

Журнал неорганической химии, 2019, T. 64, № 12, стр. 1263-1270
Синтез замещенных производных клозо-декаборатного аниона с пептидной связью – путь к созданию биологически активных борсодержащих соединений
А. В. Нелюбин 1, И. Н. Клюкин 1, А. П. Жданов 1, *, М. С. Григорьев 2, К. Ю. Жижин 1, Н. Т. Кузнецов 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
2 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр-т, 31, корп. 4, Россия
* E-mail: zhdanov@igic.ras.ru
Поступила в редакцию 24.05.2019
После доработки 31.05.2019
Принята к публикации 17.06.2019
Аннотация
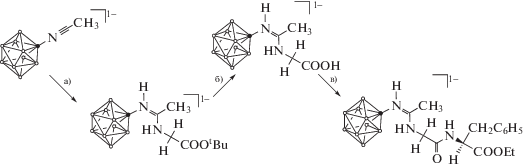
Предложен новый многостадийный синтез N-борилированного дипептида R-GlyPheOEt, в основе которого лежит реакция нуклеофильного присоединения производных аминокислот к аниону [2-B10H9NCCH3]–. Продукты каждой стадии исследованы методами ЯМР- и ИК-спектроскопии, ESI-масс-спектрометрии. Методом РСА установлена структура монокристалла (NBu4)[2-B10H9NH=C(NH2CH2COOtC4H9)CH3].
ВВЕДЕНИЕ
10B-нейтронoзахватная терапия (БНЗТ) представляет собой перспективный метод терапии для ряда опухолевых заболеваний [1]. Основной задачей при создании веществ для БНЗТ является получение соединений, сочетающих высокую селективность накопления в опухолевых тканях и низкую токсичность. История применения различных аминокислот в качестве векторных групп насчитывает уже более пятидесяти лет [2].
Наиболее изучены борсодержащие аминокислоты на основе бороновых кислот [3–7]. Низкое содержание бора в пересчете на одну молекулу действующего вещества в подобных соединениях требует использования больших количеств препарата для достижения терапевтических концентраций (15–30 мкг на 1 г опухолевой ткани) [8]. Использование в качестве борсодержащей группы производных карборанов позволяет преодолеть этот недостаток [9–12]. Высокая липофильность карборановых фрагментов требует введения в транспортные группы различных гидрофильных доменов. Для повышения селективности доставки и снижения токсичности подобные соединения часто используют в виде различных липосомальных форм [13–15]. Препараты на основе кластерных анионов бора не имеют указанных недостатков [16–18]. Особый интерес для потенциального применения в качестве агентов для БНЗТ представляют производные клозо-декаборатного аниона. Это связано с их низкой токсичностью, стабильностью в биологических средах и широко развитыми методами функционализации [19–26]. Удобный метод получения замещенных клозо-декаборатов может быть основан на реакциях нуклеофильного присоединения к экзополиэдрическому нитрилиевому заместителю [27–38] и [2+3]-циклоприсоединения [39, 40].
Цель настоящей работы – получение конъюгатов нитрилиевого производного клозо-декаборатного аниона с глицином и дальнейшее использование полученного производного как стартового соединения для создания веществ для БНЗТ методами пептидного синтеза.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Элементный анализ на углерод, водород и азот осуществляли на автоматическом газовом анализаторе CHNS-3 FA 1108 Elemental Analyser (Carlo Erba). Определение бора методом ICP MS выполнено на атомно-эмиссионном спектрометре с индуктивно-связанной плазмой iCAP 6300 Duo в ЦКП “Научно-аналитического центра ФГУП “ИРЕА” Национального исследовательского центра Курчатовский институт”.
ИК-спектры соединений записывали на ИК-фурье-спектрофотометре Инфралюм ФТ-08 (НПФ АП “Люмекс”) в области 4000–400 см–1 с разрешением 1 см–1. Образцы готовили в виде раствора в хлороформе.
Cпектры11 ЯМР 1H, 11B, 13C растворов исследуемых веществ в CD3CN записывали на импульсном фурье-спектрометре Bruker MSL-300 (Германия) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешних стандартов использовали тетраметилсилан или эфират трехфтористого бора.
Препаративную высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на изократической ВЭЖХ-системе “СТАЙЕР” (НПКФ “Аквилон”) со спектрофотометрическим детектором UVV 104 с переменной длиной волны (200–360 нм) на колонке Knauer Eurospher 110 Si.
Рентгеноструктурный анализ соединения 2 выполнен в ЦКП ИФХЕ РАН на автоматическом четырехкружном дифрактометре с двумерным детектором Bruker KAPPA APEX II (излучение MoKα) [41] (использовали фрагмент кристалла размерами 0.12 × 0.08 × 0.04 мм) при температуре 100 K.
Параметры элементарной ячейки уточнены по всему массиву данных [42]. Структура расшифрована прямым методом [43] и уточнена полноматричным методом наименьших квадратов [44] по F 2 по всем данным в анизотропном приближении для всех неводородных атомов (кроме разупорядоченных, если такие имеются). Атомы H кластера бора локализованы из разностного Фурье-синтеза электронной плотности и уточнены изотропно без каких-либо ограничений. Атомы H групп NH, CH, CH2 и CH3 размещены в геометрически вычисленных позициях и уточнены с изотропными температурными параметрами, равными 1.2 Uэкв атома N или C для NH, CH, CH2 и 1.5 Uэкв атома C для CH3.
Координаты атомов и кристаллографические данные для соединения 2 депонированы в Кембриджском банке структурных данных (CCDC № 1918914).
Растворители марки “х. ч.” и “ос. ч.”, а также эфиры аминокислот GlyOtBu, PheOEt (Sigma-Аldrich и Panreac (99.7%)) использовали без дополнительной очистки.
Синтез (NBu4)[2-B10H9(NCCH3)] (1). Соединение 1 получали по методике [45]. Навеску 10.00 г (0.017 моль) (NBu4)2[B10H10] растворяли в 50 мл CH3CN и добавляли 5 мл CF3COOH. Раствор нагревали до 60°C в атмосфере сухого аргона при перемешивании в течение 2 ч до прекращения газовыделения. Охлажденный до комнатной температуры раствор концентрировали на роторном испарителе. Концентрированный раствор разбавляли 20 мл ледяной уксусной кислоты и фильтровали через фильтр Шотта. Осадок промывали 50 мл ледяной уксусной кислоты и 50 мл диэтилового эфира и высушивали в эксикаторе над P2O5. Получено 6.58 г (0.019 моль) (NBu4)[2-B10H9(NCCH3)] (98.9%).
Синтез (NBu4)[2-B10H9NHC(CH3)HNCH2COOС(СH3)3] (2). Готовили суспензию HCl ⋅ GlyOtBu (0.25 г, 1.5 ммоль) в 15 мл CH2Cl2 и добавляли 210 мкл (1.5 ммоль) триэтиламина. После растворения осадка добавляли соединение 1 (0.400 г, 1 ммоль). Реакционную массу кипятили в течение 4 ч в колбе, снабженной магнитной мешалкой и обратным холодильником. После охлаждения до комнатной температуры реакционную массу экстрагировали 15 мл 0.1 н раствора соляной кислоты, а затем трижды промывали дистиллированной водой. Органический слой отделяли, сушили над безводным Na2SO4 и упаривали на роторном испарителе. Получено 0.516 г (NBu4)[2-B10H9NHC(CH3)HNCH2COOС(СH3)3] (97%).
ИК-спектр (CHCl3, см–1): 3417, 3296, 3242 (ν(N–H)), 2470 (ν(B–H)), 1746 (ν(C=O)), 1641 (ν(С=N)), 1055 (δ(B–B–H)).
11B-{1H} ЯМР (CD3CN, м.д.): 2.9 (д, 1B, B(10), JB–H = 147 Гц), –4.4 (д, 1B, B(1), JB–H = 142 Гц), ‒17.6 (с, 1B, B(2)), –26.4 (д, 3B, B(4,7,8), JB–H = = 125 Гц), –29.6 (д, 4B, B(3,5,6,9), JB–H = 131 Гц); 1Н ЯМР (CD3CN, м.д.), δ: –1.01–1.55 (м, 9Н, В10Н9), 8.56 (с, 1H, NH–C=NH), 6.37 (с, 1H, NH–C=NH), 4.15 (д, 2H, NH–CH2–COO, J = 5.9 Гц), 3.15 (м, 8H, NBu4), 2.02 (с, 3H, NH=C–CH3), 1.64 (м, 8H, NBu4), 1.50 (с, 9H С(СH3)3), 1.40 (м, 8H, NBu4), 1.33 (т, 3H, COO–CH2–CH3 J = 7.15 Гц), 1.00 (м, 12H, NBu4); 13С ЯМР (CD3CN, м.д.), δ: 168.5 (СOO), 166.6 (С=NH), 83.2 (COO–C(CH3)3), 59.2 (NBu4), 46.2 (CH2–COO), 28.2 (COO–C(CH3)3), 24.3 (NBu4), 20.2 (NBu4), 13.2 (CH3–C=NH), 13.8 (NBu4).
Синтез (NBu4)[2-B10H9NHC(CH3)HNCH2COOH] (3). Навеску 0.500 г соединения 2 растворяли в 30 мл смеси CH3CN и концентрированной соляной кислоты в соотношении 4 : 1. Реакционную массу кипятили в течение 2 ч в колбе, снабженной магнитной мешалкой и обратным холодильником. После охлаждения до комнатной температуры реакционную массу концентрировали на роторном испарителе и трижды экстрагировали 15 мл CH2Cl2. Объединенные экстракты сушили над безводным Na2SO4 и упаривали на роторном испарителе. Получено 0.383 г (NBu4)[2-B10H9NHC(CH3)HNCH2COOСH] (85.6%).
ИК-спектр (CHCl3, см–1): 3407, 3295, 3238 (ν(N–H)), 2470 (ν(B–H)), 1732 (ν(C=O)), 1632 (ν(С=N)), 1055 (δ(B–B–H)).
11B–{1H} ЯМР (CD3CN, м.д.): 2.7 (д, 1B, B(10), JB–H = 155 Гц), –4.1 (д, 1B, B(1), JB–H = 139 Гц), –15.0 (с, 1B, B(2)), –23.6 (д, 3B, B(4,7,8), JB–H = 110 Гц), –27.1 (д, 4B, B(3,5,6,9), JB–H = 123 Гц); 1Н ЯМР (CD3CN, м.д.), δ: –1.01–1.55 (м, 9Н, В10Н9), 8.55 (с, 1H, NH–C=NH), 6.31 (с, 1H, NH–C=NH), 4.02 (д, 2H, NH–CH2–COO, J = 4.40 Гц), 3.12 (м, 8H, NBu4), 2.02 (с, 3H, NH=C–CH3), 1.64 (м, 8H, NBu4), 1.40 (м, 8H, NBu4), 1.00 (м, 12H, NBu4); 13С ЯМР (CD3CN, м.д.), δ: 178.8 (СOO), 165.7 (С=NH), 58.7 (NBu4), 47.2 (CH2–COO), 23.7 (NBu4), 19.7 (NBu4), 19.0 (CH3–C=NH), 13.2 (NBu4).
Синтез (NBu4)[2-B10H9NHC(CH3)HNCH2COHNCH(CH2C6H5)COOCH2CH3] (4). К 0.237 г (0.5 ммоль) соединения 3 в 10 мл CH2Cl2 при активном перемешивании добавляли 0.110 г (0.53 ммоль) N,N'-дициклогексилкарбодиимида, 0.115 (0.5 ммоль) HCl ⋅ PheOEt и 0.122 г (1 ммоль) 4-диметиламинопиридина. Реакционную массу перемешивали в течение 3 ч при комнатной температуре. После завершения реакции отфильтровывали осадок. Фильтрат экстрагировали 5 мл 0.1 н раствора соляной кислоты, затем трижды промывали дистиллированной водой. Органический слой отделяли, сушили над безводным Na2SO4 и упаривали на роторном испарителе. Полученный продукт очищали с помощью препаративной ВЭЖХ в системе CHCl3–CH3CN в соотношении 10 : 1. Получено 0.08 г (NBu4)[2-B10H9NHC(CH3)HNCH2COHNCH(CH2C6H5)COOCH2CH3] (24.3%).
ИК-спектр (CHCl3, см–1): 3414, 3290, 3248 (ν(N–H)), 2455 (ν(B–H)), 1746 (ν(C=O)), 1641 (ν(С=N)), 1055 (δ(B–B–H)).
11B–{1H} ЯМР (CD3CN, м.д.): 2.6 (д, 1B, B(10), JB–H = 145 Гц), –4.0 (д, 1B, B(1), JB–H = 140 Гц), ‒15.5 (с, 1B, B(2)), –23.6 (д, 3B, B(4,7,8), JB–H = = 112 Гц), –27.1 (д, 4B, B(3,5,6,9), JB–H = 123 Гц); 1Н ЯМР (CD3CN, м.д.), δ: –1.01–1.55 (м, 9Н, В10Н9), 8.49 (с, 1H, NH–C=NH), 7.47–6.96 (м, 5H, CH2–C6H5), 6.31 (с, 1H, NH–C=NH), 5.97 (с, 1H, –CO–NH–), 4.83 (м, 1H, NH–CH–COO), 4.20 (кв, 2H, COO–CH2–CH3, J = 7.15 Гц), 3.95 (д, 2H, NH–CH2–CONH, J = 4.40 Гц), 3.25 (кв, 2H, CH2–C6H5, J = 7.15 Гц), 3.12 (м, 8H, NBu4), 2.02 (с, 3H, NH=C–CH3), 1.64 (м, 8H, NBu4), 1.40 (м, 8H, NBu4), 1.32 (т, 3H, COO–CH2–CH3, J = 6.97Гц) 1.00 (м, 12H, NBu4); 13С ЯМР (CD3CN, м.д.), δ: 171.6 (СOO), 166.8 (СO–NH), 166.7 (С=NH), 136.7, 129.9, 129.2, 127.6 (–CH2–Ph), 62.1 (COO–CH2–CH3), 58.7 (NBu4), 56.2 (NH–CH–COO) 47.2 (CH2–COO), 46.9 (NH–CH2–CONH), 38.2 (CH2–C6H5), 23.7 (NBu4), 19.7 (NBu4), 20.2 (CH3–C=NH), 14.5 (COO–CH2–CH3), 13.2 (NBu4).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Как было показано ранее [31, 46–48], нитрилиевые производные клозо-декаборатного аниона проявляют высокую реакционную способность к органическим аминам. Природные аминокислоты также содержат функциональную аминогруппу, поэтому нами были предложены методы получения конъюгатов клозо-декаборатного аниона и производных аминокислот.
Аминокислоты плохо растворяются в органических растворителях, а использование в качестве растворителя водных буферных растворов приводит к образованию продуктов гидролиза исходного нитрилиевого производного, что снижает выход целевых соединений и требует трудоемкой хроматографической очистки получаемых продуктов.
Альтернативным методом синтеза подобных соединений является использование сложных эфиров аминокислот, хорошо растворимых в органических растворителях, с последующим гидролизом. Незначительная гидролитическая стабильность производных клозо-декаборатного аниона амидинового типа в водных растворах щелочей накладывает определенные ограничения на строение защитной сложноэфирной группы. В качестве таковой в данной работе использовали трет-бутильную защитную группу, которая может быть удалена в мягких кислотных условиях [49].
Процесс присоединения трет-бутилового эфира глицина к ацетонитрильному производному клозо-декаборатного аниона представлен на схеме 1 а. Контроль за ходом реакции осуществляли при помощи 11В ЯМР-спектроскопии. В спектрах продукта сигналы от апикальных атомов бора находятся в области 2.9 м.д. [B(10), I = 1] и –4.4 м.д. [B(1), I = 1], что характерно для монозамещенных амидинов [46]. Сигнал от замещенного атома бора B(2) наблюдается при –17.5 м.д., сигналы от незамещенных экваториальных вершин борного кластера – при –26.4, –29.6 м.д.
Строение синтезированных конъюгатов клозо-декаборатного аниона и трет-бутилового эфира глицина устанавливали с помощью мультиядерной ЯМР-спектроскопии. Так, в спектре ЯМР 1Н соединения 2 амидиновый фрагмент представлен двумя сигналами: от протона иминогруппы при 6.37 м.д. и протона аминогруппы при 8.56 м.д. Аминокислотный остаток представлен сигналами от протонов при α-атоме углерода при 4.15 м.д. Сигнал метильных протонов трет-бутильной группы представляет собой синглет при 1.5 м.д., сигналы протонов от заместителя нитрилиевой группы наблюдаются при 2.02 м.д. Также в спектрах присутствуют сигналы от н-тетрабутиламмониевого катиона.
В спектре ЯМР 13C аминокислотный фрагмент представлен сигналами от карбоксильного атома углерода при 168.5 м.д. и сигналом α-атома углерода при 46.2 м.д., амидиновая функциональная группа – сигналами атома углерода, связанного с азотом иминогруппы, при 166.6 м.д. и сигналом атома углерода метильной группы при 13.2 м.д. Сигналы атомов углерода трет-бутильной группы наблюдаются при 83.2 и 28.2 м.д.
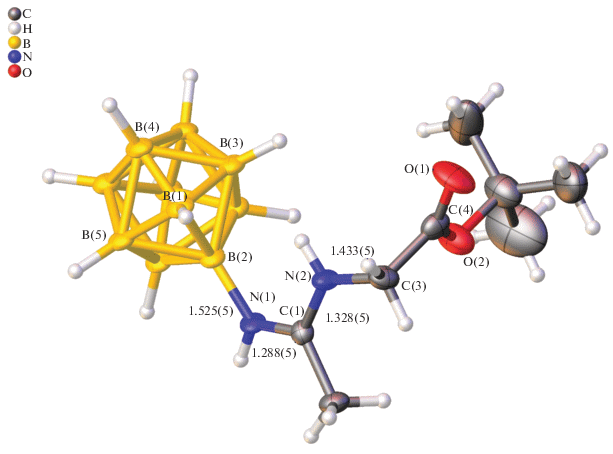
Строение полученного производного подтверждено с помощью рентгеноструктурного анализа (рис. 1, рис. 1 и 2 выполнены с использованием программного пакета OLEX2 [50]). В полученном амидине заместитель находится в Z-конфигурации и располагается в экваториальном поясе клозо-декаборатного аниона. Длина связи бор–азот составляет 1.525 Å, что соответствует длине ординарной связи [51–53]. Амидиновый фрагмент N(1)C(1)N(2)C(3) плоский (стандартное отклонение атомов от плоскости составляет 0.13°), а связи C(1)–N(1) и C(1)–N(2) существенно укорочены (1.287(5) и 1.328(5) Å соответственно), что указывает на наличие сопряжения и частичной делокализации положительного заряда на атомах N(1)C(1)N(2). Атом C(1) находится в sp2-гибридизации, углы N(1)C(1)N(2), N(1)C(1)C(2) и N(2)C(1)C(2) составляют 120.0(4)°, 120.7(4)° и 119.4(3)° соответственно. Длины связей трет-бутоксикарбонильной функции хорошо согласуются с таковыми для сложных эфиров [51]. Карбонильный атом углерода находится в sp2-гибридизации, на что указывают значение угла, близкое к 120°.
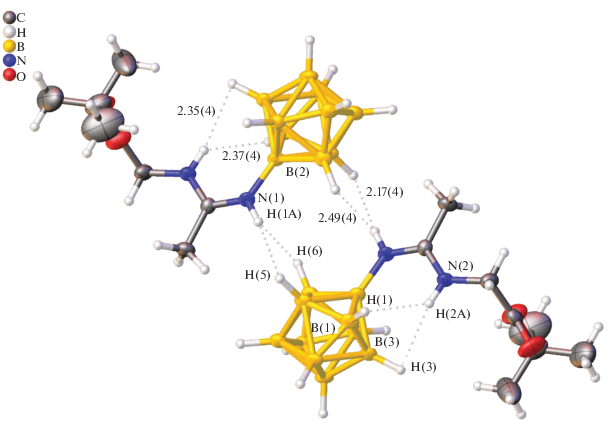
Конфигурация заместителя стабилизирована внутримолекулярными диводородными связями между атомом водорода аминогруппы N(2)H(2) и атомами водорода апикального ребра кластера H(1)B(1) и H(3)B(3). Длины связей составляют: N(2)H(2) … H(3)B(3) 2.35 Å и N(2)H(2) … H(1)B(3) 2.37 Å. Межмолекулярные диводородные связи объединяют анионы в центросимметричные димеры (рис. 2).
Дальнейшим шагом для получения конъюгатов клозо-декаборатного аниона с глицином является гидролиз полученных сложных эфиров (схема 1 б). Использование для удаления трет-бутильной группы широко известной методики [54] невозможно, так как образующийся трет-бутильный катион взаимодействует с борным остовом кластера, в результате чего существенно снижается выход целевого продукта и возникает необходимость его очистки. Нами предложена методика удаления трет-бутильной группы соляной кислотой в смеси с ацетонитрилом. Установлено, что данная реакция протекает с количественным выходом и без образования побочных продуктов. Контроль за ходом реакции осуществляли при помощи ТСХ в системе CH2Cl2–CH3CN в соотношении 1 : 2.
Строение синтезированного конъюгата клозо-декаборатного аниона и глицина устанавливали с помощью мультиядерной ЯМР-спектроскопии.
В спектрах ЯМР 11В сигналы от апикальных атомов бора лежат при 2.7 м.д. [B(10), I = 1] и –4.1 м.д. [B(1), I = 1], сигнал от замещенного атома бора B(2) наблюдается при –15.0 м.д., сигналы от незамещенных экваториальных вершин борного кластера – при –23.6, –27.1 м.д. В спектре ЯМР 1Н амидиновый фрагмент представлен двумя сигналами: от протона иминогруппы при 6.31 м.д. и протона аминогруппы при 8.55 м.д. Аминокислотный отстаток представлен сигналами от протонов при α-атоме углерода при 4.02 м.д. Сигналы протонов от заместителя нитрилиевой группы наблюдаются при 2.02 м.д. В спектрах также присутствуют сигналы от н-тетрабутиламмониевого катиона (3.15, 1.65, 1.40, 1.00 м.д.). Отсутствие сигналов протонов метильных групп трет-бутильной группы свидетельствует о полном деблокировании карбоксильной группы.
В спектре ЯМР 13C аминокислотный фрагмент представлен сигналами от карбоксильного атома углерода при 178.8 м.д. и сигналом α-атома углерода при 47.2 м.д. Амидиновая функциональная группа представлена сигналами атома углерода, связанного с азотом иминогруппы, при 165.7 м.д. и сигналом атома углерода метильной группы при 19.0 м.д. Аналогично спектрам ЯМР 1Н после гидролиза не наблюдаются сигналы углерода трет-бутильной группы.
Полученный конъюгат клозо-декаборатного аниона с глицином является стартовым соединением для получения широкого спектра агентов для БНЗТ с различными векторными группами, которые могут быть получены с использованием методик пептидного синтеза [55].
Для получения дипептидного коньюгата использован стандартный подход с применением дициклогексилкарбодиимида в качестве сшивающего реагента (cхема 1в). Выбор в качестве аминокомпонента этилового эфира фенилаланина обусловлен тем, что сложные эфиры аминокислот хорошо растворяются в органических растворителях и не требуют использования дорогостоящих реагентов, применяемых для образования амидных связей в водной среде. Реакцию проводили в среде CH2Cl2, контроль за ходом процесса осуществляли при помощи тонкослойной хроматографии (ТСХ) в системе CH2Cl2–CH3CN (соотношение 1 : 2). Установлено, что данная реакция протекает с образованием побочных продуктов сложного состава и требует хроматографической очистки целевых соединений.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры
| Соединение | (NBu4)[2-B10H9NHC(CH3)NHCH2COOtBu] |
|---|---|
| Эмпирическая формула | C24H61B10N3O2 |
| М | 531.87 |
| T, K | 100(2) |
| Крист. гр. | Моноклинная |
| Пр. гр. | P21/c |
| a, Å | 9.889(4) |
| b, Å | 18.418(7) |
| c, Å | 19.876(7) |
| α, град | 90 |
| β, град | 101.912(10) |
| γ, град | 90 |
| V, Å3 | 3542.17 |
| Z | 4 |
| Dx, мг/м3 | 0.997 |
| μ, мм–1 | 0.057 |
| Размер кристалла, мм | 0.300 × 0.120 × 0.020 |
| Интервал сканирования θ, град | 4.179–27.495 |
| 25 746 | |
| Общее число рефлексов независимых (N) [Rint] в том числе с I > 2σ(I) (No) |
8020 [0.2732] 2486 |
| Tmax, Tmin | – |
| Данные/ограничения/параметры | 8020/0/380 |
| GOOF | 0.930 |
| R1, wR2 для No | 0.1020, 0.1742 |
| R1, wR2 для N | 0.3088, 0.2496 |
| Δρmax/Δρmin, e/Å3 | 0.297/–0.240 |
Строение полученного коньюгата установлено при помощи мультиядерной ЯМР-спектроскопии. В спектрах ЯМР 11В сигналы от апикальных атомов бора лежат при 2.6 м.д. [B(10), I = 1] и ‒4.0 м.д. [B(1), I = 1], сигнал от замещенного атома бора B(2) – при –15.5 м.д., сигналы от незамещенных экваториальных вершин борного кластера – при –23.6, –27.1 м.д.
В спектре ЯМР 1Н наблюдаются сигналы от трех протонов, связанных с тремя атомами азота: протона аминогруппы при 8.49 м.д., протона иминогруппы при 6.31 м.д. и протона амидной группы при 5.97 м.д. Аминокислотный фрагмент глицина представлен сигналом протонов при α-атоме углерода в области 3.95 м.д., аминокислотный фрагмент фенилаланина – сигналами фенильной группы в диапазоне 7.47–6.96 м.д., сигналом протонов при α-атоме углерода в области 4.83 м.д. и сложным сигналом метиленовой группы при 3.25 м.д. Этоксикарбонильный фрагмент представлен сигналами при 4.20 и 1.32 м.д. Сигналы протонов от заместителя нитрилиевой группы лежат в области 2.02 м.д.
В спектре ЯМР 13C аминокислотный фрагмент глицина представлен сигналами от амидного атома углерода при 166.8 м.д. и α-атома углерода при 47.2 м.д. Аминокислотный фрагмент фенилаланина представлен сигналами от карбоксильного атома углерода при 171.6 м.д., сигналами фенильной группы в диапазоне 125–137 м.д., α-атома углерода при 56.2 м.д., метиленовой группы при 38.2 м.д. Этоксикарбонильному фрагменту отвечают сигналы при 62.1 и 14.5 м.д. Сигнал метильной группы заместителя нитрилиевой группы наблюдается при 20.2 м.д.
Таким образом, в работе предложен метод получения производных клозо-декаборатного аниона с экзополиэдрическими заместителями, содержащими пептидную связь. Показано, что нуклеофильное присоединение трет-бутилового эфира глицина к ацетонитрильному производному [2-B10H9NCCH3]– приводит к стерео- и региоселективному образованию амидин-клозо-декаборатов. В результате кислотного гидролиза N-борилированного трет-бутилового эфира получено производное свободной аминокислоты с высоким выходом.
Список литературы
Bregadze V., Sivaev I. Boron Sci., CRC Press, 2011. 181 p. https://doi.org/10.1201/b11199-14
Snyder H.R., Reedy A.J., Lennarz W.J. et al. // J. Am. Chem. Soc. 1958. V. 80. № 4. P. 835. https://doi.org/10.1021/ja01537a021
Díaz S., González A., González de Riancho S. et al. // J. Organomet. Chem. 2000. V. 610. № 1–2. P. 25. https://doi.org/10.1016/S0022-328X0000363-6
Srivastava R.R., Singhaus R.R., Kabalka G.W. // J. Org. Chem. 1999. V. 64. № 23. P. 8495. https://doi.org/10.1021/jo990878c
Kabalka G.W., Yao M.-L., Wu Z. // Org. Process Res. Dev. 2006. V. 10. № 5. P. 1059. https://doi.org/10.1021/op060052u
Ryynänen P., Kangasmäki A., Hiismäki P. et al. // Phys. Med. Biol. 2002. V. 47. № 4. P. 737. https://doi.org/10.1088/0031-9155/47/5/304
Kaiser P.F., Churches Q.I., Hutton C.A. // Aust. J. Chem. 2007. V. 60. № 4. P. 799. https://doi.org/10.1071/CH07103
Kabalka G.W., Yao M.L. // Anticancer. Agents Med. Chem. 2006. V. 6. № 2. P. 111. https://doi.org/10.2174/187152006776119144
Leukart O., Caviezel M., Eberle A. et al. // Helv. Chim. Acta. 1976. V. 59. № 6. P. 2184. https://doi.org/10.1002/hlca.19760590630
Srivastava R.R., Singhaus R.R., Kabalka G.W. // J. Org. Chem. 1997. V. 62. № 13. P. 4476. https://doi.org/10.1021/jo970148+
Kahl S.B., Kasar R.A. // J. Am. Chem. Soc. 2002. V. 118. № 5. P. 1223. https://doi.org/10.1021/ja9534260
He T., Musah R.A. // ACS Omega. 2019. V. 4. № 2. P. 3820. https://doi.org/10.1021/acsomega.8b03407
Perugini P., Pavanetto F. // J. Microencapsul. 1998. V. 15. № 4. P. 473. https://doi.org/10.3109/02652049809006874
Pavanetto F., Perugini P. // Drug Deliv. 2002. V. 7. № 2. P. 97. https://doi.org/10.1080/107175400266669
Martini S., Ristori S., Pucci A. et al. // Biophys. Chem. 2004. V. 111. № 1. P. 27. https://doi.org/10.1016/j.bpc.2004.03.010
Bregadze V., Semioshkin A., Sivaev I. // Appl. Radiat. Isot. 2011. V. 69. № 12. P. 1774. https://doi.org/10.1016/j.apradiso.2011.01.043
Kusaka S., Hattori Y., Uehara K. et al. // Appl. Radiat. Isot. 2011. V. 69. № 12. P. 1768. https://doi.org/10.1016/j.apradiso.2011.03.042
Hattori Y., Kusaka S., Mukumoto M. et al. // J. Med. Chem. 2012. V. 55. № 15. P. 6980. https://doi.org/10.1021/jm300749q
Zhizhin K.Y., Zhdanov A.P., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2010. V. 55. № 15. P. 2089. https://doi.org/10.1134/S0036023610140019
Клюкин И.Н., Жданов А.П., Разгоняева Г.А и др. // Журн. неорган. химии. 2013. Т. 58. № 12. С. 1559. [Klyukin I.N., Zhdanov A.P., Razgonyaeva G.A. et al. // Russ. J. Inorg. Chem. 2013. V. 58. № 12. P. 1395.] https://doi.org/10.1134/S0036023613120140
Klyukin I.N., Kubasov A.S., Limarev I.P. et al. // Polyhedron. 2015. V. 101. P. 215. https://doi.org/10.1016/j.poly.2015.09.025
Klyukin I.N., Zhdanov A.P., Matveev E.Y. et al. // Inorg. Chem. Commun. 2014. V. 50. P. 28. https://doi.org/10.1016/j.inoche.2014.10.008
Матвеев Е.Ю., Кубасов А.С., Разгоняева Г.А. и др. // Журн. неорган. химии. 2015. Т. 60. № 7. С. 858. [Matveev E.Y., Kubasov A.S., Razgonyaeva G.A. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 7. P. 776.] https://doi.org/10.1134/S0036023615070104
Кубасов А.С., Матвеев Е.Ю., Турышев Е.С. и др. // Докл. АН. 2017. Т. 477. № 3. С. 303. Kubasov A.S., Matveev E.Y., Turyshev E.S. et al. // Dokl. Chem. 2017. V. 477. № 1. P. 257. https://doi.org/10.1134/S0012500817110088
Zhdanov A.P., Zhdanova K.A., Bykov A.Y. et al. //Polyhedron. 2018. V. 139. P. 125. https://doi.org/10.1016/j.poly.2017.09.050
Клюкин И.Н., Жданов А.П., Быков А.Ю. и др. // Журн. неорган. химии. 2018. Т. 63. № 2. С. 200. [Klyukin I.N., Zhdanov A.P., Bykov A.Yu. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 2. P. 213.] https://doi.org/10.1134/S0036023618020110
Жданов А.П., Лисовский М.В., Гоева Л.В и др. // Изв. АН. Сер. хим. 2009. № 8. С. 1643. [Zhdanov A.P., Lisovsky M. V., Goeva L.V. et al. // Russ. Chem. Bull. 2009. V. 58. № 8. P. 1694]. https://doi.org/10.1007/s11172-009-0234-9
Mindich A.L., Bokach N.A., Kuznetsov M.L. et al. // Chempluschem. 2012. V. 77. № 12. P. 1075. https://doi.org/10.1002/cplu.201200257
Жданова К.А., Жданов А.П., Ежов А.В. и др. // Изв. АН. Сер. хим. 2014. № 1. С. 194. [Zhdanova K.A., Zhdanov A.P., Ezhov A.V. et al. // Russ. Chem. Bull. 2014. V. 63. № 1. P. 194.] https://doi.org/10.1007/s11172-014-0413-1
Zhdanova K.A., Zhdanov A.P., Ezhov A.V. et al. // Macroheterocycles. 2014. V. 7. № 4. P. 394. https://doi.org/10.6060/mhc140494z
Losytskyy M.Y., Kovalska V.B., Varzatskii O.A. et al. // J. Lumin. 2016. V. 169. P. 51. https://doi.org/10.1016/j.jlumin.2015.08.042
Bolotin D.S., Burianova V.K., Novikov A.S. et al. // Organometallics. 2016. V. 35. P. 3612. https://doi.org/10.1021/acs.organomet.6b00678
Zhdanov A.P., Klyukin I.N., Bykov A.Y. et al. // Polyhedron. 2017. V. 123. P. 176. https://doi.org/10.1016/j.poly.2016.11.035
Болотин Д.С., Демакова М.Я., Дайнес Е.А. и др. // Журн. общ. химии. 2017. Т. 87. № 1. С. 41. [Bolotin D.S., Demakova M.Y., Daines E.A. et al. // Russ. J. Gen. Chem. 2017. V. 87. № 1. P. 37.] https://doi.org/10.1134/S107036321701008X
Жданов А.П., Быков А.Ю., Кубасов А.С. и др. // Журн. неорган. химии. 2017. Т. 62. № 4. С. 467. [Zhdanov A.P., Bykov A.Y., Kubasov A.S. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 4. P. 468.] https://doi.org/10.1134/S0036023617040210
Burianova V.K., Mikherdov A.S., Bolotin D.S. et al. // J. Organomet. Chem. 2018. V. 870. P. 97. https://doi.org/10.1016/j.jorganchem.2018.06.017
Daines E.A., Bolotin D.S., Bokach N.A. et al. // Inorg. Chim. Acta. 2018. V. 471. P. 372. https://doi.org/10.1016/j.ica.2017.11.054
Burianova V.K., Bolotin D.S., Mikherdov A.S. et al. // New J. Chem. 2018. V. 42. № 11. P. 8693. https://doi.org/10.1039/c8nj01018h
Mindich A.L., Bokach N.A., Dolgushin F.M. et al. // Organometallics. 2012. V. 31. № 5. P. 1716. https://doi.org/10.1021/om200993f
Mindich A.L., Bokach N.A., Kuznetsov M.L. et al. // Organometallics. 2013. V. 32. № 1. P. 6576. https://doi.org/10.1021/om400892x
SAINT. Version 7.23A. Bruker AXS Inc., 2003.
SADABS-2004/1. Bruker AXS Inc., 2004.
Sheldrick G.M. //Acta Crystallogr. A. 2008. V. 64. P. 112. https://doi.org/10.1107/S0108767307043930
Sheldrick G.M. // Acta Crystallogr. Sect. A. Found. Crystallogr. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053273314026370
Sivaev I.B., Votinova N.A., Bragin V.I. et al. // J. Organomet. Chem. 2002. V. 657. № 1–2. P. 163. https://doi.org/10.1016/S0022-328X0201419-5
Жданов А.П., Полякова И.Н., Разгоняева Г.А. и др. // Журн. неорган. химии. 2011. Т. 56. № 6. С. 903. [Zhdanov A.P., Polyakova I.N., Razgonyaeva G. A. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 6. P. 847.] https://doi.org/10.1134/S003602361106026X
Losytskyy M.Y., Kovalska V.B., Varzatskii O.A. et al. // J. Lumin. 2016. V. 169. P. 51. https://doi.org/10.1016/j.jlumin.2015.08.042
Ezhov A.V., Vyal’ba F.Y., Kluykin I.N. et al. // Macroheterocycles. 2017. V. 10. № 4–5. P. 505. https://doi.org/10.6060/mhc171254z
McOmie J.F.W. Protective Groups in Organic Chemistry, Springer US, Boston, MA, 1995. https://doi.org/10.1007/978-1-4684-7218-9
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. № 2. P. 339. https://doi.org/10.1107/S0021889808042726
Allen F.H., Kennard O., Watson D.G. et al. // J. Chem. Soc., Perkin Trans. 1987. V. 2. № 12. S1. https://doi.org/10.1039/p298700000s1
Konieczka S.Z., Himmelspach A., Hailmann M. et al. // Eur. J. Inorg. Chem. 2013. № 1. P. 134. https://doi.org/10.1002/ejic.201200969
Li F., Shelly K., Knobler C.B. et al. // Inorg. Chem. 1999. V. 38. № 22. P. 4926. https://doi.org/10.1021/ic990744h
Bryan D.B., Hall R.F., Holden K.G. et al. // J. Am. Chem. Soc. 1977. V. 99. № 7. P. 2353. https://doi.org/10.1021/ja00449a063
Alam F., Soloway A.H., Barth R.F. et al. // J. Med. Chem. 1989. V. 32. № 10. P. 2326. https://doi.org/10.1021/jm00130a017
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии