

Журнал неорганической химии, 2019, T. 64, № 5, стр. 545-552
Синтез и сравнительные исследования микроструктуры и свойств керамики LiNbO3 и LiNbO3:Zn, полученной с использованием золь-гель методов
М. Н. Палатников 1, О. Б. Щербина 1, *, В. В. Ефремов 1, С. М. Маслобоева 1, С. В. Владимирова 1, Д. В. Иваненко 1
1 Институт химии и технологии редких элементов и минерального сырья им. И.В. Тананаева ФИЦ КНЦ РАН
184209 Мурманская обл., Апатиты, Академгородок, 26а, Россия
* E-mail: shcerbina@chemy.kolasc.net.ru
Поступила в редакцию 06.04.2018
После доработки 26.10.2018
Принята к публикации 12.11.2018
Аннотация
Исследовано получение двумя вариантами золь-гель метода высокодисперсных порошков LiNbO3 и LiNbO3:Zn. Изучена микроструктура, механические и электрические свойства керамики LiNbO3 и LiNbO3:Zn, приготовленной из порошков различного генезиса. Керамика, полученная из порошков, фракционированных методом отмучивания, имеет равномерную микро- и субмикроструктуру, улучшенные прочностные характеристики и ионную проводимость по границам зерен керамики. Легирование LiNbO3 катионами Zn2+ снижает электронную проводимость керамики LiNbO3:Zn, что позволяет эффективно провести поляризацию керамики и улучшить ее пьезоэлектрические свойства.
ВВЕДЕНИЕ
Ниобат лития (LiNbO3) привлекает внимание специалистов интегральной и нелинейной оптики, акустоэлектроники, квантовой электроники, физики твердого тела [1–3]. Активный поиск стойких к оптическому повреждению материалов инициировал интерес к исследованию сильно легированных кристаллов LiNbO3:Zn (~4.0–9.0 мол. % ZnО в расплаве) [3]. Влияние легирующего катиона на свойства кристаллов ниобата лития носит скачкообразный характер, что определяется термином “концентрационный порог” [3]. В общем случае в легированных кристаллах ниобата лития может быть несколько концентрационных порогов, в области которых характеристики кристаллов испытывают аномальное поведение [3].
Для акустоэлектронных и пьезоэлектрических приложений широкое распространение получила керамика LiNbO3. Развитие таких приложений предъявляет жесткие требования к вновь синтезируемым и уже известным материалам, что стимулирует поиск новых методик и способов стабилизации их механических, электрических и акустических свойств [4]. Наряду с методами традиционной керамической технологии, основанной на твердофазном высокотемпературном синтезе исходной порошковой шихты, широко распространены золь-гель методы, которые позволяют при сравнительно низких температурах получать мелкодисперсные монофазные порошки высокой степени химической гомогенности [5]. Кроме того, легирование ниобата лития нефоторефрактивнымии примесями (Mg, Zn, Gd, Sс), помимо повышения стойкости кристаллов LiNbO3 к оптическому повреждению, может существенно изменять проводимость материалов на его основе, в частности керамики. Так, при концентрации цинка в ниобате лития ниже “пороговой” концентрации (~5.0 мол. % [3, 6, 7]) основные акцепторные центры (переходные металлы, антиструктурные дефекты NbLi) действуют как ловушки электронов. Например, происходит перезарядка ионов железа Fe3+ + e– = Fe2+ или образование поляронов при захвате электронов дефектами NbLi. Эти перезаряженные дефекты участвуют в поляронной или прыжковой проводимости. При “пороговых” и “послепороговых” концентрациях цинка (≥5 мол. %) в ниобате лития катионы Fe3+ локализуются в Nb-позициях [8] и дефект ${{[{\text{Fe}}_{{{\text{Nb}}}}^{{{\text{3 + }}}}]}^{{{\text{2}} - }}}$ не может быть электронной ловушкой и участвовать в прыжковой проводимости. В то же время такие ловушки электронов, как антиструктурные дефекты NbLi, в ниобате лития с концентрацией цинка выше “пороговой” практически отсутствуют [3, 9]. В этом смысле интересно оценить влияние легирования нефоторефрактивными катионами (Zn2+), вносящими изменения в электронную подсистему ниобата лития, на структуру керамики и ее электрические свойства, поскольку существенное снижение проводимости позволяет более эффективно проводить поляризацию керамики и значительно улучшать ее пьезоэлектрические свойства.
В настоящей работе исследованы процессы получения мелкодисперсных порошков LiNbO3 и LiNbO3:Zn двумя разновидностями золь-гель метода. Методами электронной и зондовой микроскопии, импеданс-спектроскопии проведено сравнительное исследование микроструктуры, механических характеристик, диэлектрических свойств и проводимости керамики LiNbO3 и LiNbO3:Zn, приготовленной на основе мелкодисперсных монофазных порошков различного генезиса.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Традиционная схема получения функциональной керамики, как правило, включает в себя синтез порошка (шихты), его компактирование и высокотемпературное спекание. При этом синтезированные порошки должны удовлетворять целому ряду требований по морфологии, агломерированности, примесному и фазовому составу [10].
В настоящей работе синтез мелкодисперсных порошков номинально чистого LiNbO3 (образец 1) и ниобата лития с содержанием Zn ≥ 5 мол. % (образцы 2 и 3) осуществляли золь-гель методом [5] с применением гидратированного ниобата аммония, растворов нитрата лития, нитрата цинка и гидроокcида лития (порошки первого типа). При синтезе мелкодисперсных порошков номинально чистого ниобата лития и ниобата лития с содержанием Zn ≥ 5 мол. % (Li0.9Zn0.05NbO3) первого типа исходными реагентами служили: гидратированный ниобат аммония с содержанием Nb2O5 40.0–42.0%, раствор нитрата лития с концентрацией по [Li+] 32.2 г/л, раствор нитрата цинка с концентрацией по [Zn2+] 105.5 г/л и раствор гидроокcида лития с концентрацией по [Li+] 6.9 г/л.
Для получения гидратированного ниобата аммония использовали кислый фторидный раствор ниобия (СNb(V) = 2.24 моль л–1, ${{С }_{{{{{\text{F}}}^{ - }}}}}$ = 24.18 моль л–1), полученный растворением Nb2O5 (ч) в 40%-ной плавиковой кислоте (ос. ч.).
Свежеосажденный гидратированный ниобат аммония получали путем постепенного добавления подогретого исходного кислого фторидного раствора ниобия(V) к концентрированному раствору аммиака, взятому в избытке (∼30%) от стехиометрии. Синтез проводили при постоянном перемешивании и при pH не ниже 10–11 в соответствии с уравнением реакции:
После выдерживания суспензии в течение ∼30 мин образовавшуюся твердую фазу отфильтровывали и промывали репульпацией в ∼3%-ном растворе аммиака для полного замещения в твердой фазе фторид-ионов на гидроксильные группы. Полученный гидратированный ниобат аммония герметизировали для предотвращения высыхания. Оптимальными условиями получения Li0.9Zn0.05NbO3 ⋅ aq в виде гидратированных сложных оксидов с заданным соотношением Li : Nb(V) и Zn : Nb(V) является гидрохимическая обработка в течение 2 ч при температуре 90–95°C с сотношениями Li : Nb(V) = 3.5–3.7 и Zn : Nb(V) = 0.05 моль/моль.
Схема синтеза ниобата лития, модифицированного цинком, отличалась тем, что непосредственно при легировании не добавляли раствор гидрооксида аммония, поскольку Zn(ОН)2 растворяется в избытке осаждающего раствора при рН 10.5, вследствие чего осаждение перестает быть полным [11].
К навеске ниобата аммония приливали раствор нитрата лития в мольном соотношении Li+:Nb5+ = 3.1–3.8 м/м и репульпировали на магнитной мешалке при комнатной температуре в течение 0.5 ч (т : ж = 1 : 7–8; рНпульпы 8). С помощью рН-метра раствором гидроксида лития доводили рНпульпы до 9–9.5. Дальнейший синтез проводили в термостате при температуре 95°С в течение 1.5–2 ч. Затем в пульпу по каплям добавляли нитратный раствор легирующего металла. Синтез прекурсора вели в течение 30 мин. Сырой осадок отфильтровывали и сушили при температуре 70°С в течение ~24 ч. Прокаливанием уже сухого прекурсора в течение 2 ч при температурах 500 и 700°С были получены порошки с удельной площадью поверхности Sуд = 15.03 и 7.2 м2/г соответственно, которые служили шихтой для керамических образцов 2 и 3 LiNbO3:Zn (табл. 1). Разница в удельной площади поверхности порошков для приготовления образцов 2 и 3 LiNbO3:Zn, по-видимому, обусловлена разным отношением Li/Nb (табл. 1). Керамику LiNbO3:Zn готовили по традиционной керамической технологии – спеканием при температуре 1050°С в течение 2 ч (образцы 2 и 3). Структуру и свойства керамического ниобата лития, модифицированного цинком, сравнивали с характеристиками керамического образца LiNbO3, спеченного из номинально чистого монофазного порошка ниобата лития (карточка 88-0289 базы данных JCPDS) при температуре 1100°С в течение 2 ч (образец 1). Порошок был синтезирован по описанной выше золь-гель технологии без введения цинка. Согласно химическому анализу, прокаленный при 500°С порошок прекурсора LiNbO3 содержал 4.56 мас. % Li и 65.3 мас. % Nb, что соответствует мольному отношению [Li]/[Nb] ≈ 0.935. Удельная площадь поверхности такого порошка составляет Sуд = 10.67 м2/г.
Таблица 1.
Удельная площадь поверхности исходных порошков и механические характеристики керамики LiNbO3 и LiNbO3:Zn
| Параметр | Образец | |||
|---|---|---|---|---|
| 1 LiNbO3 | 2 LiNbO3:Zn | 3 LiNbO3:Zn | 4 LiNbO3 | |
| Отношение Li/Nb | 0.935 | 1.07 | 1.09 | 1.04 |
| Sуд частиц шихты ниобата лития, м2/г | 10.67 | 7.20 | 15.03 | 5.26 |
| Микротвердость, H, ГПа Среднее значение |
5.0 ± 0.8 | 4.37 ± 0.65 | 4.18 ± 1.05 | 5.0 ± 0.7 |
| Модуль Юнга, E, ГПа | 175.7 ± 4.0 | 162.6 ± 1.0 | 169.4 ± 2.9 | 188.7 ± 2.9 |
Номинально чистый монофазный, узкой гранулометрической фракции (удельная площадь поверхности частиц Sуд = 5.26 м2/г) мелкодисперсный порошок ниобата лития (образец 4) с отношением [Li]/[Nb] = 1.04 был получен с использованием второго варианта золь-гель метода по технологической схеме, описанной в работе [12], путем синтеза ацетатного прекурсора, дальнейшего его прокаливания и выделения узкой гранулометрической фракции с помощью отмучивания [13]. Для синтеза мелкодисперсного порошка номинально чистого ниобата лития второго типа был использован высокочистый фторидный ниобийсодержащий раствор состава Nb2O5 – 127.4, F– – 132.3 г/л. По данным масс-спектрометрического анализа с индуктивно-связанной плазмой, содержание микропримесей в нем (Mg, Al, Ti, Fe, Cu, Pb, Sn, Ni, Cr, Co) было на уровне (1–2) × 10–4 г/л и менее, Та – 0.008 г/л. Синтез порошков проводили в едином технологическом цикле по схеме, подробно описанной в работе [12]. Для осаждения гидроксида ниобия в раствор вводили 25%-ный NH4OH (ос. ч.) до значения рН 8–9. Осадок отфильтровывали на нутч-фильтре, затем репульпацией трехкратно промывали деионизированной водой при соотношении объемов твердой и жидкой фаз т : ж = 1 : 2. После сушки влажный (~65%) очищенный гидроксид ниобия смешивали при соотношении объемов твердой и жидкой фаз т : ж = 1 : 1 с раствором ацетата лития H3COOLi. Перемешивание осуществляли в течение 3 ч. Полученную пульпу упаривали до вязкого состояния, сушили при 140°С, остаток прокаливали при температуре 1100°С в течение 6 ч. H3COOLi готовили из карбоната лития Li2CO3 (ос. ч.) и раствора уксусной кислоты СН3СООН (ос. ч.) с концентрацией Li, соответствующей мольному отношению [Li]/[Nb] = 1.04. Результаты рентгенофазового анализа свидетельствовали, что при выбранных условиях эксперимента получен монофазный продукт (карточка 88-0289 базы данных JCPDS). При исследовании морфологии такого порошка LiNbO3 установлено, что он представляет собой конгломераты, состоящие из кристаллических частиц различной величины с развитой поверхностью, для которых характерна сглаженная ограненность с нечетким проявлением габитуса. Для разрушения образовавшихся конгломератов порошок LiNbO3 размалывали и разделяли на узкие гранулометрические фракции, используя метод отмучивания [13]. Значения Sуд для частиц выделенной фракции (<3 мкм) были выше Sуд исходного порошка (<140 мкм) более чем в 30 раз, что должно сказываться на кинетике спекания, однородности, мезоструктуре и свойствах приготовленных из нее керамических образцов за счет возрастания удельной реакционной способности частиц шихты с увеличением их дисперсности.
Химический состав продуктов синтеза контролировали гравиметрическим методом (определение Nb), а также методом атомно-эмиссионной спектрометрии с индуктивно-связанной плазмой на спектрометре Optima 8300 ИСП-АЭС. Фазовый состав определяли на дифрактометре ДРОН-2 со скоростью движения счетчика 2 град/мин (CuKα-излучение, графитовый монохроматор). Для идентификации фаз использовали базу данных JCPDS.
Удельную площадь поверхности порошков измеряли методом низкотемпературной адсорбции азота (БЭТ; FlowSorb II 2300; TriStar 3020 V1. 03).
Микроструктуру керамических образцов LiNbO3 и LiNbO3:Zn изучали с помощью сканирующего электронного микроскопа SEM LEO 420.
Механические характеристики образцов исследовали контактным методом с помощью зондового микроскопа-нанотвердомера NANOSKAN. Микротвердость (H, ГПа) керамик определяли методом сравнительной склерометрии [14], а модуль Юнга (E, ГПа) – по кривым подвода [15].
Для изучения диэлектрических свойств и проводимости на специально подготовленные керамические образцы диаметром ~10 и толщиной ~1.5 мм методом магнетронного напыления наносили платиновые электроды. Измерения выполняли на приборе Solartron 1260 в диапазоне частот 0.1–107 Гц в режиме ступенчатого нагрева.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
При исследовании микроструктуры керамики было установлено, что в керамическом образце 1 присутствуют в основном крупные зерна от ~3 до 50 мкм (рис. 1а, 1б), что, по-видимому, обусловлено быстрыми процессами перекристаллизации при 1100°С образца, приготовленного из ультрадисперсной шихты со средним размером частиц около 121.5 нм. Кроме того, в зернах керамики (образец 1) образовалось большое количество пор (рис. 1а, 1б). Несмотря на то, что керамические образцы LiNbO3:Zn (образцы 2 и 3) спекали из порошков, дисперсность которых различалась более чем в 2 раза, по своей структуре они практически не различались и состояли из зерен размером для образца 2 ~3–35 мкм (рис. 1в, 1г), а для образца 3 – от 2 до 30 мкм (рис. 1д, 1е). Для керамики, полученной из мелкодисперсных порошков первого типа (образцы 1–3), наилучшими механическими свойствами обладают дефицитные по литию образцы керамики номинально чистого LiNbO3 (образец 1), имеющие более высокую микротвердость и прочность (модуль Юнга), чем образцы 2 и 3.
Рис. 1.
Микроструктура (а, в, д, ж) и размерный состав (б, г, е, з) керамических образцов ниобата лития: LiNbO3 (образец 1) – а, б; LiNbO3:Zn (образец 2) – в, г; LiNbO3:Zn (образец 3) – д, е; LiNbO3 (образец 4) – ж, з.
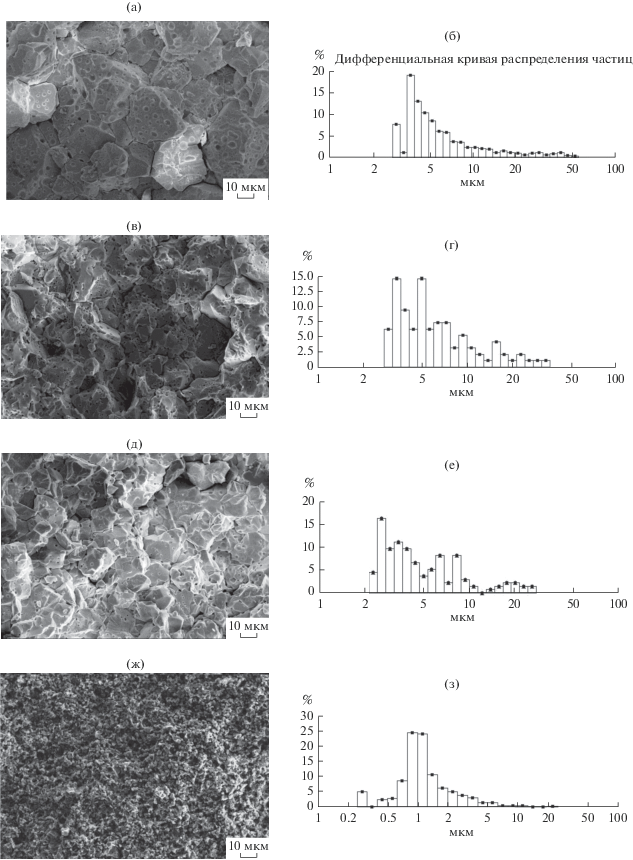
Керамический образец 4, приготовленный из мелкодисперсного порошка второго типа, характеризуется гораздо более равномерной микро- и субмикроструктурой, укладывающейся в диапазон размеров зерен ~ 0.25–5 мкм со средним значением ~1 мкм (рис. 1ж, 1з). Столь радикальное отличие от микроструктуры керамических образцов, полученных из порошков первого типа (образцы 1–3), по-видимому, объясняется тем, что при помоле исходного крупнозернистого порошка (<140 мкм) и последующем фракционировании его методом отмучивания резко уменьшается способность порошка к проявлению аномального прерывистого роста отдельных зерен при спекании, в результате получается керамика с более однородной мелкозернистой структурой (рис. 1ж, 1з). Размерность и однородность микроструктуры приводят к существенному улучшению прочностных характеристик (увеличению модуля Юнга) керамического образца 4 по сравнению с образцами 1–3. Кроме того, мелкозернистость, а значит бóльшая, чем в образцах 1–3, доля объема керамического образца, занимаемая границами зерен, приводит к отличию в типе ионной проводимости.
Использованная методика исследования комплексного импеданса позволяет корректно разделить вклад различных релаксационных процессов в измеряемые параметры и рассчитать значения статической удельной проводимости [16–18]. Дисперсия комплексного импеданса Z *(ω) керамических образцов LiNbO3:Zn (2 и 3) и LiNbO3 (1 и 4) была исследована в интервале температур от комнатной до ~800 K в диапазоне частот 0.1–107 Гц. Z ''–Z ' диаграммы для каждого образца во всем исследованном температурном диапазоне качественно подобны. На комплексных диаграммах импеданса обнаруживается один хорошо разрешенный релаксационный процесс дебаевского типа в виде полуокружности (рис. 2).
Рис. 2.
Диаграммы комплексного импеданса керамических образцов: а – LiNbO3 образец 1 (747 K); б – LiNbO3:Zn образец 2 (750 K), в – LiNbO3:Zn образец 3 (749 K), LiNbO3 образец 4 (791 K), частоты указаны в Гц.
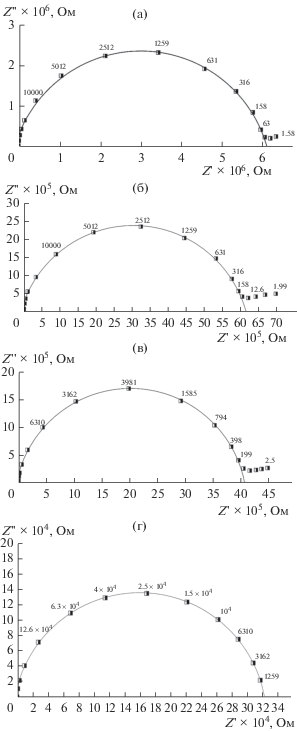
В низкочастотной области для всех образцов наблюдается проявление второго релаксационного процесса в виде “хвоста” (рис. 2), характеризующего электрические свойства приэлектродных областей исследуемой керамики. Наличие низкочастотных “хвостов” является следствием образования двойного электрического слоя вблизи электродов. Поскольку релаксационный процесс ионной миграции в приэлектродном слое протекает достаточно медленно, он доминирует в импедансе при низких частотах.
В результате анализа диаграмм комплексного импеданса для каждого образца были определены значения удельной статической проводимости σsv(Т) во всем исследованном диапазоне температур. Температурные зависимости удельной статической проводимости представлены на рис. 3. Следует обратить внимание на то, что проводимость для LiNbO3:Zn образцов 2 и 3 в высокотемпературной области почти в 2 раза выше, чем для LiNbO3 (образец 1), что, по-видимому, обусловлено избыточным содержанием лития в структуре ниобата лития (Li/Nb > 1). Это возможно, учитывая существование широкой области гомогенности ниобата лития [2, 3]. Для керамического образца 4, приготовленного из мелкодисперсного порошка второго типа, при температуре ~800 K проводимость в 2–4 раза выше, чем у образцов 1–3 (рис. 3), причем зависимость σsv(Т) в высокотемпературной области подчиняется закону Аррениуса и имеет при высоких температурах монотонный вид. По температурным зависимостям удельных статических проводимостей были определены энтальпии активации носителей заряда. Величины энтальпии активации для образцов 1–3 характерны для объемной ионной проводимости по литию [19, 20]. Энтальпия активации статической проводимости для керамического образца 1 LiNbO3 в области проводимости по литию при температуре Т ≥ 550 K (На ≈ 1.08 эВ) несколько ниже, чем для образцов 2 и 3 керамики LiNbO3:Zn (при Т ≥ ≥ 550 K: На ≈ 1.2 и 1.23 эВ соответственно). Это может быть связано с затруднением ионного транспорта из-за возникновения сложных полярных кластеров [3, 21, 22] в структуре LiNbO3:Zn, что не наблюдается в структуре номинально чистого LiNbO3 [22].
Рис. 3.
Температурные зависимости статической удельной проводимости керамических образцов: 1 – образец 1 LiNbO3, 2 – образец 2 LiNbO3:Zn; 3 – образец 3 LiNbO3:Zn; 4 – образец 4 LiNbO3.
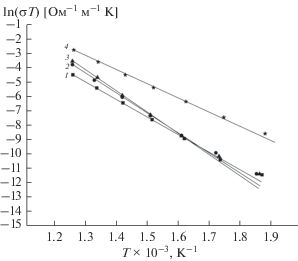
Величина энтальпии активации носителей заряда для керамического образца 4 LiNbO3На ≈ ≈ 0.88 эВ типична для ионной проводимости по границам зерен керамики. Преобладание проводимости по границам зерен однозначно обусловлено микро- и субмикроструктурой образца 4, в котором доля объема керамического образца, занимаемая границами зерен существенно выше, чем в образцах 1–3 (рис. 1).
Изучена дисперсия комплексной диэлектрической проницаемости образцов. Реальная часть температурной зависимости ε(Т) представлена на рис. 4. Дисперсия диэлектрической проницаемости керамических образцов LiNbO3 и LiNbO3:Zn в диапазоне температур ~300–550 K незначительна. Резкий рост реальной части диэлектрической проницаемости и существенная дисперсия в области низких частот начинаются лишь после ~600 K (рис. 4). Причем значения диэлектрической проницаемости керамических образцов 2 и 3 LiNbO3:Zn при температуре 800 K в два раза больше, чем у образца 1 LiNbO3 (рис. 4а–4в). Это обусловлено как более высокой проводимостью этих образцов, так и большей дефектностью их структуры. В то же время диэлектрическая проницаемость керамического образца 4 LiNbO3 при температуре 800 K еще в два раза больше, чем образцов 2 и 3 LiNbO3:Zn (рис. 4б–4г). Это объясняется существенно большей проводимостью этой керамики и существенным вкладом границ зерен в диэлектрическую релаксацию (рис. 1, 3). Высокие значения ε вблизи комнатной температуры и ее последующее уменьшение при нагревании связано с наличием в образцах воды и ее испарением при высокой температуре (рис. 4).
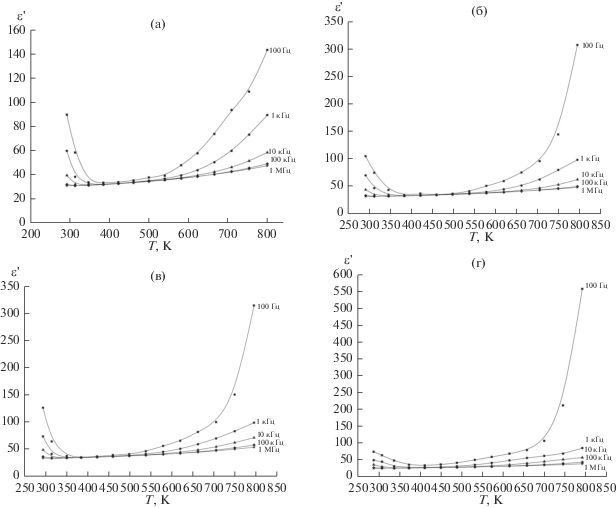
ЗАКЛЮЧЕНИЕ
Исследованы процессы получения двумя вариантами золь-гель метода мелкодисперсных порошков номинально чистого LiNbO3 и LiNbO3:Zn. Оценено влияние легирования нефоторефрактивным катионом Zn2+ на электрические свойства керамики LiNbO3:Zn. Присутствие катионов Zn2+ в структуре вносит изменения в электронную подсистему ниобата лития и существенно снижает низкотемпературную электронную проводимость, что позволяет более эффективно проводить поляризацию керамики и значительно улучшать ее пьезоэлектрические свойства. Методами электронной, зондовой микроскопии и импеданс-спектроскопии исследованы микроструктура, механические характеристики, диэлектрическая дисперсия и проводимость керамики LiNbO3 и LiNbO3:Zn, приготовленной на основе мелкодисперсных монофазных порошков различного генезиса.
Установлено, что только из порошка второго типа, подвергнутого размолу и затем разделению на узкие гранулометрические фракции методом отмучивания, может быть получена керамика с равномерной микро- и субмикроструктурой размером ~0.25–5 мкм, причем основная доля зерен такой керамики имеет размер ~1 мкм (образец 4 LiNbO3). При помоле исходного крупнозернистого порошка и последующем фракционировании его методом отмучивания резко снижается способность порошка к рекристаллизации, получается керамика с однородной мелкозернистой структурой, что приводит к существенному улучшению ее прочностных характеристик и изменению типа ионной проводимости. Энтальпия активации На ≈ 0.88 эВ образца 4 LiNbO3 типична для ионной проводимости по границам зерен керамики, что обусловлено его микро- и субмикроструктурой, в которой доля объема, занимаемая границами зерен, существенно выше, чем в образцах, для которых характерна объемная ионная проводимость с энтальпией активации На > 1 эВ (образцы 1–3).
Список литературы
Lines M.E., Glass A.M. Principles and application of ferroelectrics and related materials Oxford: Clarendon Press, 1977.
Кузьминов Ю.С. Ниобат и танталат лития – материалы для нелинейной оптики. М.: Наука, 1975.
Сидоров Н.В., Волк Т.Р., Маврин Ю.Н. и др. Ниобат лития: дефекты, фоторефракция, колебательный спектр, поляритоны. М.: Наука, 2003.
Wang F., Grinberg I., Rappe A.M. // Appl. Phys. Lett. 2014. V. 104. P. 15290. doi https://doi.org/10.1063/1.4871707
Иваненко В.И., Локшин Э.П., Громов О.Г., Калинников В.Т. Синтез сегнетоэлектрических и люминесцентных сложных оксидов редких элементов. Апатиты: Изд-во КНЦ РАН, 2009.
Palatnikov M.N., Sidorov N.V., Manukovskaya D.V. et al. // J. Am. Ceram. Soc. 2017. V. 100. № 8. P. 3703. doi https://doi.org/10.1111/jace.14851
Chernaya T.S., Volk T.R., Verin I.A., Simonov V.I. // Crystallog. Rep. 2008. T. 53. № 4. C. 573.
Feng H., Wen J., Wang H. et al. // J. Phys. Chem. Solids. 1990. V. 51. № 5. P. 397. doi https://doi.org/10.1016/0022-3697(90)90173-D
Симонов В.И. // Природа. 2003. № 11. С. 4.
Garanin S.G., Dmitryuk A.V., Zhilin A.A. et al. // J. Opt. Technol. 2010. V. 77. № 9. P. 565. doi 10.1364/JOT.77.000565 [Гаранин С.Г., Дмитрюк А.В., Жилин А.А. и др. // Оптический журнал. 2010. Т. 77. № 9. С. 52.]
Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1979.
Маслобоева С.М., Палатников М.Н., Арутюнян Л.Г. и др. // Сб. науч. тр. “Физико-химические аспекты изучения кластеров, наноструктур и наноматериалов”. Тверь: Твер. гос. ун-т, 2016. Вып. 8. С. 239.
Буланов В.Я., Кватер Л.И., Долгаль Т.В. и др. Диагностика металлических порошков. М.: Наука, 1983.
Oliver W.C., Pharr G.M. // J. Mater. Res. 1992. V. 6. № 7. P. 1564. doi https://doi.org/10.1557/JMR.1992.1564
Усейнов А.С. // Приборы и техника эксперимента. 2004. № 1. С. 134.
Tsai Y.-T., Whitmore D.H. // Solid State Ionics. 1982. V. 7. № 2. P. 129. doi https://doi.org/10.1016/0167-2738(82)90006-6
Jonscher A.K. Dielectric Relaxation in Solids. London: Chelsea Dielectrics Press, 1983. V. 16.
Хладик Дж М.: Мир, 1978 / Физика электролитов. Процессы переноса в твердых электролитах и электродах.
Niitsu G.T., Nagata H., Rodrigúes A.C.M. // J. Appl. Phys. 2004. V. 95. № 6. P. 3116. doi https://doi.org/10.1063/1.1647263
Rahn J., Hüge E., Dörrer L. et al. // J. Phys. Chem. 2012. V. 14. № 7. P. 2427. doi https://doi.org/10.1039/C2CP23548J
Kimura H. et al. // J. Cryst. Growth. 2009. V. 311. № 6. P. 1553. doi https://doi.org/10.1016/j.jcrysgro.2008.09.178
Palatnikov M.N., Sandler V.A., Sidorov N.V. et al. // Inorg. Mater. 2016. V. 52. № 2. P. 147. [Палатников М.Н., Сандлер В.А., Сидоров Н.В. и др. // Неорган. материалы. 2016. Т. 52. № 2. С. 180. doi 10.7868/S0002337X16020111]
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии