

Журнал неорганической химии, 2019, T. 64, № 1, стр. 15-22
Комплексные соединения сурьмы: {[2,6-(OMe)2C6H3]3SbCH2C(O)OEt}$_{2}^{ + }$[Hg2I6]2– и {[2,6-(OMe)2C6H3]3SbMe}$_{2}^{ + }$[HgI4]2– ⋅ ДМСО. Синтез и строениеИ. В. Егорова, В. В. Жидков, И. П. Гринишак, И. Ю. Багрянская, Н. В. Первухина, И. В. Ельцов, Н. В. Куратьева
И. В. Егорова 1, *, В. В. Жидков 1, И. П. Гринишак 1, И. Ю. Багрянская 2, 3, Н. В. Первухина 3, 4, И. В. Ельцов 3, Н. В. Куратьева 3, 4
1 Благовещенский государственный педагогический университет
675000 Благовещенск, ул. Ленина, 104, Россия
2 Новосибирский институт органической химии им. Н.Н. Ворожцова СО РАН
630090 Новосибирск, пр-т Академика Лаврентьева, 9, Россия
3 Новосибирский национальный исследовательский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
4 Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, пр-т Академика Лаврентьева, 3, Россия
* E-mail: bgpu.chim.egorova@mail.ru
Поступила в редакцию 07.12.2017
После доработки 04.07.2018
Принята к публикации 19.02.2018
Аннотация
Установлено, что этилиодацетат и 1,4-дииодбутан алкилируют триарилcурьму Ar3Sb с образованием [Ar3SbCH2C(O)OEt]+I– и [Ar3Sb(CH2)4I]+I–, [Ar3Sb(CH2)4SbAr3]2+${\text{I}}_{2}^{ - },$ где Ar = 2,6-(OMe)2C6H3. Взаимодействием [Ar3SbCH2C(O)OEt]+I– и [Ar3SbMe]+I– с дииодидом ртути получены и исследованы методом РСА $\left[ {{\text{A}}{{{\text{r}}}_{{\text{3}}}}{\text{SbC}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}\left( {\text{O}} \right){\text{OEt}}} \right]_{2}^{ + }$[Hg2I6]2– и $\left[ {{\text{A}}{{{\text{r}}}_{{\text{3}}}}{\text{SbMe}}} \right]_{2}^{ + }$[HgI4]2– ⋅ ДМСО. Атомы сурьмы и иода имеют искаженную тетраэдрическую координацию. Углы CSbC и IHgI находятся в интервалах 103.28(14)°–116.68(14)°, 103.7(4)°–115.5(4)° и 98.122(9)°–125.590(12)°, 102.66(2)°–115.64(2)°.
ВВЕДЕНИЕ
Присоединение иодистого алкила к алкильным моно- и биядерным производным сурьмы(III), сопровождаемое образованием связей Sb–C, приводит к получению широкого ряда иодидов тетраалкилстибония общей формулы [R3R'Sb]+I– (R = Me, Еt, н-Pr, н-Bu, изо-Bu, н-Am; R' = Me, Еt) и [R2SbMe(CH2)nMeSbR2]2+${\text{I}}_{2}^{ - }$ (R = Еt, трет-Bu; n = 4, 6) соответственно [1–5]. Взаимодействием бромистого алкила и трибутилсурьмы получены сурьмаорганические соединения общей формулы [Alk3SbСН2Е]+Br–, где Alk = н-Bu; E = Ph, CH=CH2, CH=CHC(O)OEt, C(O)OMe, CN, CH=CHR; R = Me, н-Pr, изо-Pr. Комплексы применяют в синтезе вторичных спиртов из соответствующих альдегидов R'CHO (R' = Ph, 4-ClC6H4, 4-MeC6H4, PhCH=CH, пиридин-2-ил, 4-BrC6H4) [6, 7]. Проведено алкилирование триэтил- и триметилсурьмы метиловым эфиром бромуксусной кислоты [8, 9].
Менее изучено взаимодействие галоидных алкилов с арильными соединениями сурьмы(III). Алкилирование диметил- и диэтилфенилсурьмы иодистым метилом приводит к образованию иодистого триалкилфенилстибония [10]. Долгое время не удавалось присоединить галоидные алкилы к соединениям сурьмы(III) c двумя или тремя ароматическими заместителями [3]. Поэтому для получения стибониевых солей [Аr3RSb]+[BF4]– (Аr = Ph, o-Tol, м-Tol, п-Tol, (3,4-Me)2C6H3, (2,4-Me)2C6H3, (2,4,6-Me)3C6H2; R = Me, Ph) используют более активные алкилирующие агенты: борфториды триметилоксония или дифенил-иодония [11–14]. Последующее действие иодида натрия приводит к соответствующим иодидным комплексам [7, 10]. Тетрафторборат триметилоксония и метилтрифторметансульфонат алкилируют биядерное сурьмаорганическое соединение 1,2-(Ph2Sb)2C6H4 с образованием комплексов [1,2-(Ph2MeSb)2C6H4][BF4]2 и [1,2-(Ph2MeSb)2C6H4][OTf], эффективно катализирующих гидросилилирование бензальдегида в мягких условиях [15]. Известен метод синтеза [Ph3MeSb]+Cl– взаимодействием бис(хлорметил)цинка и трифенилсурьмы с последующим гидролизом образующегося цинкорганического соединения [16]. Описано получение трифенилалкилстибониевых цвиттер-ионов, где алкилирующими агентами являлись дибромид бромметилиндия(III) или хлорангидрид фенилметансульфоновой кислоты в присутствии пероксида водорода [17, 18]. Присоединением галоидных алкилов к триарилсурьме получены галогениды тетраорганилсурьмы [Аr3RSb]+X– [Аr = 2,6-(OMe)2C6H3; R = Me, Et, н-Bu, СН2СН=СН2; X = Cl, Br, I] [19].
Цель работы – исследование направленного синтеза стибониевых комплексов, содержащих потенциальные координирующие центры – атомы кислорода карбонильных и/или метоксильных групп – в органических заместителях при атоме сурьмы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Изучена возможность алкилирования трис(2,6-диметоксифенил)сурьмы этиловым эфиром иодуксусной кислоты и 1,4-дииодбутаном. Синтезированы и изучены методом ЯМР-спектроскопии иодид трис(2,6-диметоксифенил)(этоксикарбонилметил)сурьмы {[2,6-(OMe)2C6H3]3SbCH2C(O)OEt}+I– (1), иодид трис(2,6-диметоксифенил)(4-иодбутил)сурьмы {[2,6-(OMe)2C6H3]3Sb(CH2)4I}+I– (2), иодид 1,4-ди[трис(2,6-диметоксифенил)антимонил]бутана {[2,6-(OMe)2C6H3]3Sb(CH2)4Sb[C6H3(OMe)2-2,6]3}2+${\text{I}}_{2}^{ - }$ (3).
Взаимодействием дииодида ртути с соединениями 1, {[2,6-(OMe)2C6H3]3SbMe}+I– получены и исследованы методом РСА комплексные соединения ртути(II) $\left\{ {{{{\left[ {{\text{2,6 - (OMe}}{{{\text{)}}}_{{\text{2}}}}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{3}}}}} \right]}}_{{\text{3}}}}{\text{SbC}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}\left( {\text{O}} \right){\text{OEt}}} \right\}_{2}^{ + }$[Hg2I6]2– (4) и $\left\{ {{{{\left[ {2,6 - {{{\left( {{\text{OMe}}} \right)}}_{2}}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{3}}} \right]}}_{3}}{\text{SbMe}}} \right\}_{2}^{ + }$[HgI4]2– ⋅ ДМСО (5).
Реакции проводили при комнатной температуре (рис. 1).
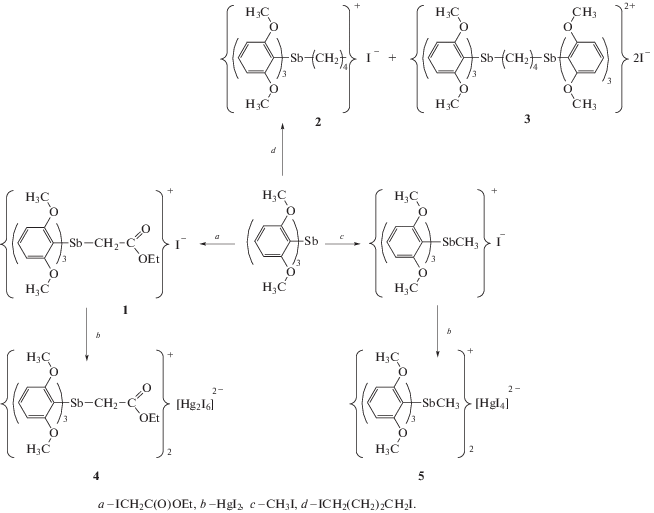
Синтез 1. К 1.03 г трис(2,6-диметоксифенил)сурьмы (1.93 ммоль) в 10 мл хлороформа при перемешивании приливали 0.41 г (1.93 ммоль) этилового эфира моноиодуксусной кислоты. Реакционную смесь оставляли на 12 ч. Растворитель испаряли, твердый остаток промывали диэтиловым эфиром (5 × 10 мл). Получили 1.40 г (97%) соединения 1, tпл = 184°С (разл.). ИК-спектр (ν, см–1): 3059, 3000, 2973, 2941, 2835, 1728, 1716, 1587, 1576, 1475, 1429, 1400, 1388, 1363, 1305, 1256, 1184, 1173, 1126, 1102, 1022, 891, 866, 804, 792, 775, 740, 713, 615, 594, 499.
Спектр ЯМР 1H, δ, м.д.: 0.88 т (3H, CH2–CH3, J 7.2 Гц), 3.63 с (18H, CH3–O), 3.65 с (2H, CH2–Sb), 3.85 к (2H, CH2–CH3, J 7.2 Гц), 6.65 д (6H, 3,5-Ph, J 8.4 Гц), 7.49 т (3H, 4-Ph, J 8.4 Гц).
Спектр ЯМР 13C{1H}, δ, м.д.: 13.65 (CH3–CH2), 29.74 (CH2–Sb), 56.52 (CH3–O), 61.69 (CH3–CH2), 104.77 (1-Ph), 104.87 (3,5-Ph), 135.78 (4-Ph), 162.15 (2,5-Ph), 167.32 (C=O).
Синтез 2, 3. К 0.60 г (1.94 ммоль) 1,4-дииодбутана в 70 мл хлороформа по каплям при перемешивании в течение 30 мин приливали раствор 1.00 г (1.88 ммоль) трис(2,6-диметоксифенил)сурьмы в 100 мл хлороформа. Раствор перемешивали 15 мин. Растворитель испаряли, остаток промывали диэтиловым эфиром (5 × 20 мл). Выделили 1.42 г бесцветного мелкокристаллического продукта, представляющего собой, по данным ЯМР, смесь двух веществ: ~90% 2 и ~10% 3.
Для 2. Спектр ЯМР 1H, δ, м.д.: 1.74 м (2H, 2-Bu), 1.88 м (2H, 3-Bu), 2.77 м (2H, 1-Bu), 3.12 т (2H, 4-Bu, J 6.6 Гц), 3.62 с (18H, CH3–O), 6.64 д (6H, 3,5-Ph, J 8.3 Гц), 7.47 т (3H, 4-Ph, J 8.4 Гц).
Спектр ЯМР 13C{1H}, δ, м.д.: 6.11 (1-Bu), 25.65 (2-Bu), 25.73 (1-Bu), 35.00 (3-Bu), 56.52 (CH3–O), 104.29 (1-Ph), 104.73 (3,5-Ph), 135.43 (4-Ph), 162.40 (2,5-Ph).
Для 3. Спектр ЯМР 1H, δ, м.д.: 1.73 м (4H, 2,3-Bu), 2.77 м (4H, 1,4-Bu), 3.57 с (18H, CH3–O), 6.63 д (6H, 3,5-Ph), 7.47 т (3H, 4-Ph).
Спектр ЯМР 13C{1H}, δ, м.д.: 26.00 (1,4-Bu), 27.32 (2,3-Bu), 56.71 (CH3–O), 104.13 (1-Ph), 104.87 (3,5-Ph), 135.52 (4-Ph), 162.40 (2,5-Ph).
Синтез 4, 5. К 0.50 г (0.66 ммоль) соединения 1 в 20 мл ДМСО приливали раствор 0.30 г (0.66 ммоль) дииодида ртути в 10 мл ДМСО при перемешивании. Растворитель испаряли. Получили 0.78 г (97%) светло-зеленых кристаллов комплекса 4, tпл = 185°С.
ИК-спектр (ν, см–1): 3086, 3069, 3001, 2972, 2937, 2834, 1724, 1587, 1576, 1471, 1427, 1385, 1362, 1306, 1286, 1257, 1180, 1102, 1032, 1017, 889, 869, 804, 800, 781, 771, 740, 715, 708, 617, 596, 584, 499.
Соединение 5 получали аналогично из 0.50 г (0.74 ммоль) иодида трис(2,6-диметоксифенил)метилстибония, синтезированного в соответствии с [19], и 0.17 г (0.37 ммоль) дииодида ртути. Получили 0.65 г (97%) светло-зеленых кристаллов комплекса 5, tпл = 168°С.
ИК-спектр (ν, см–1): 3151, 3066, 3024, 3006, 2991, 2970, 2937, 2848, 2835, 1587, 1576, 1471, 1427, 1303, 1257, 1172, 1151, 1102, 1035, 1020, 950, 891, 829, 781, 773, 740, 717, 696, 594.
| С | Н | O | |
| Найдено, %: | 33.07; | 3.61; | 10.70. |
| Для C50H60HgI4O12Sb2 ⋅ 0.92C2H6SO (C51.84H65.52HgI4O12.92S0.92Sb2) | |||
| вычислено, %: | 33.18; | 3.53; | 11.02. |
ИК-спектры соединений снимали на Фурье-спектрометре ФСМ 2202 в интервале 500–7000 см–1 в таблетках с KBr. Элементный анализ выполняли на CHN-анализаторе Carlo Erba (модель 1106).
Спектры ЯМР регистрировали на спектрометре Bruker Avance III 500 с рабочей частотой 500.03 МГц для ядер 1H и 125.73 МГц для ядер 13С. Спектры записывали в растворе 25 мг соединения в 0.6 мл CDCl3. В качестве стандартов использовали сигналы растворителя: δ = 7.26 м.д. для остаточных протонов в спектре ЯМР 1H и δ = 77.23 м.д. для спектров ЯМР 13С. Отнесение сигналов проводили на основании гомоядерной 1H,1H-COSY, гетероядерных 13C,1H-HMBC и 13C,1H-HSQC двумерных корреляций.
РСА соединений 4, 5 выполнен на дифрактометре KAPPA APEX II (Bruker) с двухкоординатным CCD-детектором (MoKα-излучение, графитовый монохроматор, ω–φ-сканирование). Структуры расшифрованы прямым методом и уточнены полноматричным методом наименьших квадратов в анизотропном приближении по программе SHELXL-97 [20]. Положение атомов водорода рассчитано геометрически и уточнено в модели “наездника” (параметры атомов водорода рассчитаны в каждом цикле уточнения по координатам соответствующих атомов углерода). В соединении 5 частично разупорядочены анион [HgI4]2– и молекула ДМСО (второе положение молекулы растворителя не определено). Основные кристаллографические характеристики соединений 4, 5, результаты эксперимента и параметры уточнения структур приведены в табл. 1. Данные РСА соединений 4, 5 депонированы в Кембриджском центре кристаллографических данных (CCDC, № 1549982, 1549983).
Таблица 1.
Кристаллографические характеристики соединений 4 и 5, данные эксперимента и параметры уточнения структур
| Параметр | 4 | 5 |
|---|---|---|
| Формула | C56H68Hg2I6O16Sb2 | C51.84H65.52HgI4O12.92S0.92Sb2 |
| М | 2403.18 | 1876.57 |
| Температура, K | 200(2) | 173(2) |
| Сингония | Моноклинная | |
| Пр. гр. | C2/c | P21/n |
| a, Å | 28.2103(12) | 17.8459(9) |
| b, Å | 11.2798(5) | 30.1024(16) |
| c, Å | 23.1211(8) | 24.3373(13) |
| β, град | 104.929(1) | 106.236(2) |
| V, Å3 | 7108.9(5) | 12552.7(11) |
| Z | 4 | 8 |
| ρвыч, г/cм3 | 2.245 | 1.986 |
| μ(MoKα), мм–1 | 7.721 | 5.347 |
| F(000) | 4448 | 7109 |
| Размеры кристалла, мм | 0.60 × 0.45 × 0.20 | 0.65 × 0.45 × 0.35 |
| Диапазон углов θ, град | 2.07–30.07 | 1.26–26.37 |
| Число измеренных рефлексов | 81829 | 295857 |
| Число независимых рефлексов | 10421 (Rint = 0.0386) | 25677 (Rint = 0.0626) |
| Пропускание, min/mаx | 0.0903/0.3073 | 0.1287/0.2562 |
| GOOF по F 2 | 1.012 | 1.089 |
| R-фактор по I > 2σ(I) | R1 0.0305, wR2 0.0799 | R1 0.0524, wR2 0.0967 |
| R-фактор по всем отражениям | R1 = 0.0376, wR2 = 0.0839 | R1 = 0.1002, wR2 = 0.1092 |
| Остаточная электронная плотность, max/min, е/Å3 |
2.491/–2.252 | 1.773/–2.342 |
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
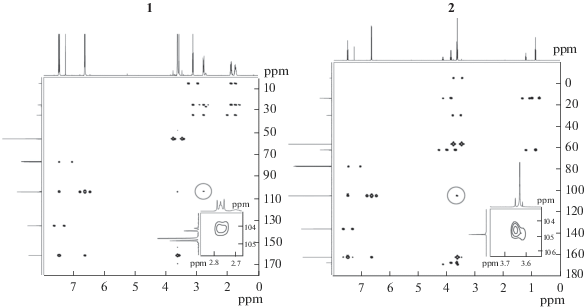
Одномерные спектры ЯМР на ядрах 1H и 13С соединений 1–3 по положению и интенсивности сигналов не противоречили предполагаемому присоединению этилиодацетата и 1,4-дииодбутана к трис(2,6-диметоксифенил)сурьме, но и не подтверждали его однозначно. Для адресного отнесения сигналов и подтверждения структур стибониевых соединений были записаны двумерные 13C,1H-корреляционные спектры. Анализ полученных данных свидетельствует об образовании в 1 и 2 новой связи Sb–CAlk. В двумерных спектрах соединений отчетливо наблюдаются кросс-пики, соответствующие взаимодействию присоединенной к атому сурьмы метиленовой группы с ипсо-углеродом фенильного фрагмента (рис. 2). Для соединения 3 данный сигнал зарегистрировать не удалось ввиду того, что в составе анализируемой смеси содержание продукта незначительно (~10%). Однако отсутствие принадлежащих данному соединению сигналов в высоком поле в спектре ЯМР 13С, характерных для фрагмента со связью C–I, позволяет предположить замещение обоих атомов галогена в 1,4-дииодбутане. Другим подтверждением симметричности структуры 3 служит наличие всего двух сигналов алифатического фрагмента в спектрах ЯМР 1H и 13С.
Состав комплексов 4 и 5, образующихся в реакциях HgI2 с 1 и {[2,6-(OMe)2C6H3]3SbMe}+I–, определяется мольным соотношением реагентов (1 : 1, 1 : 2). Согласно данным РСА, в центросимметричном биядерном анионе [Hg2I6]2– атомы ртути имеют искаженную тетраэдрическую координацию (углы IHgI составляют 98.122(9)°–125.590(12)°, рис. 3).
Рис. 3.
Общий вид катиона {[2,6-(OMe)2C6H3]3SbCH2C(O)OEt}+ и аниона [Hg2I6]2– соединения 4. Атомы водорода не показаны.
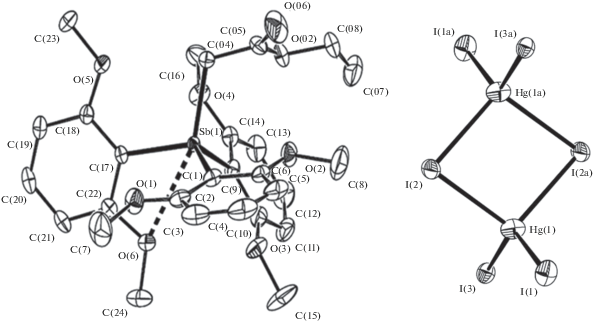
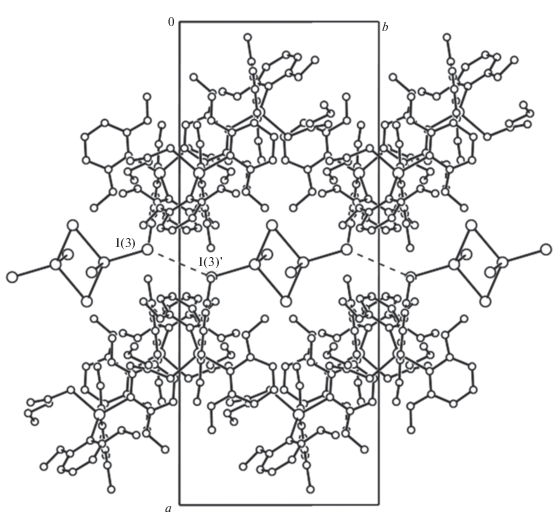
Концевые атомы иода более прочно связаны с атомами ртути (Hg–Iконц 2.6874(4), 2.6945(3) Å) по сравнению с двухкоординированными мостиковыми атомами иода (Hg–Iмост 2.9246(4), 2.9260(4) Å). Эти значения сравнимы с параметрами комплексов $\left[ {{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb}}} \right]_{2}^{ + }$[Hg2I6]2–, $\left[ {n{\text{ - To}}{{{\text{l}}}_{4}}{\text{Sb}}} \right]_{2}^{ + }$[Hg2I6]2– (Hg–Iконц 2.6910(4) и 2.6996(4); 2.7028(3) и 2.7222(3) Å; Hg–Iмост 2.8250(4) и 3.0748(5); 2.8539(3) и 2.9163(3) Å) [21]. Плоскости Hg(1)I(2)Hg(1а)I(2а) и I(1)I(3)I(1а)I(3а) практически взаимно перпендикулярны, угол между ними составляет 87.47°. Концевые атомы иода посредством взаимодействий I…I (3.944 Å) формируют вдоль оси с цепочки из анионов [Hg2I6]2– (рис. 4).
В элементарной ячейке комплекса 5 присутствуют четыре структурно неэквивалентных катиона {[2,6-(OMe)2C6H3]3SbMe}+ и два аниона [HgI4]2– (рис. 5). В тетраэдрических анионах углы IHgI составляют 102.66(2)°–115.64(2)°. Расстояния Hg–I неравноценны (2.670(5)–2.956(7) Å), что также имело место в комплексе $\left[ {n{\text{ - To}}{{{\text{l}}}_{4}}{\text{Sb}}} \right]_{2}^{ + }$[HgI4]2– (2.7719(13)–2.7908(12) Å) [21].
Рис. 5.
Независимая часть структуры соединения 5. Атомы водорода, вторая компонента разупорядочения анион–растворитель и некоторые обозначения атомов не показаны.
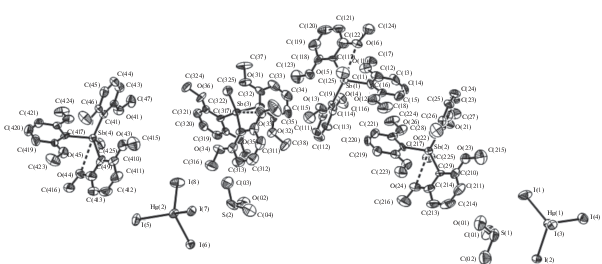
В катионах соединений 4, 5 между атомами сурьмы и кислорода метоксигрупп имеет место дополнительная координация (расстояния Sb…O 2.853–2.895 Å, сумма ван-дер-ваальсовых радиусов Sb и O 3.7 Å [22]), приводящая к появлению в тетраэдрической структуре вклада тригонально-бипирамидальной составляющей (рис. 3, 5). Наблюдается отклонение валентных углов CSbC (103.28(14)°–116.68(14)° в 4, 103.7(4)°–115.5(4)° в 5) от идеального для тетраэдра значения. Длины связей равны: Sb–CAlk 2.140(4), Sb–CAr 2.085(3)–2.093(3) Å в 4; Sb–CMe 2.085(10)–2.113(9), Sb–CAr 2.051(9)–2.114(8) Å в 5 (сумма ковалентных радиусов Sb и C составляет 2.18 Å [22]).
Отметим близость длин связей Sb−СAr, валентных углов CSbC в катионах 4, 5 и оксиде трис(2,6-диметоксифенил)сурьмы (2.073(4)–2.098(5) Å, 107.47(18)°–116.24(18)° [23]).
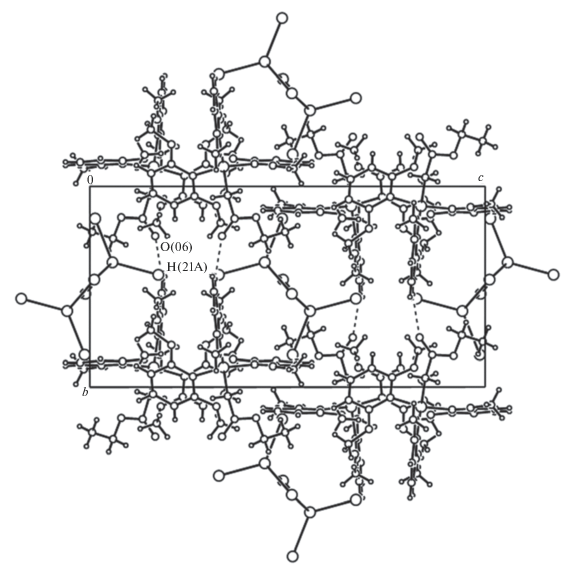
Карбонильные атомы кислорода участвуют в образовании водородных связей СAr(21)–H(21)…O(06) (H…O 2.493 Å), объединяя катионы соединения 4 в цепочку вдоль оси b (рис. 6). Включение в координационную сферу атома ртути молекулы растворителя имеет место в анионе [HgI3(ДМСО)]– [24]. Однако в соединении 5 молекулы ДМСО участвуют лишь в образовании слабых водородных связей O…H–CAr (O…H 2.48–2.67 Å) с катионом стибония.
Рис. 6.
Фрагмент упаковки структуры соединения 4. Пунктиром показаны водородные связи, формирующие цепочки катионов вдоль оси b.
Полосы поглощения в ИК-спектрах соединений 1–5 отнесены в соответствии с данными [23, 25]. Частоты колебаний связей в спектрах 1, 4, 5 находятся при 1102 см−1 [νs(О−СAlk)]; 1022, 1256 (1), 1017, 1257 (4), 1020, 1257 (5) см−1 [νаs(СМе−О−СAr)]; 2835, 2941 (1), 2834, 2937 (4), 2835, 2937 (5) см−1 [νs,аs(С−Н)]; 1716, 1728 (1), 1724 (4) см−1 [νas(OCO)].
ВЫВОДЫ
Изучено алкилирование трис(2,6-диметоксифенил)сурьмы, позволяющее расширить ряд стибониевых солей. Состав стибониевых катионов {[2,6-(OMe)2C6H3]3SbCH2C(O)OEt}+, {[2,6-(OMe)2C6H3]3Sb(CH2)4I}+, {[2,6-(OMe)2C6H3]3Sb(CH2)4Sb[C6H3(OMe)2-2,6]3}2+ определяется природой алкилирующего агента: этилиодацетата и 1,4-дииодбутана. Предложен синтетический подход и установлена структура неизвестных ранее меркуратов триарилалкилстибония: $\left\{ {{{{\left[ {{\text{2,6 - (OMe}}{{{\text{)}}}_{{\text{2}}}}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{3}}}}} \right]}}_{{\text{3}}}}{\text{SbC}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}\left( {\text{O}} \right){\text{OEt}}} \right\}_{2}^{ + }$[Hg2I6]2– и $\left\{ {{{{\left[ {{\text{2,6 - (OMe}}{{{\text{)}}}_{{\text{2}}}}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{3}}}}} \right]}}_{{\text{3}}}}{\text{SbMe}}} \right\}_{2}^{ + }$[HgI4]2–, содержащих потенциальные координирующие центры в органических заместителях при атоме сурьмы.
Список литературы
Doering W.E., Hoffmann A.K. // J. Am. Chem. Soc. 1955. V. 77. № 3. P. 521. doi 10.1021/ja01608a003
Brinnand M.E., Dyke W.J.C., Jones W.H., Jones W.J. // J. Chem. Soc. 1932. P. 1815. doi 10.1039/JR9320001815
Кочешков К.А., Сколдинов А.П., Землянский Н.Н. Методы элементоорганической химии. Сурьма, висмут. М.: Наука, 1976. С. 184.
Issleib K., Hamann B. // Z. Anorg. Allg. Chem. 1965. V. 339. № 5–6. P. 289. doi 10.1002/zaac.19653390509
Issleib K., Hamann B., Schmidt L. // Z. Anorg. Allg. Chem. 1965. V. 339. № 5–6. P. 298. doi 10.1002/zaac.19653390510
Huang Y.-Z., Liao Y. // J. Org. Chem. 1991. V. 56. № 4. P. 1381. doi 10.1021/jo00004a010
Huang Y.-Z., Zhang L.-J., Chen C., Guo Z.-G. // J. Organomet. Chem. 1991. V. 412. № 1–2. P. 47. doi 10.1016/0022-328X(91)86040-W
Issleib K., Linder R. // Justus Liebigs Annalen der Chemie. 1967. V. 707. № 1. P. 120. doi 10.1002/jlac.19677070119
Balàzs G., Balàzs L., Breunig H.J., Lork E. // Appl. Organomet. Chem. 2002. V. 16. № 1. P. 155. doi 10.1002/aoc.255
Grüttner G., Wiernik M. // Chem. Ber. 1915. V. 48. № 2. P. 1759. doi 10.1002/cber.19150480271
Henry M.C., Wittig G. // J. Am. Chem. Soc. 1960. V. 82. № 3. P. 563. doi 10.1021/ja01488a017
Henning D., Kempter G., Ahrens E. et al. // Z. Chem. 1967. V. 7. № 12. P. 463. doi 10.1002/zfch.19670071213
Henning D., Kempter G., Worlitzer K.-D. // Z. Chem. 1969. V. 9. № 8. P. 306. doi 10.1002/zfch.19690090813
Макарова Л.Г., Несмеянов А.Н. // Изв. АН СССР. ОХН. 1945. № 6. С. 617.
Hirai M., Cho J., Gabbai F.P. // Chem. Eur. J. 2016. V. 22. № 19. P. 6537. doi 10.1002/chem.201600971
Wittig G., Schwarzenbach K. // Justus Liebigs Annalen der Chemie. 1961. V. 650. № 1. P. 1. doi 10.1002/ jlac.19616500102
Felix L.A., Oliveira C.A.F., Kross R.K. et al. // J. Organomet. Chem. 2000. V. 603. № 1–2. P. 203. 10.1016@S0022-328X(00)00178-9
Пакусина А.П. Автореф. дис. … докт. хим. наук. Иркутск, 2006. 36 с.
Wada M., Miyake S., Hayashi S. et al. // J. Organomet. Chem. 1996. V. 507. № 1–2. P. 53. doi 10.1016/0022-328X(95)05716-3
Sheldrick G.M. SHELX-97, Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1997.
Cambridge Structural Database System, V. 5.38. 2016.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. № 12. С. 3015 [Batsanov S.S. // Russ. J. Inorg. Chem. 1991. V. 36. P. 1694].
Егорова И.В., Жидков В.В., Гринишак И.П., Родионова Н.А. // Журн. общ. химии. 2016. Т. 86. № 11. С. 1841 [Egorova I.V., Zhidkov V.V., Grinishak I.P., Rodionova N.A. // Russ. J. Gen. Chem. V. 86. № 11. P. 2484. doi 10.1134/S1070363216110141].
Cramer R.E., Carrie M.J.J. // Inorg. Chem. 1990. V. 29. P. 3902. doi 10.1021/ic00344a052
Smith B.C. Infrared Spectral Interpretation: A Systematic Approach. Boca Raton, FL. CRC Press, 1998. P. 288.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии