфер и ДМСО) заметного разложения наших комплексов не происходит, и, таким образом, была показана приемлемость данной методики для биологических испытаний.
Поскольку основное биологическое применение галогеновых комплексов благородных металлов – токсичность в отношении раковых клеток, нами изучена цитотоксическая активность соединений 1, 3. Вещества не проявляют цитотоксического действия как в отношении клеток рабдомиосаркомы (RD TE 32), так и в отношении клеток карциномы толстой кишки (HCT116), так как рассчитанные для них концентрации IC50 превышают 200 мкM.
ЗАКЛЮЧЕНИЕ
Найдены синтетические подходы к трем типам комплексов катиона адамантилимидазолия с анионами Pd(ДМСО)(Hal3)– (1–4), ${\text{P}}{{{\text{d}}}_{2}}{\text{Br}}_{6}^{{2 - }}$ (5, 6) и ${\text{PdCl}}_{4}^{{2 - }}$ (7). Строение комплексов подтверждено методом РСА. Установлено, что МАО-ингибирующая активность находится на уровне препаратов сравнения. Комплексы с анионом трибромпалладия обладают большей МАО-ингибирующей активностью, чем трихлорзамещенные.
БЛАГОДАРНОСТЬ
Работа поддержана РФФИ (грант № 17-03-00456-а) и комплексной программой УрО РАН “Фундаментальные науки – медицине” (№ 18-7-3-4).
Авторы выражают благодарность инженеру И.А. Борисовой за съемку ИК-спектров, научному сотруднику Е.В. Байгачевой за выполнение элементного анализа, ведущему инженеру О.А. Майоровой, к.х.н. И.Г. Мокрушину и А.Р. Галееву – за запись ЯМР 1Н и 13С-спектров.
Список литературы
Molnar A. (Ed.) Palladium-Catalysed Coupling Reactions: Practical Aspects and Future Developments, Weinheim: Wiley-VCH, 2013. 511 p. doi 10.1002/ 9783527648283
Hongbo Li, Seechurn C.C.C.J., Colacot T.J. // ACS Catal. 2012. V. 2. P. 1147. doi 10.1021/cs300082f
Nolan S.P. // N-Heterocyclic Carbenes Effective Tools for Organometallic Synthesis, Weinheim: Wiley-VCH, 2014. 543 p. doi 10.1002/9783527671229
Kantchev E.S.B., O’Brien C.J., Organ M.G. // Angew. Chem. Int. Ed. 2007. V. 46. № 16. P. 2768. doi 10.1002/anie.200601663
Джеваков П.Б., Асаченко А.Ф., Кашин А.Н. и др. // Изв. АН, сер. хим. 2014. Т. 64. № 4. С. 890 [Dzhevakov P.B., Asachenko A.F., Nechaev M.S. et al. // Russ. Chem. Bull. 2014. V. 64. № 4. P. 890. doi 10.1007/s11172-014-0525-7]
Herrmann W.A., Schwarz J., Gardiner M.G. // Organometallics. 1999. V. 18. P. 4082. doi 10.1021/om990326k
Herrmann W.A., Resinger C.P., Spiegler M. // J. Organomet. Chem. 1998. V. 557. № 1. P. 93. doi 10.1016/S0022-328X(97)00736-5
Linningher C.S., Herdtweck E., Hoffmann S.D., Herrmann W.A. // J. Mol. Struct. 2008. V. 890. № 1–3. P. 192. doi 10.1016/j.molstruc.2008.05.037
Sureshbabu B., Ramkumar V., Sankararaman S. // Dalton Trans. 2014. V. 43. P. 10710. doi 10.1039/C4DT01112K
Hohloch S., Deibel N., Schweinfurth D. et al. // Eur. J. Inorg. Chem. 2014. V. 2014. № 12. P. 2131. doi 10.1002/ejic.201301339
Bernhammer J.C., Chong N.X., Jothibasu R. et al. // Organometallics. 2014. V. 33. № 13. P. 3607. doi 10.1021/om500566n
Huynh H.V., Lee. C.-S. // Dalton Trans. 2013. V. 42. P. 6803. doi 10.1039/C3DT50237F
Heckenroth M., Kluzer E., Neels A., Albrecht M. // Angew. Chem. Int. Ed. 2007. V. 46. № 33. P. 6293. doi 10.1002/anie.200702199
Heckenroth M., Kluser E., Neels A., Albrecht M. // Dalton Trans. 2008. P. 6242. doi 10.1039/B812405A
Danopoulos A.A., Braunstein P., Stylianides N., Wesolek M. // Organometallics. 2011. V. 30. № 24. P. 6514. doi 10.1021/om200951m
Song H., Yan N., Fei Z. et al. // Catalysis Today. 2012. V. 183. № 1. P. 172. doi 10.1016/j.cattod.2011.12.008
Lu Z., Williams T.J. // ACS Catalysis. 2016. V. 6. № 10. P. 6670. doi 10.1021/acscatal.6b02101
Кукушкин В.Ю., Власова Р.А., Палзухина Ю.Л. // Журн. прикл. химии. 1968. Т. 41. С. 2381.
Hazell A., McKenzie C.J., Nielsen L.P. // Polyhedron. 2000. V. 19. № 11. P. 1333. doi 10.1016/S0277-5387(00)00409-5
Meyer D., Taige M.A., Zeller A. et al. // Organometallics. 2009. V. 28. № 7. P. 2142. doi 10.1021/om8009238
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Журн. неорган. химии. 2013. Т. 58. № 5. С. 616 [Sharutin V.V., Senchurin V.S., Sharutina O.K. // Russ. J. Inorg. Chem. 2013. V. 58. № 5. P. 543. doi 10.1134/S0036023613050203]
Lang C., Pahnke K., Kiefer C. et al. // Polym. Chem. 2013. V. 4. P. 5466. doi 10.1039/C3PY90071A
Guest D., Menezes da Silva V.H., de Lima Batista A.P. at al. // Organometallics. 2015. V. 34. № 11. P. 2463. doi 10.1021/om5012038
Huynh H.V., Han Y., Ho J.H.H., Tan G.K. // Organometallics. 2006. V. 25. № 13. P. 3267. doi 10.1021/ om060151w
Liu Q.-X., Chen A.-H., Zhao X.-J. et al. // CrystEngComm. 2011. V. 13. P. 293. doi 10.1039/C0CE00142B
Buchalski P., Pacholski R., Chodkiewicz K. et al. // Dalton Trans. 2015. V. 44. P. 7169. doi 10.1039/C4DT03786C
Yen S.K., Koh L.L., Huynh H.V., Hor T.S.A. // Aust. J. Chem. 2009. V. 62. № 9. P. 1047. doi 10.1071/ CH09196
Meyer D., Taige M.A., Zeller A. et al. // Organometallics. 2009. V. 28. № 7. P. 2142. doi 10.1021/om8009238
Schroeter F., Soellner J., Strassner T. // ACS Catal. 2017. V. 7. № 4. P. 3004. doi 10.1021/acscatal.6b03655
Schroeter F., Strassner T. // Eur. J. Inorg. Chem. 2017. V. 2017. P. 4231. doi 10.1002/ejic.201701000
Baquero E.A., Silbestri G.F., Gómez-Sal P. et al. // Organometallics. 2013. V. 32. № 9. P. 2814. doi 10.1021/om400228s
Baquero E.A., Flores J.C., Perles J. et al. // Organometallics. 2014. V. 33. № 19. P. 5470. doi 10.1021/ om500753v
Buhl H., Ganter C. // J. Organomet. Chem. 2016. V. 809. P. 74. doi 10.1016/j.jorganchem.2016.02.034
Borrè E., Dahm G., Aliprabdi A. et al. // Organometallics. 2014. V. 33. P. 4374. doi 10.1021/om5003446
Velders A.H., Bergamo A., Alessio E. et al. // J. Med. Chem. 2004. V. 47. № 5. P. 1110. doi 10.1021/ jm030984d
Musumeci D., Rozza L., Merlino A. et al. // Dalton Trans. 2015. V. 44. P. 13914. doi 10.1039/C5DT01105A
Schleicher D., Leopold H., Strassner T. // J. Organomet. Chem. 2017. V. 829. P. 101. doi 10.1016/j.jorganchem.2016.10.036
Gautier A., Cisnetti F. // Metallomics. 2012. V. 4. P. 23. doi 10.1039/c1mt00123j
Karaca Ö., Meier-Menches S.M., Casini A., Kühn F.E. // Chem. Commun. 2017. V. 53. P. 8249. doi 10.1039/C7CC03074F
Schmidt A., Molano V., Hollering M. et al. // Chem. Eur. J. 2016. V. 22. № 7. P. 2253. doi 10.1002/chem.201504930
Тихомиров А.Г., Иванова Н.А., Ерофеева О.С. и др. // Коорд. химия. 2003. Т. 29. № 7. С. 525.
Горбачева Л.Б., Тихомиров А.Г., Дедер Л.Ю. и др. // Хим.-фарм. журн. 2008. Т. 42. № 2. С. 3 [Gorbacheva P.B., Tikhomirov A.G., Dederer L.Yu. // Pharm. Chem. J. V. 42. № 2. P. 53. 10.1007/s11094-008-0058-1]
Касьяненко Н.А., Левыкина Е.В., Ерофеева О.С. и др. // Журн. структур. химии. 2009. Т. 50. № 5. С. 1034 [Kas’yanenko N.A., Levykina E.V., Erofeeva O.S. et al. // J. Struct. Chem. 2009. V. 50. № 5. P. 996. 10.1007/s10947-009-0148-2]
Ефименко А., Добрынина Н.А., Шишилов О.Н. и др. // Коорд. химия. 2012. Т. 38. № 4. С. 243 [Efimenko I.A., Shishilov O.N., Ivanova N.A. et al. // Russ. J. Coord. Chem. 2012. V. 38. № 4. P. 233. doi 10.1134/ S1070328412020029]
Грехова А.К., Горбачева Л.Б., Иванова Н.А. и др. // Биомедицинская химия. 2013. Т. 59. № 1. С. 107. [Grekhova A.K., Gorbacheva L.B., Ivanova N.A. et al. // Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry. 2013. V. 7. №. 3. P. 226. 10.1134/S1990750813030050]
Nosova Y.N., Zenin I.V., Maximova V.P. et al. // Bioinorg. Chem. Appl. 2017. V. 2017. In press. doi 10.1155/2017/4736321
Государственный реестр лекарственных средств. Официальное издание: в 2 т. М.: Медицинский совет, 2009. Т. 2. Ч. 1. 568 с.; ч. 2 560 с.
Денисов М.С. Дис. … канд. хим. наук. Пермь, 2015. 164 с.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339. doi 10.1107/S0021889808042726
Sheldrick G.M. // Acta Crystallogr. 2008. V. A64. P. 112. doi 10.1107/S0108767307043930
Sheldrick G.M. // Acta Crystallogr. 2015. V. C71. P. 3. doi 10.1107/S2053229614024218
Mercury 3.3 (Build RC5) [Электронный ресурс] – прикладная прогр. (132Мб). Cambridge: Cambridge Crystallographic Data Centre, 2013. http://www.ccdc.cam.ac.uk/mercury/.
Thull U., Testa B. // Biochem. Pharmacol. 1994. V. 47. № 12. P. 2307. doi 10.1016/0006-2952(94)90271-2
Andrade J.M., Passos C.S., Dresch R.R. et al. // Brazil. Phcog. Mag. 2014. V. 10. № 1. P. s100. doi 10.4103/0973-1296.127354
Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. // J. Biol. Chem. 1951. V. 193. № 1. P. 265. http://www.jbc.org/content/193/1/265.short
Gonçalves B.M.F., Salvador J.A.R., Marín S., Cas-cante M. // Eur. J. Med. Chem. 2016. V. 114 P. 101. doi 10.1016/ j.ejmech.2016.02.057
Литвин Е.А., Колыванов Г.Б., Жердев В.П. // Фармокинетика и фармодинамика. 2012. V. 1. P. 18.
Neumüller B., Chitsaz S., Dehnicke K. // Z. Anorg. Allg. Chem. 2002. V. 628. № 3. P. 523. doi 10.1002/1521-3749(200203)628:3<523:AID-ZAAC523>3.0.CO;2-C
Kuhn N., Göhner M., Steimann M., Nachti. // Z. Kristallogr NCS. 1999. V. 214. № 4. P. 565. doi 10.1515/ncrs-1999-0483
Wang X., Fei Z., Geldbach T.J. et al. // Organometallics. 2008. V. 27. № 15. P. 3971. doi 10.1021/om800355g
Silarska E., Trzeciak A.M., Pernak J., Skrzypcza A. // Appl. Catal. A: General. 2013. V. 466. P. 216. doi j.apcata.2013.06.046
Huang Z., Li F., Chen B. et al. // ChemSusChem. 2013. V. 6. № 8. P. 1337. doi 10.1002/cssc.201300289
Adams C.J., Lusi M., Mutambi E.M., Orpen A.G. // Chem. Commun. 2015. V. 51. P. 9632. doi 10.1039/ C5CC02924D
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Ильченко И.А. // Коорд. химия. 2015. Т. 41. № 7. С. 462 [Sharutin V.V., Sharutina O.K., Senchurin V.S., Il’chenko I.A. // Russ. J. Coord. Chem. 2015. V. 41. № 7. P. 262. doi 10.1134/S1070328415070088
Ливингстон С. // Химия рутения, родия, палладия, осмия, иридия, платины. М.: Мир, 1978. С. 215.
Лященко А.К., Логинова Д.В., Лилеев А.С. и др. // Коорд. химия. 2009. Т. 35. № 9. С. 643. [Lyashchenko A.K., Loginova D.V., Lileev A.S. // Russ. J. Coord. Chem. 2009. V. 35. № 9. P. 633. doi 10.1134/S1070328409090012]
Huang H., Humbert N., Bizet V. et al. // J. Organomet. Chem. 2017. V. 839. P. 15. doi 10.1016/j.jorganchem.2016.12.010
Ефименко И.А., Иванова Н.А., Ерофеева О.С. и др. // Коорд. химия. 2009. Т. 35. № 4. С. 276. [Efimenko I.A., Ivanova N.A., Erofeeva O.S. et al. // Russ. J. Coord. Chem. 2009. V. 35. № 4. P. 272. doi 10.1134/S107032840904007.]
Ефименко И.А., Чураков А.В., Иванова Н.А. и др. // Журн. неорган. химии. 2017. Т. 62. № 11. С. 1476. [Efimenko I.A., Churakov A.V., Ivanova N.A. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 11. P. 1469. doi 10.1134/S0036023617110043]
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии