

Журнал неорганической химии, 2019, T. 64, № 2, стр. 190-195
Формирование координационных соединений ионов металлов с анионами яблочной кислоты: квантово-химическое моделирование
Н. С. Панина 1, *, М. К. Давыдова 2, Е. М. Никандров 1, Д. О. Рузанов 1, А. Н. Беляев 1
1 Санкт-Петербургский государственный технологический институт (технический университет),
190013 Санкт-Петербург, Московский пр-т, 26, Россия
2 Санкт-Петербургский государственный педиатрический медицинский университет
194353 Санкт-Петербург, Литовская ул., 2, Россия
* E-mail: nataliepanina2707@gmail.com
Поступила в редакцию 07.05.2018
После доработки 06.07.2018
Принята к публикации 10.05.2018
Аннотация
Методом DFT M06/631G(d,p) проведено квантово-химическое исследование анионных форм яблочной кислоты H2mal и модельного комплекса [VV(O)2(mal)]–, в котором лиганд mal2– координирован к иону металла атомами кислорода карбоксильной и гидроксильной групп. Установлено, что образование малатных координационных соединений в кислой среде за счет гидроксильной группы, теряющей протон только в щелочной среде, обусловлено не ее кислотной диссоциацией, а процессом конкурентного замещения на более электроположительный ион металла. Показано, что одновременное отсутствие протонов у карбоксильной и гидроксильной групп приводит к электронной избыточности в области связывания –CH2–CHO–. При образовании связей с электроположительными ионами металлов происходит отток электронной плотности из этой области, что приводит к стабилизации двухзарядного аниона.
ВВЕДЕНИЕ
Яблочная кислота HOOCCH2CH(OH)COOH (H2mal, соли – малаты) относится к биологически важным гидроксикислотам. Ее разнообразные свойства, в том числе антиоксидантные, кислотные и способность к комплексообразованию, широко используются в медицине и фармакологии. В связи с поиском новых лекарственных препаратов большой интерес представляет способность анионов яблочной кислоты образовывать координационные соединения с ионами биологически активных d-элементов. Для малатного комплекса ванадия(IV) подтверждена выраженная гипогликемическая активность [1, 2], что делает соединения такого типа чрезвычайно перспективными для создания антидиабетических средств. Однако в связи с малой устойчивостью полученного соединения, описанного в [1] как моноядерный комплекс бис(L-малато)оксованадия(IV), его структура не установлена методом РСА. При синтезе подобных координационных соединений часто существует проблема получения устойчивых интермедиатов с ионами металлов.
Возможности координации с ионами металлов яблочной кислоты и ее анионных форм связаны с наличием трех функциональных центров – двух карбоксильных и одной гидроксильной групп. Несимметричное расположение гидроксильной группы в молекуле относительно карбоксильных групп приводит к поляризации анионных форм и создает возможность их различного ион-дипольного ориентирования при образовании координационных соединений с ионами металлов. Это позволяет анионам яблочной кислоты в качестве лигандов образовывать как моно-, так и полиядерные координационные соединения. При определенном расположении функциональных групп в этих лигандах легко образуются хелатные комплексы с ионами металлов. Хелатные структуры с катионами металлов представляют большой интерес в связи с их ролью в кислотном катализе [3].
Некоторые вопросы, связанные с получением устойчивых координационных соединений с анионами яблочной кислоты, требуют детального рассмотрения. Один из таких вопросов относится к влиянию пространственного расположения функциональных карбоксильных и гидроксильных групп на хелатирующие возможности лигандов при образовании комплексов с ионами металлов. Другой связан с формированием координационных соединений анионами яблочной кислоты, в том числе и за счет атомов кислорода гидроксильной группы, которое проводят в кислой среде. Это условие не согласуется с ограничением депротонирования гидроксильной группы, происходящим только в сильнощелочной среде.
Задачей настоящей работы являлось исследование особенностей координации анионных форм яблочной кислоты к ионам металлов-комплексообразователей методами квантовой химии с целью интерпретации экспериментальных данных.
РАСЧЕТНАЯ ЧАСТЬ
Квантово-химическое моделирование соединений с расчетами термодинамических характеристик и учетом влияния растворителя в рамках модели поляризуемого континуума (PCM) [4, 5] выполнено по программному комплексу GAUSSIAN’09 [6] методом DFT M06 [7] в базисе 631G(d,p) [8, 9].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В результате квантово-химического моделирования получены четыре устойчивых геометрических изомера яблочной кислоты (I–IV) с разным взаимным расположением О= и Н–О-групп двух карбоксильных и одного гидроксильного центров и рассчитанными значениями энергии Гиббса с учетом растворителя (∆G° + ∆Esolv), различающимися в пределах 16 кДж/моль (табл. 1). Три изомера (I, II, IV) имеют хелатное строение за счет сближения атомов кислорода концевых карбоксильных групп и один – цепочечное строение (III). Пространственное расположение функциональных групп этой кислоты, по всей видимости, должно влиять на координационные возможности ее анионов в комплексах.
Таблица 1.
Структурные и энергетические характеристики геометрических изомеров I–IV кислоты Н(1)O(1)OCCH2CHO(3)H(3)COO(2)H(2) и ее анионов в газовой фазе и водном растворе: G° – энергия Гиббса, Esolv – энергия сольватации
| Изомер, анион | R(C–C)*, Å | G°, a.е. | ∆Esolv, кДж/моль |
|---|---|---|---|
| I | 1.538 | –531.845211 | –48.37 |
| II | 1.530 | –531.846032 | –38.22 |
| III | 1.523 | –531.854535 | –36.74 |
| IV | 1.525 | –531.856177 | –35.27 |
| I-Н1 | 1.538 | –531.314887 | –246.54 |
| I-Н2 | 1.523 | –531.293537 | –277.89 |
| I-Н3 | 1.722 | –531.266543 | –258.96 |
| II-Н1 | 1.527 | –531.312914 | –250.93 |
| II-Н2 | 1.530 | –531.332550 | –217.59 |
| II-Н3 | 1.560 | –531.276539 | –249.55 |
| III-Н1 | 1.537 | –531.314806 | –243.30 |
| III-Н2 | 1.521 | –531.297049 | –276.06 |
| III-Н3 | 1.679 | –531.265606 | –263.41 |
| IV-Н1 | 1.526 | –531.312919 | –250.93 |
| IV-Н2 | 1.533 | –531.312654 | –257.73 |
| IV-Н3 | 1.701 | –531.265022 | –265.04 |
| II-Н2–Н3 | 1.572 | –530.589276 | –810.80 |
Обычно полагают, что кислотные свойства яблочной кислоты обусловлены преимущественно диссоциацией карбоксильных групп. Поэтому ее часто относят к двухосновным гидроксикарбоновым кислотам и обозначают H2mal, включая протон гидроксильной группы в анион mal2–. Авторами [10] уточнены экспериментальные значения рKа для кислотной диссоциации этой кислоты: рKа1 = 3.26, рKа2 = 4.78. Диссоциация протона гидроксильной группы, по результатам этой работы, характеризуется значением рKа3 = 12.34.
Для простоты изложения присвоим следующие номера центрам депротонирования яблочной кислоты: Н(1)O(1)OCCH2CHO(3)H(3)COO(2)H(2). Согласно полученным квантово-химическим данным, в реакции кислотной диссоциации наименьшее значение (∆G° + ∆Esolv) = 146 кДж/моль соответствует переносу протона $Н _{{({\text{1}})}}^{ + }$ к молекуле растворителя Н2О от карбоксильной группы хелатного изомера II. В результате образуется анионная система (II-H1), наиболее стабилизированная водородными связями (рис. 1).
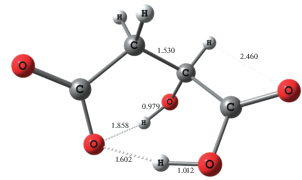
В анионе (II-H1) расстояния между атомами кислорода внутри каждой карбоксильной группы составляют 2.22–2.26 Å. Близкое расположение лигандных атомов, а также большая подвижность протона ${\text{H}}_{{\left( {\text{2}} \right)}}^{ + },$ позволяющая ему мигрировать между атомами O(1) и O(2), способствуют координации аниона Hmal– к металлу-комплексообразователю как одним, так и двумя атомами кислорода любой карбоксильной группы. В результате с различными ионами металлов, как в случае обычных карбоновых кислот [11–14], могут формироваться координационные соединения, в которых анионы яблочной кислоты могут выступать в роли моно- и бидентатных лигандов.
Процесс отрыва протона ${\text{H}}_{{\left( {\text{3}} \right)}}^{ + }$ от гидроксильной группы нуждается в более значительных энергетических затратах. Образование модельного аниона (II-H3) в этом процессе требует 261 кДж/моль, а для анионов других молекулярных изомеров это значение увеличивается до 290–300 кДж/моль. В структурах последних следует отметить неожиданный эффект существенного увеличения межатомных расстояний между атомами углерода в центральной части анионов –CH2–CHO(3)– по сравнению с таковыми в нейтральных геометрических изомерах I–IV и анионе (II-H1) (табл. 1), который может оказывать влияние на устойчивость этих лигандов в составе координационных соединений.
В анионах [H(1)O(1)OC–CH2–CHO(3)–COO(2)–]2–, происходящих от молекулярных изомеров I, III и IV, при одновременном отсутствии протонов ${\text{H}}_{{\left( {\text{2}} \right)}}^{ + }$ и ${\text{H}}_{{\left( {\text{3}} \right)}}^{ + }$ у близко расположенных карбоксильной и гидроксильной групп следует отметить большую электронную избыточность в области нахождения трех атомов кислорода. По данным распределения электронной плотности, там локализовано ~80% отрицательного заряда аниона. Протон ${\text{H}}_{{\left( {\text{1}} \right)}}^{ + }$ карбоксильной группы аниона, происходящего от хелатных изомеров указанной группы, развернут в противоположную сторону от области избыточного отрицательного заряда. Поэтому единственным путем для выравнивания электроотрицательности атомов [15] в анионной системе является перемещение избыточной электронной плотности вдоль линии связи –CH2 ← CHO(3)COO(2)–. Однако по мере накопления отрицательного заряда в области связывания начинают возрастать силы электростатического отталкивания групп атомов. Это приводит к увеличению межатомных расстояний, уже отмеченному ранее для однозарядных анионов (табл. 1). В случае анионов [H(1)O(1)OC–CH2–CHO(3)–COO(2)–]2– квантово-химическое моделирование указанных изомерных форм показало, что еще бóльшая избыточность электронной плотности может приводить к распаду двухзарядного аниона на два фрагмента – [H(1)O(1)OCCH2]– и [CH(O(3))COO(2)]–.
Вместе с тем структура модельного аниона с депротонированными карбоксильной и гидроксильной группами (II-H2–H3) (рис. 2), ведущего свое происхождение от хелатного молекулярного изомера II, оказалась устойчивой лишь при немного увеличенном значении R(C–C) = 1.572 Å. В этом анионе благодаря миграции подвижного протона ${\text{Н }}_{{({\text{1}})}}^{ + }$ в область наибольшего отрицательного заряда за счет Н-связи достигается более равномерное распределение электронной плотности. Миграции протона особенно способствует хелатная конформация (II) яблочной кислоты, в которой расстояние между атомом Н(1) карбоксильной группы и атомом О(2) другой карбоксильной группы минимально.
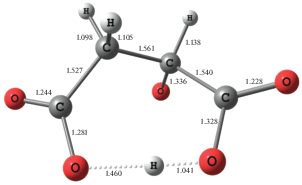
Анион mal2– потенциально обладает высокой комплексообразующей способностью за счет трех атомов кислорода. Экспериментально доказано, что в координационных соединениях с ионами металлов он образует бидентатные связи обеими карбоксильными группами, карбоксильной и гидроксильной группами одновременно [16–19], а также тридентатные связи за счет двух карбоксильных и одной гидроксильной групп [20–24]. Ранее было отмечено, что депротонирование яблочной кислоты по всем трем кислородным центрам возможно только при высоких значениях рН. При этом в большинстве случаев ион металла-комплексообразователя будет образовывать малорастворимые гидроксосоединения различного типа, в том числе и полимерные. Поэтому обычно синтез координационных соединений с анионами яблочной кислоты проводят в кислой среде [19]. Однако, исходя из величины экспериментальной характеристики рKа3 = 12.34 для яблочной кислоты [10], процесс комплексообразования одновременно карбоксильной и гидроксильной группами кажется нереализуемым в кислой среде.
Очевидно, что в условиях кислой среды образование координационных соединений ионов металла за счет их донорно-акцепторной связи с атомом кислорода гидроксильной группы яблочной кислоты не может быть связано с обычным процессом кислотной диссоциации ее третьего протона. Оно происходит только в процессе конкурентного замещения более электроположительным, чем протон, ионом металла-комплексообразователя. Ион металла снимает электростатическое напряжение в центральной части лиганда, образуя донорно-акцепторные связи с атомами кислорода, и предотвращает его распад, оттягивая на себя излишнюю электронную плотность. Внедрение иона металла в область наибольшего отрицательного заряда в данном случае ингибирует распад аниона яблочной кислоты [3].
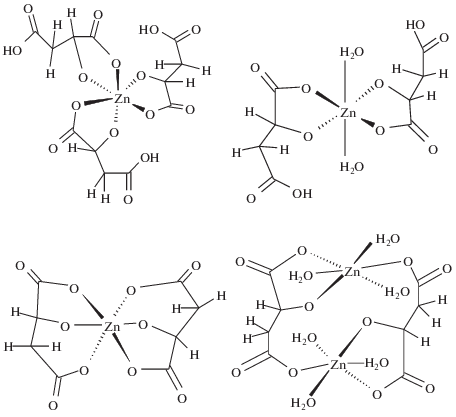
В качестве примера устойчивой структуры с бидентатной координацией аниона mal2– карбоксильной и гидроксильной группами была получена оптимизированная модельная структура комплексного аниона [VV(O)2(mal)]– (рис. 3). Этот анион является прототипом окисленной части биядерного смешановалентного соединения [(O)(phen)2VIV(μ-O)VV(O)(phen)(mal)]+, полученного и исследованного методом РСА в работе [19]. В свободном катионе диоксованадила (VO2)+ рассчитанный положительный заряд на ванадии составляет 1.54 е, что делает ион ванадия(V) более электроположительным центром, чем протон. Образование донорно-акцепторных связей (VO2)+ с атомами кислорода приводит к оттягиванию электронной плотности из центральной области –CH2–CH(O)– лиганда mal2– и формированию устойчивого хелатного пятичленного цикла. При образовании комплексов этого типа процессу конкурентного замещения третьего протона кислоты ионом металла способствуют близость расположения координирующихся атомов кислорода карбоксильной и гидроксильной групп и хелатная структура самого аниона, в которой электронное распределение более равномерно за счет образования водородной связи между атомами кислорода карбоксильных групп. Примеры координационных соединений, в которых анион mal2– действует как би- и тридентатный лиганд, образуя устойчивые пятичленные циклы с ионами металлов, а также благодаря своей протяженности связывает различные координационные полиэдры [25], представлены на рис. 4.
Рис. 3.
Структура модельного комплекса [VV(O)2(mal)]– с бидентатной координацией аниона mal2– атомами кислорода карбоксильной и гидроксильной групп.
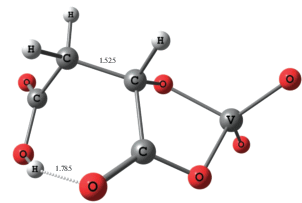
Рис. 4.
Схематическое представление би- и тридентатной координации анионов яблочной кислоты к иону металла [25].
ЗАКЛЮЧЕНИЕ
Проведенное в работе квантово-химическое моделирование яблочной кислоты и ее анионных форм показало, что перенос протона гидроксильной группы, происходящий только в щелочной среде, требует больших энергетических затрат. При отсутствии протонов одновременно у карбоксильной и гидроксильной групп выявлен эффект значительного ослабления связи С–С в центральной части аниона, обусловленный большой электронной избыточностью в этой области. Установлено, что в стабилизации анионных форм кислоты большую роль играет образование водородных связей.
Экспериментально доказанная способность формирования яблочной кислотой координационных соединений ионов металлов в кислой среде посредством гидроксильной группы объясняется тем, что образованию донорно-акцепторных связей предшествует не диссоциация протона, а процесс его конкурентного замещения более электроположительным катионом металла. Комплексообразование снимает электростатическое напряжение в центральной части аниона mal2– посредством оттягивания избытка электронной плотности. На примере модельного соединения [VV(O)2(mal)]– с бидентатной координацией аниона mal2– атомами кислорода карбоксильной и гидроксильной групп продемонстрирована возможность образования устойчивого пятичленного цикла: металл–анион яблочной кислоты. Процессу конкурентного замещения протона гидроксильной группы яблочной кислоты ионом металла способствует близость расположения координирующихся атомов кислорода карбоксильной и гидроксильной групп в хелатной структуре аниона.
Список литературы
Gorodetskii V.K., Tochilkin A.I., Belayeva N.F. et al. // Biochemistry (Moscow). Supplement Series B: Biomedical Chemistry. 2011. V. 5. № 3. P. 260. doi 10.1134/S1990750811030048
Коровкин Б.Ф., Арчаков А.И., Сергеев П.Б. и др. Оксованадиевые комплексы L-яблочной кислоты, проявляющие гипогликемическую активность. ФИПС. Открытые реестры. Пат. № 2101287 (1998). Россия.
Басоло Ф., Пирсон Р. Механизмы неорганических реакций. Изучение комплексов металлов в растворе / Пер. c англ. под ред. Ермакова А.Н. М.: Мир, 1971. С. 592. [Basolo F., Pearson R.G. // Mechanisms of Inorganic Reactions. A Sudy of Metal Complexes in Solution. N.Y.–London–Sydney: J. Wiley and Sons, Inс. 1967.]
Miertus S., Scrocco E., Tomasi J. // Chem. Phys. 1981. V. 55. № 1. P. 117. doi 10.1016/0301-0104(81)85090-2
Tomasi J., Persico M. // Chem. Rev. 1994. V. 94. № 7. P. 2027. doi 10.1021/cr00031a013
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2010.
Zhao Y., Truhlar D.G. // Theor. Chem. Account. 2008. V. 120. № 1. P. 215. doi 10.1007/s00214-007-0310-x
Hehre W.J., Ditchfield R., Pople J.A. // J. Chem. Phys. 1972. V. 56. P. 2257. doi 10.1063/1.1677527
Rassolov V.A., Pople J.A., Ratner M., Windus T.L. // J. Chem. Phys. 1998. V. 109. P. 1223. doi 10.1063/1.476673
Сухно И.В., Гаврилюк М.Б., Бузько В.Ю., Панюшкин В.Т. // Коорд. химия. 2004. Т. 30. № 7. С. 555. [Sukhno I.V., Gavrilyuk M.B., Buzko V.Y. Panyushkin V.T. // Russ. J. Coord. Chem. 2004. V. 30. № 7. P. 520. doi 10.1023/B:RUCO.0000034794.85192.45].
Черкашина Н.В., Нефедов С.Е., Уварова М.А. и др. // Журн. неорган. химии. 2014. Т. 59. № 5. С. 612. [Cherkashina N.V., Nefedov S.E., Uvarova M.A. et al. // Russ. J. Inorg. Chem. 2014. V. 59. №5. P. 446. doi 10.1134/S0036023614050076]. doi 10.7868/S0044457X14050080
Уварова М.А., Агешина А.А., Голубничая М.А., Нефедов С.Е. // Журн. неорган. химии. 2015. Т. 60. № 8. С. 1032. [Uvarova M.A., Ageshina A.A., Golubnichaya M.A., Nefedov S.E. // Russ. J. Inorg. Chem. 2015. V. 60. № 8. P. 934. doi 10.1134/S0036023615080215]. doi 10.7868/ S0044457X1508022X
Сережкина Л.Б., Вологжанина А.В., Карасев М.О. и др. // Журн. неорган. химии. 2015. Т. 60. № 1. С. 41. [Serezhkina L.B., Vologzhanina A.V., Karasev M.O. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 1. P. 38. 10.1134/S0036023615010155]. doi 10.7868/ S0044457X1501016X
Сережкин В.Н., Григорьев М.С., Абдульмянов А.Р., Сережкина Л.Б. // Журн. неорган. химии. 2016. Т. 61. № 1. С. 26. [Serezhkin V.N., Grigor’ev M.S. Abdul’myanov A.R., Serezhkina L.B. // Russ. J. Inorg. Chem. 2016. V. 61. № 1. P. 24. doi 10.1134/S0036023616010228]. doi 10.7868/S0044457X16010220
Sanderson R.T. Chemical Bonds and Bond Energy. N.Y.: Acad. Press, 1971. P. 222.
Karipides A. // Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1981. V. 37. P. 1115. doi 10.1107/S0567740881005190
Gudavarthy R.V., Burla N., Kulp E.A. et al. // J. Mater. Chem. 2011. V. 21. P. 6209. doi 10.1039/c0jm03423a
Fleck M., Tillmanns E., Bohaty L., Held P. // Z. Kristallogr. 2004. V. 219. P. 101. doi 10.1524/zkri.219.2.101.26323
Никандров Е.М., Рузанов Д.О., Панина Н.С., Беляев А.Н. // Журн. общ. химии. 2017. Т. 87. № 11. С. 1872. [Nikandrov E.M., Ruzanov D.O., Panina N.S., Belyaev A.N. // Russ. J. Gen. Chem. 2017. V. 87. № 11. P. 2612. doi 10.1134/S1070363217110160].
Kaliva M., Giannadaki T., Salifoglou A. et al. // Inorg. Chem. 2001. V. 40. P. 3711. doi 10.1021/ic000894o
Takuma M., Ohki Y., Tatsumi K. // Organometallics. 2005. V. 24. P. 1344. doi 10.1021/om049072f
Lin D.-D., Xu D.-J. // Acta Crystallogr., Sect. E: Struct. Rep. Online. 2005.V. 61. P. m1215. doi 10.1107/S1600536805016004
Liu Q.-X., Ni L.-B., Liang F.-P. et al. // Polyhedron. 2009. V. 28. P. 917. doi 10.1016/j.poly.2009.01.017
He L. // Acta Crystallogr., Sect. E: Struct. Rep. Online. 2005. V. 61. P. m1752. doi 10.1107/S1600536805025274
Zhang R.-H., Hong Q.-M., Yang J.-M. et al. // Inorg. Chim. Acta. 2009. V. 362. P. 2643. doi 10.1016/j.ica.2008.12.001
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии