

Журнал неорганической химии, 2019, T. 64, № 12, стр. 1239-1245
Синтез одномерных наноструктур оксида CeO2-10%Y2O3 методом программируемого соосаждения в присутствии поливинилового спирта
Т. Л. Симоненко 1, *, Н. П. Симоненко 1, Е. П. Симоненко 1, В. Г. Севастьянов 1, Н. Т. Кузнецов 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: egorova.offver@gmail.com
Поступила в редакцию 12.08.2019
После доработки 20.08.2019
Принята к публикации 22.08.2019
Аннотация
С использованием метода программируемого прямого соосаждения гидроксидов металлов в присутствии поливинилового спирта получены иерархические наноструктуры на основе стержневидных частиц состава CeO2-10%Y2O3. Показано влияние концентрации структурообразующего полимерного темплата на характер морфологии формирующихся оксидов. Установлена зависимость дисперсности и микроструктуры продуктов от концентрации поливинилового спирта в реакционной системе. С помощью синхронного термического и рентгенофазового анализа установлено, что во время осаждения полимер внедряется в структуру получаемого нанопорошка, предотвращая укрупнение частиц при термообработке.
ВВЕДЕНИЕ
Благодаря особым физико-химическим свойствам нанокристаллический диоксид церия представляет собой уникальный материал с широким спектром областей практического применения: энергетика, хемосенсорика, оптика, катализ, биомедицина [1–5]. Это связано со способностью иона церия переходить из одного валентного состояния (Ce4+) в другое (Ce3+), что обусловливает возможность кристаллической решетки CeO2 участвовать в обратимых окислительно-восстановительных реакциях. В восстановительных средах происходит выделение кислорода с образованием кислородных вакансий (согласно уравнению (1) в обозначениях Крегера–Винка), а в окислительных средах протекает обратный процесс (уравнение (2)) [6, 7]:
(1)
$2{\text{C}}{{{\text{e}}}_{{{\text{Ce}}}}} + {{{\text{O}}}_{{\text{o}}}} \to {\text{V}}_{{\text{o}}}^{{\centerdot \centerdot }} + 2{\text{Ce}}_{{{\text{Ce}}}}^{'} + {1 \mathord{\left/ {\vphantom {1 {2{{{\text{O}}}_{2}}}}} \right. \kern-0em} {2{{{\text{O}}}_{2}}}},$(2)
${\text{Ce}}{{{\text{O}}}_{2}} \rightleftarrows {\text{Ce}}{{{\text{O}}}_{{2 - y}}} + {y \mathord{\left/ {\vphantom {y {2{{{\text{O}}}_{2}}}}} \right. \kern-0em} {2{{{\text{O}}}_{2}}}}.$С точки зрения энергетики и хемосенсорики число подвижных кислородных вакансий, участвующих в процессах переноса ионов кислорода, является одним из основных факторов, определяющих функциональные свойства получаемых материалов (электропроводность, газовую чувствительность, величину и скорость отклика) [8–11]. Увеличения количества кислородных вакансий в кристаллической решетке диоксида церия можно достичь допированием его элементами с меньшей степенью окисления или меньшим ионным радиусом (например, Ca2+, Mg2+, Sc2+, Y3+, Gd3+, Zr4+ и др.) [12–18]. Особый интерес в данном контексте вызывает иттрий, так как твердые растворы в системе CeO2–Y2O3 перспективны не только как рецепторные материалы резистивных газовых сенсоров, но и в качестве среднетемпературных твердых электролитов твердооксидных топливных элементов (ТОТЭ) за счет достаточно высокой кислород-ионной проводимости в интервале температур 400–700°С, а также высокой стабильности электрофизических характеристик в течение длительного времени эксплуатации [19–23]. Анализ современной литературы показал, что не только размер, но и форма частиц материалов на основе диоксида церия может в значительной степени улучшать их функциональные (электрофизические, хемосенсорные, каталитические) свойства [4, 7, 24, 25]. Данный факт обусловливает растущий интерес к получению наноматериалов различного типа с иерархической организацией составляющих их частиц, а также исследование влияния их микроструктуры на физико-химические свойства [26–30]. В настоящее время были получены различные иерархические наноструктуры диоксида церия в виде наностержней [31, 32], наносфер [28, 33], нанокубов [34], нанотрубок [35], нанооктаэдров [36], наноцветов [37] и т.п. Тем не менее до настоящего времени в литературе не были представлены работы, посвященные исследованию процессов получения иерархически организованных наноматериалов на основе диоксида церия, допированного оксидом иттрия, хотя, как было сказано выше, этот материал является весьма перспективным с практической точки зрения.
Создание иерархических наноструктур с контролируемой микроморфологией и размером частиц требует разработки новых, а также усовершенствования традиционных методов и подходов к синтезу наноматериалов. В настоящее время наиболее популярными в данном контексте являются совместное осаждение [4, 30], сольво- [32] и гидротермальный синтез [38, 39], а также золь-гель метод [40, 41]. В настоящей работе предлагается использовать метод программируемого соосаждения гидроксидов металлов, который позволяет тонко варьировать параметры синтеза – осуществлять непрерывную или дискретную подачу раствора осадителя к раствору металлсодержащих реагентов в широком диапазоне скоростей и объемов, а также автоматически изменять скорость подачи по мере приближения к точке эквивалентности или заданному значению pH, обеспечивая получение целевых продуктов с заданной дисперсностью и микроструктурой.
Цель работы – изучение процесса синтеза одномерных иерархических наноструктур состава CeO2-10%Y2O3 методом программируемого совместного осаждения гидроксидов металлов в присутствии поливинилового спирта.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При проведении синтеза применяли нитраты металлов Сe(NO3)3 · 6H2O “х. ч.” и Y(NO3)3 · 6H2O “х. ч.”, 5%-ный водный раствор NH3 · H2O “ос. ч.” и поливиниловый спирт (C2H4O)x “ч.”.
Синтез нанопорошков состава CeO2-10%Y2O3 проводили методом программируемого соосаждения гидроксидов металлов с использованием автоматического высокоточного потенциометрического титратора АТП-02 путем дозированной подачи 5%-ного водного раствора гидрата аммиака с заданной скоростью (0.017 мл/с) к раствору нитратов церия и иттрия (0.05 М), содержащему 7.46 × 10–4 (образец 1), 3.51 × 10–3 (образец 2) или 6.55 × 10–3 мас. % (образец 3) поливинилового спирта, при перемешивании до достижения значения pH 10.1–10.3. Частицы дисперсной фазы отделяли от дисперсионной среды путем центрифугирования, промывали дистиллированной водой, а затем сушили при 100°С (3 ч).
Синхронный (ТГА/ДСК) термический анализ порошков, полученных в результате осаждения и сушки при 100°С, был выполнен с использованием термоанализатора SDT Q-600 (контролируемый нагрев проводили в Al2O3-тиглях в интервале температур 20–1000°С со скоростью 10 град/мин в токе воздуха, скорость потока газа составляла 250 мл/мин, масса навески варьировалась в диапазоне 35–45 мг).
Для записи ИК-спектров пропускания были приготовлены суспензии порошков в вазелиновом масле, которые помещали в виде пленки между стеклами KBr. Спектральный анализ проводили в диапазоне волновых чисел 350–4000 см–1 (время накопления сигнала составляло 15 с, разрешение – 1 см–1) с использованием ИК-Фурье-спектрометра ИнфраЛЮМ ФТ-08.
Рентгенофазовый анализ порошков осуществляли на рентгеновском дифрактометре Bruker D8 Advance (излучение CuKα, диапазон 2θ = 15°–80°, разрешение 0.02°, время накопления сигнала в точке 0.3 с). Для оценки размеров областей когерентного рассеяния (ОКР) использовали уравнение Селякова–Шерера: DОКР = 0.9λ/(βсosθ), где λ – длина волны СuKα, β – ширина дифракционного рефлекса на полувысоте.
Микроструктуру полученных порошков изучали с помощью растровой электронной микроскопии (РЭМ) на трехлучевой рабочей станции NVision 40 (Carl Zeiss), оснащенной энергодисперсионной приставкой-микроанализатором INCA X-MAX (Oxford Instruments), а также с применением просвечивающего электронного микроскопа (ПЭМ) JEOL JEM-1011 с цифровой фотокамерой ORIUS SC1000W.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
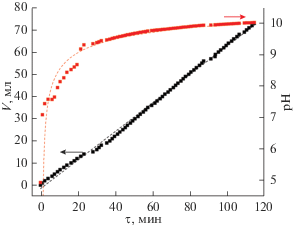
Скорость и характер изменения рН реакционной смеси при дозированном добавлении в нее 5%-ного водного раствора гидрата аммиака в процессе синтеза для всех рассматриваемых в данной работе систем носит схожий характер. В качестве примера на рис. 1 показаны зависимости рН(τ) и ${{V}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}\,\,{\cdot}\,\,{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ (τ) в ходе осаждения гидроксидов церия и иттрия в присутствии наибольшего количества поливинилового спирта (образец 3). Видно, что добавление осадителя в реакционную систему происходит линейно и равномерно со скоростью ∼0.017 мл/с. Изменение величины рН носит характер затухающей логарифмической функции. При добавлении первой порции гидрата аммиака (∼1–2 мл) в самом начале процесса осаждения происходит резкое скачкообразное увеличение величины рН с 4.9 до 7.5, наблюдается постепенное помутнение раствора и образование частиц гидроксидов церия и иттрия. При рН ∼9.3 кривая выходит в область насыщения, увеличение водородного показателя значительно замедляется, и необходимого для полноты осаждения получаемых гидроксидов уровня рН удается достичь лишь после добавления 60 мл NH3 · H2O спустя 90 мин после начала эксперимента.
Рис. 1.
Кривые временной зависимости pH и объема добавляемого к реакционной системе 5%-ного водного раствора гидрата аммиака.
Поведение полученных в результате осаждения и сушки при 100°С порошков в токе воздуха в интервале температур 20–1000°С было изучено с помощью синхронного (ТГА/ДСК) термического анализа (рис. 2). Как видно из термограмм, при нагревании порошков наблюдаются четыре основные ступени потери массы, несколько смещенные по температурам в зависимости от химического состава целевых продуктов. Потеря массы на первой ступени (в диапазоне температур 20–150°С для порошка 1, 20–180°С для порошков 2 и 3), сопровождаемая поглощением энергии, для всех образцов составила ∼3% и связана с удалением остаточного растворителя и сорбированных атмосферных газов. Вторая и третья ступени уменьшения массы для всех образцов (в диапазонах температур 150–200 и 200–400°С для порошка 1, 180–200 и 200–400°С для порошков 2 и 3) различаются по интенсивности и сопровождаются экзотермическими эффектами, что в интервале температур 150–300°С даже приводит к заметному отклонению программы нагрева от линейного закона. Вторую ступень можно отнести к первому этапу окисления поливинилового спирта, вошедшего в структуру порошка во время осаждения, а третью – к окислению образовавшегося в ходе термолиза углерода. Установлено, что с увеличением концентрации полимера в реакционной системе на стадии осаждения интенсивность теплового эффекта на второй ступени изменения массы падает, а на третьей – растет, что может быть связано как с количеством вошедшего в структуру гидроксидов полимера, так и со структурой образовавшегося органо-неорганического композита. Последняя ступень потери массы (1 – 400–800°С, 2 и 3 – 400–700°С) является наиболее растянутой по температуре и связана с окислением остаточного углерода из внутреннего объема порошков. Таким образом, результаты синхронного термического анализа порошков, полученных в результате программируемого осаждения и подвергнутых сушке при 100°С, свидетельствуют о формировании органо-неорганических композитов, содержащих полимерный компонент, количество которого растет с увеличением концентрации поливинилового спирта в реакционной системе на стадии синтеза: общая потеря массы при нагревании в диапазоне температур 20–1000°С растет с 13.1 (образец 1) до 27.6% (образец 3), т.е. практически в 2 раза. Опираясь на результаты термического анализа, был определен режим термообработки порошков гидроксидов с целью кристаллизации оксидов – выдержка при 500°С в течение 1 ч.
Рис. 2.
Кривые ТГА (а) и ДСК (б) в потоке воздуха для порошков 1, 2 и 3, полученных в результате соосаждения и сушки.

Как видно из ИК-спектров порошков (рис. 3), термообработка при 500°С приводит к уменьшению интенсивности полос поглощения в интервале 3100–3700 см–1 (валентные колебания OH-групп), а также в области 1600–1700 см–1 (деформационные колебания HOH). При этом наблюдается формирование выраженного плеча в области 430–650 см–1. ИК-спектроскопия порошков после сушки не зафиксировала функциональные группы от органических компонентов, что можно объяснить перекрыванием соответствующих полос поглощения сигналами от вазелинового масла. Отмеченные изменения спектральных характеристик порошков свидетельствуют о разложении гидроксидов металлов при 500°С и формировании целевых оксидных твердых растворов состава CeO2-10%Y2O3.
Рис. 3.
ИК-спектры пропускания порошков 1, 2 и 3 после сушки (синий) и термообработки при 500°С (красный).

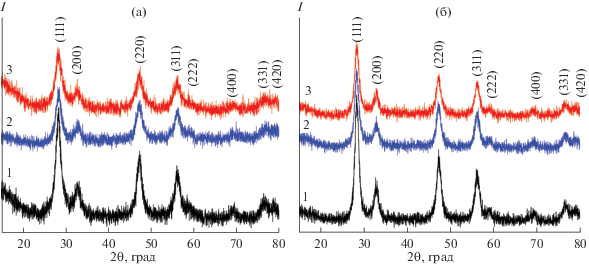
Результаты РФА полученных порошков свидетельствуют о том, что после сушки при 100°С в течение 3 ч во всех случаях формируется однофазный твердый раствор с кристаллической структурой флюорита (рис. 4а). По уширению рефлексов видно, что с увеличением концентрации поливинилового спирта в реакционной системе формируются более высокодисперсные порошки, а значения среднего размера кристаллитов D111 составляют 7.1 ± ± 0.7 (порошок 1), 6.5 ± 0.6 (2) и 5.8 ± 0.6 нм (3). В результате прокаливания данных порошков при 500°С (1 ч), как видно из соответствующих рентгенограмм (рис. 4б), тип кристаллической решетки всех продуктов сохраняется, однако наблюдается некоторое (на 25–35%) укрупнение кристаллитов – до 9.2 ± 0.9 (порошок 1), 8.1 ± 0.8 (2) и 7.9 ± 0.8 нм (3). При этом сохраняется тенденция к повышению дисперсности с ростом концентрации полимера на стадии осаждения порошков гидроксидов. Таким образом, можно предположить, что увеличение доли органического компонента, входящего на стадии синтеза в структуру порошка, вероятно, в виде оболочки, что было подтверждено ранее с помощью синхронного термического анализа, приводит к стерическим ограничениям роста оксидных частиц в ходе термообработки.
Морфология оксидных порошков, полученных в результате термообработки при 500°С, была изучена с помощью растровой электронной микроскопии (рис. 5). Как видно из микрофотографий, порошки представляют собой агломераты, пористость которых уменьшается с ростом концентрации поливинилового спирта в реакционной системе на стадии осаждения. При этом форма частиц из-за высокой степени их агломерации не может быть точно описана, но из микрофотографий видно, что в некоторых случаях частицы представляют собой одномерные структуры. Средний размер элементов частиц, выходящих на поверхность агломератов, как правило, лежит в диапазоне 15–20 нм.
Рис. 5.
Микроструктура нанопорошков 1 (а), 2 (б) и 3 (в) после термообработки при 500°С (по данным РЭМ).

Более детальное изучение микроструктуры полученных нанопорошков с помощью ПЭМ (рис. 6) позволило установить влияние концентрации полимера на иерархическую организацию оксидных наночастиц. Так, с ростом содержания поливинилового спирта в реакционной системе на стадии программируемого соосаждения гидроксидов наблюдается склонность к формированию одномерных стержневидных наноструктур, отношение длины к диаметру которых растет с 5 (образец 1) до 40 (3). Диаметр образующихся наностержней составляет ∼15–20 нм, что хорошо согласуется с данными растровой электронной микроскопии. Так, вероятно, описанные выше агломераты в значительной степени состоят из одномерных структур, торцы которых выходят на поверхность.

Таким образом, с помощью электронной микроскопии было показано, что на стадии соосаждения гидроксидов церия и иттрия поливиниловый спирт выполняет роль структурообразующего темплата и приводит к формированию оксидных наностержней состава CeO2-10%Y2O3, длина которых растет с увеличением концентрации указанного полимера в реакционной системе.
Изучение микроструктуры порошков в режиме контраста по среднему атомному номеру (детектор отраженных электронов) и результаты энергодисперсионного элементного анализа свидетельствуют о формировании однородных веществ целевого состава.
ЗАКЛЮЧЕНИЕ
Методом программируемого прямого совместного осаждения в присутствии поливинилового спирта (7.46 × 10–4–6.55 × 10–3 мас. %) как структурообразователя получены иерархические наноструктуры состава CeO2-10%Y2O3.
Полученные нанопорошки как после сушки (100°С, 3 ч), так и после проведения термообработки при 500°С (1 ч) являются однофазными и характеризуются кубической кристаллической структурой типа флюорита со средним размером ОКР 5.8–7.1 нм (100°С) и 7.9–9.2 нм (500°С).
Установлено, что с увеличением концентрации поливинилового спирта в реакционной системе наблюдается тенденция к формированию стержневидные оксидных наноструктур диаметром 15–20 нм. При этом увеличение концентрации используемого полимера приводит к увеличению отношения длины от диаметра наблюдаемых наностержней от 5 (образец 1) до 40 (образец 3).
Полученные наноструктуры по своим физико-химическим свойствам и микроструктурным особенностям представляют интерес и планируются к исследованию как перспективные компоненты среднетемпературных ТОТЭ, а также в качестве рецепторных материалов резистивных газовых сенсоров.
Список литературы
Иванов В.К., Щербаков А.Б., Баранчиков А.Е., Козик В.В. Нанокристаллический диоксид церия. Томск: Изд-во Томск. ун-та, 2013. 284 с.
Phoka S., Laokul P., Swatsitang E. et al. // Mater. Chem. Phys. 2009. V. 115. P. 423. https://doi.org/10.1016/j.matchemphys.2008.12.031
Izu N., Shin W., Murayama N., Kanzaki S. // Sens. Actuators, B. 2002. V. 87. P. 95. https://doi.org/10.1016/S0925-4005(02)00224-1
Chaudhary Y.S., Panigrahi S., Nayak S. et al. // J. Mater. Chem. 2010. V. 20. P. 2381. https://doi.org/10.1039/B922914K
Khan S.B., Akhtar K. Cerium Oxide: Applications and Attributes. IntechOpen, United Kingdom, 2019. https://www.intechopen.com/books/cerium-oxide-applications-and-attributes
Montini T., Melchionna M., Monai M., Fomasiero P. // Chem. Rev. 2016. V. 116. P. 5987. https://doi.org/10.1021/acs.chemrev.5b00603
Melchionna M., Fornasiero P. // Mater. Today. 2014. V. 17. P. 349. https://doi.org/10.1016/j.mattod.2014.05.005
Иванов-Шиц А.К., Мурин И.В. Ионика твердого тела: в 2 т. СПб.: Изд-во С.-Петерб. ун-та, 2010. Т. 2. 1000 с.
Chroneos A., Yildiz B., Tarancón A. et al. // Energy Environ. Sci. 2011. V. 4. P. 2774. https://doi.org/10.1039/C0EE00717J
Kim H.-J., Lee J.-H. // Sens. Actuators, B. 2014. V. 192. P. 607. https://doi.org/10.1016/j.snb.2013.11.005
Walker J.M., Akbar S.A., Morris P.A. // Sens. Actuators, B. 2019. V. 286. P. 624. https://doi.org/10.1016/j.snb.2019.01.049
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Ceram. Int. 2018. V. 44. P. 19879. https://doi.org/10.1016/j.ceramint.2018.07.249
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Glass. Phys. Chem. 2018. V. 44. P. 314. https://doi.org/10.1134/S1087659618040144
Egorova T.L., Kalinina M.V., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2017. V. 62. P. 1275. https://doi.org/10.1134/S0036023617100072
Egorova T.L., Kalinina M.V., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2016. V. 61. P. 1061. https://doi.org/10.1134/S0036023616090047
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Int. J. Hydrogen Energy. 2019. V. 44. P. 20345. https://doi.org/10.1016/j.ijhydene.2019.05.231
Mokrushin A.S., Simonenko E.P., Simonenko N.P. et al. // J. Alloys. Compd. 2019. V. 773. P. 1023. https://doi.org/10.1016/j.jallcom.2018.09.274
Ali A., Raza R., Khalil R.M.A. et al. // Ceram. Int. 2018. V. 44. P. 12676. https://doi.org/10.1016/j.ceramint.2018.04.068
Ma J., Zhang T.S., Kong L.B. et al. // J. Eur. Ceram. Soc. 2004. V. 24. P. 2641. https://doi.org/10.1016/j.jeurceramsoc.2003.09.023
Zhang T.S., Ma J., Kong L.B. et al. // Solid State Ionics. 2004. V. 170. P. 209. https://doi.org/10.1016/j.ssi.2004.03.003
Fu Y.-P. // Ceram. Int. 2009. V. 35. P. 653. https://doi.org/10.1016/j.ceramint.2008.01.027
Abazani M., Tsuchiya M., Ramanathan S. // J. Am. Soc. 2012. V. 95. P. 312. https://doi.org/10.1111/j.1551-2916.2011.04786.x
Izu N., Nishizaki S., Shin W. et al. // Sensors. 2009. V. 9. P. 8884. https://doi.org/10.3390/s91108884
Zhang Z., Shi H., Wu Q. et al. // Mater. Lett. 2019. V. 242. P. 20. https://doi.org/10.1016/j.matlet.2019.01.097
Michel C.R., Martínez-Preciado A.H. // Sens. Actuators, B. 2014. V. 197. P. 177. https://doi.org/10.1016/j.snb.2014.02.090
Lin K.-S., Chowdhury S. // Int. J. Mol. Sci. 2010. V. 11. P. 3226. https://doi.org/10.3390/ijms11093226
Li C.R., Cui M.Y., Sun Q.T. et al. // J. Alloys. Compd. 2010. V. 504. P. 498. https://doi.org/10.1016/j.jallcom.2010.05.151
Feng Z., Zhang M., Ren Q. et al. // Chem. Eng. J. 2019. V. 369. P. 18. https://doi.org/10.1016/j.cej.2019.03.051
Wang G., Mu Q., Chen T., Wang Y. // J. Alloys. Compd. 2010. V. 493. P. 202. https://doi.org/10.1016/j.jallcom.2009.12.053
Zhou X.D., Huebner W., Anderson H.U. // Appl. Phys. Lett. 2002. V. 80. P. 3814. https://doi.org/10.1063/1.1481244
Zhang D., Fu H., Liyi S. et al. // Inorg. Chem. 2007. V. 46. P. 2446. https://doi.org/10.1021/ic061697d
Sun C., Li H., Znahg H. et al. // Nanotechnology. 2005. V. 16. P. 1454. https://doi.org/10.1088/0957-4484/16/9/006
Deus R.C., Cilence M., Foschini C.R. et al. // J. Alloys. Compd. 2013. V. 550. P. 245. https://doi.org/10.1016/j.jallcom.2012.10.001
He L., Yu Y., Zhang C., He H. // J. Environ. Sci. China. 2011. V. 23. P. 160. https://doi.org/10.1016/S1001-0742(10)60388-9
Zhao X.B., You J., Lu X.W., Chen Z.G. // J. Inorg. Mater. 2011. V. 26. P. 159. https://doi.org/10.3724/SP.J.1077.2011.00159
Li J., Wang C., Zhu X. et al. // Mater. Lett. 2019. V. 240. P. 73. https://doi.org/10.1016/j.matlet.2018.12.081
Shen G., Liu M., Wang Z., Wang Q. // Nanomaterials. 2018. V. 8. P. 773/1. https://doi.org/10.3390/nano8100773
Cai P., Yi X., Song H., Lv Y. // Anal. Bioanal. Chem. 2018. V. 410. P. 5113. https://doi.org/10.1007/s00216-018-1141-4
Huang J., Chen F., Wang H., Yan H. // Res. Chem. Intermed. 2018. V. 44. P. 2729. https://doi.org/10.1007/s11164-018-3257-8
Tang W., Li W., Shan X. et al. // Mater. Lett. 2015. V. 140. P. 95. https://doi.org/10.1016/j.matlet.2014.11.011
Lu P., Qiao B., Lu N. et al. // Adv. Funct. Mater. 2015. V. 25. P. 4153. https://doi.org/10.1002/adfm.201501392
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии