

Журнал неорганической химии, 2019, T. 64, № 8, стр. 845-849
Синтез гидроксосолей цинка-магния-алюминия со структурой гидроталькитового типа и катализаторов окислительного дегидрирования этана
О. Н. Краснобаева 1, И. П. Беломестных 2, Т. А. Носова 1, Т. А. Елизарова 1, Д. Ф. Кондаков 1, В. П. Данилов 1, *
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
2 Институт органической химии им. Н.Д. Зелинского РАН
119334 Москва, Ленинский пр-т, 47, Россия
* E-mail: vpdanilov@igic.ras.ru
Поступила в редакцию 27.12.2018
После доработки 24.01.2019
Принята к публикации 15.02.2019
Аннотация
Разработан способ синтеза гидроксосолей гидроталькитового типа, содержащих цинк, а также магний, алюминий, ванадий, молибден, ниобий в различных сочетаниях. Методом потенциометрического титрования изучены условия образования этих гидроксосолей, синтезированы образцы и получены их рентгенограммы. Путем термической обработки гидроксосолей синтезированы образцы оксидных катализаторов окислительного дегидрирования (ОД) углеводородов. Исследована их каталитическая активность. Установлено, что ряд катализаторов проявляет высокую эффективность в реакциях ОД этана – увеличивается выход этилена и повышается селективность реакции.
ВВЕДЕНИЕ
Работа является продолжением исследований, связанных с разработкой новых катализаторов окислительного дегидрирования (ОД) органических соединений, содержащих оксиды переходных металлов в сочетании с другими каталитически активными оксидами ряда металлов (Zn, Cu, Nb, Ta, Te, V, Cr, Mo, W, Sb и др.). В качестве прекурсоров катализаторов ОД мы использовали сложные гидроксосоли со слоистой структурой гидроталькитового типа [1]. В работах [2–14] нами синтезирован ряд прекурсоров катализаторов ОД, содержащих каталитически активные оксиды различных металлов, в настоящей работе – сложные гидроксосоли, содержащие цинк, а также магний, алюминий, ванадий, молибден, ниобий в различных сочетаниях. Изучены условия образования этих гидроксосолей. Синтезированные гидроксосоли использовали в качестве прекурсоров оксидных катализаторов ОД этана с целью определения влияния добавок цинка на каталитические свойства оксидных катализаторов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез цинксодержащих гидроксосолей, используемых в качестве прекурсоров катализаторов, осуществляли по следующей методике. К раствору, содержащему нитраты цинка, магния и алюминия в заданных количествах, при температуре 60°С добавляли при перемешивании по каплям раствор, содержащий гидроксид и карбонат калия в молярном соотношении 2 : 1 до изменения рН от 1 до 10. Полученный осадок, представлявший собой тройной гидроксокарбонат цинка-магния-алюминия, отмывали водой от ионов калия по тетрафенилборату.
Для осуществления анионного обмена с целью замещения карбонат-ионов в гидроксокарбонате на декаванадат (V10O28)6–- или парамолибдат (Mo7O24)6–-ионы полученную после осаждения и промывания пасту разбавляли водой до соотношения т : ж = 1 : 2 и добавляли к ней заданное количество раствора декаванадата калия (0.15 М) и парамолибдата аммония (0.15 М). Пульпу перемешивали в течение 10 мин, затем к ней по каплям добавляли азотную кислоту (0.2 М) до рН 4.5 для обеспечения необходимых условий протекания анионного обмена [15, 16]. После выдерживания смеси в течение 10 мин при рН 4.5 осадок отфильтровывали и отмывали водой от ионов калия и аммония по тетрафенилборату.
Для введения ниобия в состав гидроксосолей сплавляли К2СО3 с Nb2O5 в мольном соотношении 10 : 1 [17], сплав растворяли в воде и полученный раствор (рН 13) использовали для проведения анионного обмена карбонат-иона на полиоксониобат-ионы [Nb6O19]8– [7, 17]. Анионный обмен проводили в течение 6 ч, после чего осадки отмывали от иона калия водой.
Для установления фазового и химического состава синтезированных гидроксосолей использовали методы химического и рентгенофазового анализа (дифрактометр ДРОН 2.0, CuKα-излучение), а для определения условий их образования – рН-метрическое титрование растворов на прецизионном цифровом рН-метре ОР-208 Раделикс (Венгрия).
Химический анализ полученных образцов осуществляли классическими методами химического анализа. Алюминий определяли титрованием избытка трилона Б раствором азотнокислого цинка в уротропиновом буфере с ксиленоловым оранжевым. При одновременном присутствии в пробе цинка и алюминия их сумму определяли по вышеописанной методике, затем в отдельной пробе тем же способом определяли цинк, предварительно связывая алюминий фтористым аммонием. При одновременном присутствии в пробе магния, алюминия и цинка сумму магния и цинка определяли в аммиачном буфере с эриохромом черным (алюминий определению не мешает). Сумму цинка и алюминия определяли в уротропиновом буфере с ксиленоловым оранжевым, цинк – тем же способом, связав алюминий фтористым аммонием. Магний этим определениям не мешает. Ванадий определяли волюмометрически с солью Мора и фенилантраниловой кислотой в качестве индикатора, молибден – весовым методом с α-бензоиноксимом, ниобий – прокаливанием при 1000°С и взвешиванием осадка, полученного после кислотного гидролиза.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Кривые потенциометрического титрования нитратов цинка, магния, алюминия и их смесей раствором гидроксида калия представлены на рис. 1. Установлено, что из растворов, содержащих один нитрат металла, цинк осаждается при рН 6.8–7.0 в виде гидроксонитрата Zn5(OH)8(NO3)2 · 2H2O (рис. 1, кривая 1) [18], алюминий – при рН 3.0–3.5 в виде гидроксосоли переменного состава Al(OH)n(NO3)3– n · mH2O, описанной в литературе [1]; магний при рН 10.0–10.5 в виде хорошо окристаллизованного Mg(OH)2 (брусита).
Рис. 1.
Кривые потенциометрического титрования раствором гидроксида калия растворов нитрата цинка (1), нитратов цинка-алюминия (2), нитратов цинка-магния-алюминия (3).
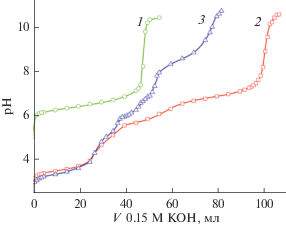
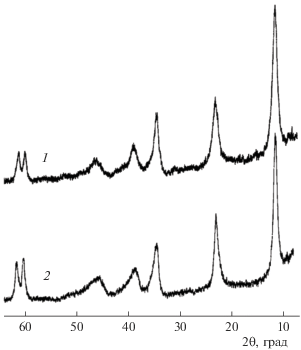
Из раствора, содержащего нитраты цинка и алюминия, сначала при рН 3.5–4.0 осаждается аморфный гидроксонитрат алюминия, затем при рН 6.0–6.5 образуется плохо окристаллизованная фаза переменного состава [ZnmAl(OH)2m+ 2][(NO3) · mH2O]. На ее рентгенограмме, которая индицируется в гексагональной сингонии с параметрами 3.16 и 26.52 Å, присутствуют рефлексы, отвечающие межплоскостным расстояниям 8.84(003), 4.44(006), 2.56(013) и 1.56(110) Å. Судя по данным РФА и химического анализа, эта фаза представляет собой двойной гидроксонитрат цинка-алюминия со слоистой гидроталькитовой структурой (рис. 2, кривая 1). Из раствора, содержащего нитраты цинка, магния и алюминия, при рН 3.0–3.5 осаждается аморфный гидроксонитрат алюминия, при рН 6.0–7.0 – гидроксонитрат цинка Zn5(OH)8(NO3)2 · 2H2О, при рН 8.0–8.5 выпадает тройной гидроталькитоподобный кристаллический гидроксонитрат цинка-магния-алюминия (рис. 2, кривая 2) переменного состава. Двойной гидроксонитрат цинка-алюминия и тройной гидроксонитрат цинка-магния-алюминия изоструктурны (судя по рентгенограммам) синтезированным нами ранее [1–14, 19, 20] сложным гидроксосолям со структурой гидроталькитового типа.
Рис. 2.
Дифрактограммы двойного гидроксонитрата цинка-алюминия (1), тройного гидроксонитрата цинка-магния-алюминия (2).
Нами получены два образца тройного гидроксонитрата состава [Zn0.16Mg1.88Al(OH)6.08][(NO3) · · mH2O] и [Zn0.10Mg1.87Al(OH)5.94][(NO3) · mH2O].
Для выяснения влияния цинка на каталитические свойства оксидных катализаторов в реакциях ОД этана нами синтезированы образцы цинксодержащих прекурсоров катализаторов. Для этого сначала получили тройной гидроксокарбонат цинка-магния-алюминия, в который методом анионного обмена ввели декаванадат- и парамолибдат-ионы (табл. 1, образцы 1, 2). Для приготовления прекурсора катализатора ОД, содержащего гексаниобат-ион вместе с декаванадат- и парамолибдат-ионами, из-за того, что последние замещают карбонат-ион в межслоевом пространстве гидроталькитоподобных гидроксосолей при рН 4.5, а гексаниобат – при рН 13.0, мы вынуждены были использовать смесь двух изоморфных фаз. Для получения прекурсора, состав которого отвечает образцу 2 в табл. 1, использовали смесь молибден-ванадийсодержащей гидроксосоли [Zn0.16Mg1.88(AlOH)6.08][(Mo7O24)0.04(V10O28)0.01(CO3)0.35 ⋅ ⋅ mH2O] и ниобийсодержащей гидроксосоли [Zn0.16Mg1.88Al(OH)6.08][(Nb6O19)0.02(CO3)0.42 ⋅ mH2O].
Таблица 1.
Состав цинксодержащих гидроталькитоподобных солей прекурсоров ОД этана
| № образца | Состав прекурсора |
|---|---|
| 1 | [Zn0.1Mg1.87Al(OH)5.94][(Mo7O24)0.02(V10O28)0.005(CO3)0.425 · mH2O] |
| 2 | [Zn0.16Mg1.88Al(OH)6.08][(Mo7O24)0.02(V10O28)0.005(Nb6O19)0.01(CO3)0.385 · mH2O] |
Указанные смеси готовили путем перемешивания в течение 2 ч (для достижения однородности) двух исходных гидроксосолей в весовом соотношении 1 : 1 в водной среде при соотношении т : ж = 1 : 2. Смеси получались достаточно однородными, о чем свидетельствовали характер седиментации осадков, а также данные химического анализа проб пульпы, отобранных в верхней и нижней частях седиментационного цилиндра (соотношения Al : Nb, Al : Mo и Al : V в этих пробах были одинаковыми). Состав полученных образцов-прекурсоров ОД представлен в табл. 1.
Для получения оксидных катализаторов синтезированные образцы прекурсоров подвергали термической обработке, которая заключалась в подсушивании осадка при 100–120°С до влажности 40%, таблетировании и прокаливании в муфеле в токе воздуха при постоянном подъеме температуры со скоростью 100 град/ч до 500°С и выдерживании при этой температуре в течение 4–5 ч.
Каталитические свойства полученных материалов изучали в кварцевом реакторе проточного типа с загрузкой 1–2 мл катализатора. В качестве окислителей использовали кислород воздуха и углекислый газ. Для поддержания изотермичности катализатор смешивали с равным объемом измельченного кварца. Температуру реакции, объемную скорость подачи углеводорода и соотношение углеводород/кислород/углекислый газ в исходной смеси варьировали в широких пределах. Продукты реакции анализировали методом жидкостной хроматографии с использованием колонки, наполненной фазой Порапак-Q.
В контактном газе определяли содержание непрореагировавшего исходного углеводорода (этан), продуктов дегидрирования (этилен, СО2, СО). По результатам опытов рассчитывали конверсию исходного соединения, селективность реакции и выход целевых продуктов [14].
На синтезированных из образцов 1 и 2 катализаторах проводили окислительное дегидрирование этана (табл. 2).
Таблица 2.
Результаты окислительного дегидрирования этана на цинксодержащих катализаторах (состав реакционной смеси C2H6 : CO2 : O2 = 1 : 0 : 0.3 моль)
| № образца |
Состав катализатора | t, °С | Конверсия С2Н6, % | Выход С2Н4, % | Селективность С2Н4, % |
|---|---|---|---|---|---|
| 1 | Zn-Mg-Al-V-Mo-O | 400 450 |
12.5 13.2 |
11.2 12.5 |
90.0 94.0 |
| 2 | Zn-Mg-Al-Nb-V-Mo-O | 400 450 450 |
17.0 19.7 20.0 |
16.5 19.0 19.6 |
96.5 96.0 95.7 |
Анализ результатов каталитических исследований показал, что на цинксодержащих катализаторах процесс превращения этана в этилен идет при низкой температуре (450°С).
На катализаторе состава Zn-Mg-Al-V-Mo-O (образец 1) ОД этана идет с высокой селективностью (94%), но с небольшим выходом (12.5%). При введении в состав катализатора ниобия (образец 2) селективность возрастает до 96%, а выход – до 20%.
Сравнение работы катализаторов, содержащих вместо Zn другие d-элементы 4-го периода (M-Al-Mg-V-Mo-O, где М = Ni, Cr, Fe), указывает на то, что у Ni,Cr,Fe-содержащих катализаторов процесс ОД этана идет при высоких температурах (700–800°С), при этом выход достигает 40–60% при селективности 40–70%.
При низких температурах (550°С) Ni- и Fe-содержащие катализаторы состава M-Al-Mg-V-Mo-O дают выход 2.6–3% при селективности 38–44%.
Результаты ОД этана, очень близкие к результатам цинксодержащих катализаторов, дают Cu-содержащие катализаторы, они также работают при низких температурах (400–450°С, табл. 3).
Таблица 3.
ОД этана на медьсодержащих катализаторах (состав реакционной смеси С2Н6 : СО2 : О2 = 1 : 0 : 0.3 моль)
| № образца |
Состав катализатора | t, °С | Конверсия, С2Н6, % | Выход С2Н4% | Селективность С2Н4, % |
|---|---|---|---|---|---|
| 1 | Cu-Mg-Al-V-Mo-O | 400 450 |
12.5 13.8 |
11.2 12.5 |
90.0 90.5 |
| 2 | Cu-Mg-Al-V-Mo-Nb-O | 400 450 |
17.6 20.0 |
17.5 19.6 |
97.2 95.7 |
Как видно из сравнения результатов ОД этана для Cu- и Zn-содержащих катализаторов, при низких температурах (400–450°C) их характеристики близки: выход – 12.5%, селективность 90–97%.
ЗАКЛЮЧЕНИЕ
Разработан способ введения цинка в состав катализаторов, содержащих Mg, Al, V, Mo, Nb в различных соотношениях.
Синтезированы новые гидроксосоли со слоистой структурой гидроталькитового типа [ZnnMgmAl(OH)2(n+m) + 2)]+[An .pH2O]–, где Аn = = ${\text{NO}}_{3}^{ - }$ (гидроксонитраты) или $\left( {{\text{С }}{{{\text{О }}}_{3}}} \right)_{{0.5}}^{--}$(гидроксокарбонаты). Гидроксокарбонаты применяли для анионного обмена (СО3) частично на (Mo7O24)6–, (V10O28)6–, (Nb6O19)8– c дальнейшим использованием полученных гидроксосолей в качестве прекурсоров катализаторов ОД этана.
При исследовании каталитических свойств полученных катализаторов в реакциях ОД этана установлено, что в присутствии как цинксодержащих, так и медьсодержащих катализаторов реакция окислительного дегидрирования этана идет при низких температурах – 400–450°С. Введение добавок ниобия в состав катализаторов приводит к повышению селективности и увеличению выхода этилена.
Список литературы
Krasnobaeva O.N., Belomestnykh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2004. V. 49. № 10. P. 1482. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган. химии. 2004. Т. 49. № 10. С. 1604.]
Krasnobaeva O.N., Belomestnykh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2005. V. 50. № 9. P. 1295. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган. химии. 2005. Т. 50. № 9. С. 1397.]
Krasnobaeva O.N., Belomestnykh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2007. V. 52. № 2. P. 141. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган. химии. 2007. Т. 52. № 2. С. 181.]
Krasnobaeva O.N., Belomestnykh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2008. V. 53. № 8. P. 1176. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган. химии. 2008. Т. 53. № 8. С. 1267.].
Krasnobaeva O.N., Belomestnykh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2009. V. 54. № 4. P. 495. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган. химии. 2009. Т. 54. № 4. С. 547.]
Krasnobaeva O.N., Belomestnyrh I.P., Isagulyants G.V. et al. // Russ. J. Inorg. Chem. 2009. V. 54. № 12. P. 1862. [Краснобаева О.Н., Беломестных И.П., Исагулянц Г.В. и др. // Журн. неорган химии. 2009. Т. 54. № 12. С. 1944.]
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 2. P. 168. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2011. Т. 56. № 2. С. 204.]
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1012. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1073.]
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2012. V. 57. № 12. P. 1540. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2012. Т. 57. № 11. С. 1639.]https://doi.org/10.1134/S003602361212011X
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2013. V. 58. № 6. P. 644. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2013. Т. 58. № 6. С. 733.]https://doi.org/10.1134/S0036023613060168
Krasnobaeva O.N., Belomestnykh I.P., Kogan V.M. et al. // Russ. J. Inorg. Chem. 2014. V. 59 № 7. P. 692. [Краснобаева О.Н., Беломестных И.П., Коган В.М. и др. // Журн. неорган. химии. 2014. Т. 59. № 7. С. 904.]https://doi.org/10.1134/S0036023614070109
Krasnobaeva O.N., Belovestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 4. P. 467. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2015. Т. 60. № 4. С. 467.]https://doi.org/10.1134/S0036023615040099
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 9. P. 942. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2016. Т. 61. № 9. С. 1129. https://doi.org/1134/S0036023616090126]
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 7. P. 879. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2017. Т. 62. № 7. С. 897.]https://doi.org/10.1134/S0036023617070129
Аналитическая химия ванадия. Сер. Аналитическая химия элементов / Под ред. Золотова Ю.А. М.: Наука, 1981. С. 216.
Tachyun Kwon, George A., Tsigdinnos G.A. et al. // J. Am. Chem. Soc. 1988. V. 110. P. 3653.
Аналитическая химия ниобия и тантала. Сер. Аналитическая химия элементов / Под ред. Виноградова А.П. М.: Наука, 1976. С. 352.
Stalin W., Oswald H.D. // Acta Crystallogr. 1970. V. 26B. P. 860.
Krasnobaeva O.N., Nosova T.A., Kondakov D.F. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 9. P. 1122. [Краснобаева О.Н., Носова Т.А., Кондаков Д.Ф. и др. // Журн. неорган. химии. 2018. Т. 63. № 9. С. 1092.]https://doi.org/10.1134/S0036023618090085
Krasnobaeva O.N., Belomestnykh I.P., Nosova T.A. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 11. P. 1419. [Краснобаева О.Н., Беломестных И.П., Носова Т.А. и др. // Журн. неорган. химии. 2018. Т. 63. № 11. С. 1394.]https://doi.org/10.1134/S0036023618110098
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии