

Журнал неорганической химии, 2019, T. 64, № 9, стр. 923-932
Включение уранила в полость полиоксометаллата. Синтез и характеризация [(UO2)8P8W48O184]24–
В. С. Коренев 1, 2, *, П. А. Абрамов 1, 2, А. Л. Гущин 1, 2, Д. В. Стась 2, 3, В. М. Бабаев 4, И. Х. Ризванов 4, М. Н. Соколов 1, 2, 5
1 Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, пр-т Академика Лаврентьева, 3, Россия
2 Новосибирский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
3 Институт химической кинетики и горения СО РАН
630090 Новосибирск, ул. Институтская, 3, Россия
4 Институт органической и физической химии им. А.Е. Арбузова, Федеральный исследовательский центр
“Казанский научный центр Российской академии наук”,
420088 Казань, ул. Академика Арбузова, 8, Россия
5 Казанский федеральный университет, Химический институт им. А.М. Бутлерова
420008 Казань, ул. Кремлевская, 29/1, Россия
* E-mail: wkorenev@niic.nsc.ru
Поступила в редакцию 28.12.2018
После доработки 17.02.2019
Принята к публикации 15.03.2019
Аннотация
Реакция нитрата уранила с полилакунарным полианионом [P8W48O184]40– приводит к образованию [(UO2)7P8W48O184]26– (1) и [(UO2)8P8W48O184]24– (2), которые были выделены в виде калиевых и аммонийных солей. Для K24[(UO2)8P8W48O184] · 50H2O · 3LiCl и (NH4)16H8[(UO2)8P8W48O184] · 50H2O выполнен рентгеноструктурный анализ. Включение уранила в [P8W48O184]40– подтверждено методами ИК- и КР-спектроскопии, а также методом масс-спектрометрии. Электрохимическое исследование соединения 2 указывает на возможность восстановления урана до U(IV). Изучена рентгенолюминесценция для калиевых солей 1 и 2.
ВВЕДЕНИЕ
Среди всех актинидов наиболее изучена координационная химия урана, которая, в свою очередь, в значительной степени является химией производных уранила (UO2)2+ из-за инертности его по существу тройных связей U–O [1, 2]. Структурное разнообразие и многообещающие фотофизические и электрохимические свойства уранила могут быть использованы для получения новых функциональных материалов. Недавно было описано получение и строение глутаросодержащих координационных полимеров уранила с органическими амидами [3]. На основе уранила можно также получать соединения с необычной топологией, как, например, высокопористые нанотрубки селената уранила, построенные из пентагональных бипирамид {UO7} и тетраэдров ${\text{SeO}}_{{\text{4}}}^{{{\text{2}}--}},$ соединенных общими вершинами [4, 5]. Особый интерес представляют оксосоединения урана из-за их значимости для переработки радиоактивных отходов и безопасной утилизации отработавшего ядерного топлива [6].
Принимая во внимание оксофильную природу урана, можно ожидать образования стабильных дискретных полиядерных оксокомплексов урана либо путем самоконденсации кислородсодержащих полиэдров, либо при включении уранила в другие полиоксометаллаты. В 2005 г. авторы [7] описали самосборку наноразмерных пероксополиуранатов в щелочной среде при нормальных условиях. Оксолиганды катионов уранила располагаются в транс-положении, а пероксолиганды связывают их в полые капсулы. Дальнейшие исследования расширили семейство пероксоуранатов, было получено более 30 комплексов с различными мостиковыми лигандами между уранильными фрагментами (пероксидные, гидроксильные, пирофосфатные, оксалатные) [8–16]. Капсулоподобная топология данных соединений приводит к интересному динамическому процессу, поскольку включенные фрагменты (вода, катионы и анионы) не только влияют на формирование капсулы [17–20], но и могут обмениваться с внешней средой [21]. Это может иметь значение при устранении последствий ядерных инцидентов на АЭС [22].
Первый пример поливольфрамата, содержащего пероксид уранила, [LiK4(H2O)4{(UO2)4(μ-O2)4(H2O)2}2(PO3OH)2P6W36O136]25−, был описан в 2008 г. [23]. Данное соединение построено из двух типов строительных блоков – двух пероксоуранильных фрагментов {(UO2)4(μ-O2)4} и трех лакунарных фосфовольфраматов {P2W12}10−, которые образуют новый анион со структурой Уэллса–Доусона {P6W36O136}26–. Эта работа показывает, что новые лакунарные полиоксовольфраматы могут быть стабилизированы необычной координацией с новыми металлокомплексными темплатами. В 2012 г. был описан циклический полиоксометаллат [(W5O21)3{(UO2)2(μ-O2)}3]30−, состоящий из димерных пероксоуранилатных {(UO2)2(μ-O2)}2+ и пентавольфраматных фрагментов [W5O21]12− [24]. В недавней работе описаны гибридные уранил-ванадатные полиоксометаллаты с металлоостовами {U20V20} и {U2V16} [25].
Известны уранилсодержащие полиоксометаллаты без пероксидных лигандов. В 1999 г. авторы [26] описали первый пример уранилсодержащего полиоксоаниона [Na2(UO2)2(PW9O34)2]12– сэндвичевого типа. Впоследствии были получены и другие уранилсодержащие гетерополивольфраматы с лакунарными структурами Кеггина и Уэллса–Доусона: [(UO2)3(H2O)5As3W29O104]19–, [(UO2)3(H2O)6As3W30O105]15– [27], [Na2(UO2)2(SiW9O34)2]14‒, [{M(OH2)}4(UO2)4(OH)2(SiW10O36)4]22− (M = Na+, K+), [(UO2)3(H2O)4As3W26O94]17–, [Na(UO2)3(OH)(H2O)6As4W40O140(WO)]18− [28], [(UO2)2(H2O)2(SbW9O33)2]14–, [(UO2)2(H2O)2(TeW9O33)2]12– [29], [(UO2)12(μ3‑O)4(μ2-H2O)12(P2W15O56)4]32– [30], [Na2(UO2)2 As2W18O68]12–, [M(UO2)2As2W18O68]13– (M = ${\text{NH}}_{{\text{4}}}^{ + },$ K+) [31], A-α,β-[Na2(UO2)2(GeW9O34)2]14–, [32] [(UO2)3As2W18O68]12– [33] и [Na(UO2)2(H2O)4(BiW9O33)2]13‒ [34]. Также известны два уранилсодержащих полиоксометаллата с дополнительным включением одного атома ванадия – [(UO2)3(H2O)6As3W29VO105]17– [27] и [(VO)(UO2)2 As2W18O68]12– [33].
Еще одна малоизученная возможность – включение урана в полость циклических полиоксовольфраматов. Пример такого взаимодействия был описан для аниона [P5W30O110]15–, в который методом гидротермального катионного обмена был введен U4+ [35, 36]. Другим кандидатом на включение урана является циклический “суперлакунарный” полиоксоанион [P8W48O184]40–, выделенный в 1985 г. в виде смешанной литий-калиевой соли в [37]. Он способен включать в свою большую анионную полость различные катионы металлов. Координационная химия типа “хозяин–гость” для [P8W48O184]40– представлена большой группой комплексов с переходными металлами [38–44] и лантаноидами [45].
В настоящей работе мы сообщаем о получении и характеризации двух новых уранилсодержащих полианионов – [(UO2)7P8W48O184]26– (1) и [(UO2)8P8W48O184]24– (2), а также о структурах калиевой K24[(UO2)8P8W48O184] · 50H2O · 3LiCl и аммонийной (NH4)16H8[(UO2)8P8W48O184] · 50H2O солей аниона 2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Циклические полиоксовольфраматы K28Li5H7[P8W48O184] · 92H2O (K28-P8W48) и Li17(NH4)21H2[P8W48O184] · 85H2O ((NH4)21-P8W48) синтезировали по известным методикам [37, 46] и идентифицировали методом ИК- и ЯМР 31P спектроскопии. Уранил азотнокислый гексагидрат (ч.), литий хлористый моногидрат (ос. ч.), калий хлористый (х. ч.), аммоний хлористый (х. ч.) и соляную кислоту (ос. ч.) получали из коммерческих источников и использовали без дополнительной очистки.
ИК-спектры (4000–400 см–1) записывали на спектрофотометре Scimitar FTS 2000 (Digilab LLC, USA) с образцов, запрессованных в таблетки KBr. Спектры КР записывали на спектрометре LabRAM HR Evolution (Horiba, Japan) с линией 632.8 нм He–Ne-лазера. Спектры ЯМР 31P исходных поливольфраматов записывали при комнатной температуре в насыщенных водных растворах в 5 мм ампулах на спектрометре Bruker Avance 500. Химические сдвиги отнесены по 85%-ной H3PO4. Элементный анализ на азот и водород выполняли на C,H,N,S-анализаторе Vario MICRO cube. Содержание лития и калия определяли методом атомно-абсорбционной спектроскопии (ААС) на спектрофотометре Z8000 (Hitachi, Japan). Термогравиметрический анализ (ТГА) выполняли на NETZSCH thermobalance TG 209 F1 Iris в инертной атмосфере (He).
Циклические вольтамперограммы (ЦВА) записывали на электрохимическом анализаторе 797 VA Computrace (Metrohm, Switzerland). Все измерения выполняли в стандартной трехэлектродной ячейке, состоящей из стеклоуглеродного рабочего и платинового вспомогательного электродов и хлорсеребряного электрода сравнения Ag/AgCl/KCl. В качестве растворителя использовали дистиллированную дегазированную воду, в качестве электролита – хлорид лития (1 М раствор) или сульфат лития (0.5 М раствор). Концентрация комплексов составляла ∼10–3 моль/л. Значения окислительно-восстановительных потенциалов (E1/2) определяли как (Ea + Ec)/2, где Ea и Ec – анодный и катодный потенциалы соответственно.
Масс-спектрометрию с ионизацией электрораспылением (ИЭР-МС) проводили на масс-спектрометре AmazonX (Bruker Daltonics, Germany) типа “ионная ловушка” в отрицательном режиме в диапазоне m/z от 500 до 6000. Использовали следующие условия: напряжение на капилляре 2.5 кВ, газ-осушитель азот, 10 л/мин, 250°C. Данные обрабатывали с помощью программного обеспечения DataAnalysis (Bruker Daltonik GmbH, Version 4.0 SP4).
Спектры рентгеновской индуцированной твердотельной люминесценции регистрировали из чистых порошков исследуемых соединений в условиях окружающей среды на специально сконструированном спектрометре [47], следуя протоколу для порошковых образцов, описанному ранее [48]. Образцы готовили в виде открытых островков размером 3 × 8 мм и толщиной ∼0.1 мм, содержащих несколько миллиграмм исследуемого сухого вещества, нанесенного через трафарет на алюминиевую пластину, покрытую двухсторонней скотч-пленкой на основе полипропилена. Открытый образец устанавливали вертикально под углом 45° к направлениям возбуждающего рентгеновского излучения и оси светособирающей части системы детектирования, которые были горизонтальны и нормальны друг к другу. Рентгеновские лучи генерировались рентгеновской трубкой БСВ-27-Mo (“Светлана”, Санкт-Петербург, Россия, 40 кВ × 20 мА). Для возбуждения всю полоску образца подвергали воздействию нефильтрованного тормозного излучения. Канал детектирования состоял из кварцевой оптической системы формирования изображений, монохроматора с решеткой (MDR-206, LOMO Photonics, Санкт-Петербург, Россия, объективная фокусная длина 180 мм, решетка 1200 линий/мм, обратная линейная дисперсия 4.3 нм × мм–1) с входной/выходной щелями, установленными на 0.5/0.5 мм (спектральное разрешение ∼2 нм) и модуля фотодатчика Hamamatsu H10493-012, выходной сигнал которого был оцифрован и передан на управляющий компьютер. Для получения представленных спектров рентгенолюминесценции исходные спектры были скорректированы по базовой линии детектора, нормированы на максимум, а затем дополнительно скорректированы на спектральную чувствительность установки детектирования. Каждый представленный спектр содержал 256 точек длины волны и представлял собой результат усреднения четырех независимых сканов по 9 мин каждый, что было более чем достаточно для этих легко испускающих образцов. Показанный диапазон длин волн (450–650 нм) полностью охватывал область излучения образцов, за пределами этого диапазона от 250 до 1000 нм никакого излучения не наблюдалось. Все спектры записывали при комнатной температуре.
Кристаллографические данные и результаты уточнения структуры для K24[(UO2)8P8W48O184] · ⋅ 50H2O · 3LiCl и (NH4)16H8[(UO2)8P8W48O184] · ⋅ 50H2O приведены в табл. 1. Все измерения проводили на дифрактометре New Xcalibur (Agilent Technologies) (MoKα-излучение, λ = 0.71073) методом φ-сканирования узких (0.5°) фреймов при 130 K. Поглощение учитывали эмпирически по программе SCALE3 ABSPACK (CrysAlisPro, Agilent Technologies, Version 1.171.37.35 (release 13-08-2014 CrysAlis171 NET)). Структуры расшифровывали прямым методом и уточняли полноматричным МНК относительно |F|2 в анизотропном приближении для неводородных атомов с использованием алгоритма SHELX 2014/7 [49] в программе ShelXle [50]. Кристаллы обоих соединений проявляли слабую дифракцию, что препятствовало использованию очень маленьких кристаллов для сбора данных. Оба эксперимента проводили на крупных кристаллах с сильным поглощением рентгеновского излучения, что привело к появлению некоторой искусственной электронной плотности вокруг тяжелых атомов. Координационное окружение U (OH– или H2O) не было однозначно идентифицировано из-за разупорядочения групп {UO2}2+ с низкой степенью занятости позиций. Атомы водорода молекул кристаллизационной воды не уточняли. Обе структуры имеют пустоты, поэтому точный катионный состав и количество гидратных молекул воды определяли из данных элементного анализа и ТГА. CIF-файл, содержащий полную информацию о структурах, депонировали в базу ISDB (Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany, e-mail: crysdata@ fiz-karlsruhe.de) под номером CSD 431742.
Таблица 1.
Кристаллографические характеристики и детали дифракционного эксперимента
| Параметр | K24 · 2 · 50H2O · 3LiCl | (NH4)16H8 · 2 · 50H2O |
|---|---|---|
| Формула | H0K16.80O246P8U8.32W48 | O249.70P8U8W48 |
| Mr | 15 677.13 | 14 972.00 |
| Сингония, пр. гр. | Тетрагональная, I4/mmm | Тетрагональная, I4/m |
| a, c, Å | 26.8224(3), 21.1281(4) | 25.6334(4), 22.1597(4) |
| V, Å3 | 15200.4(4) | 14560.5(5) |
| µ, мм–1 | 22.87 | 23.44 |
| Размер кристалла, мм | 0.30 × 0.30 × 0.30 | 0.20 × 0.20 × 0.20 |
| Tmin, Tmax | 0.407, 1.000 | 0.619, 1.000 |
| Число измеренных, независимых и наблюдаемых [I > 2σ(I)] рефлексов | 30 107, 5341, 4661 | 21 294, 8848, 5641 |
| Rint | 0.026 | 0.038 |
| Область θ, град | θmax = 29.6, θmin = 3.4 | θmax = 29.6, θmin = 3.3 |
| (sin θ/λ)max, Å–1 | 0.695 | 0.695 |
| Интервалы индексов h, k, l | –35 ≤ h ≤ 36, –36 ≤ k ≤ 22, –23 ≤ l ≤ 29 | –33 ≤ h ≤ 25, –18 ≤ k ≤ 32, –15 ≤ l ≤ 29 |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.064, 0.140, 1.16 | 0.065, 0.177, 1.02 |
| Число независимых рефлексов, уточняемых параметров | 5341, 180 | 8848, 271 |
| Весовая схема | w = 1/[σ2$(F_{{\text{o}}}^{{\text{2}}})$ + (0.0193P)2 + + 2308.0291P] where P = ($F_{{\text{o}}}^{{\text{2}}}$ + ${\text{2}}F_{{\text{c}}}^{{\text{2}}}$)/3 | w = 1/[σ2$(F_{{\text{o}}}^{{\text{2}}})$ + (0.0685P)2 + + 965.5565P] where P = ($F_{{\text{o}}}^{{\text{2}}}$ + ${\text{2}}F_{{\text{c}}}^{{\text{2}}}$)/3 |
| Δρmax, Δρmin, e/Å3 | 4.15, –4.94 | 3.17, –5.59 |
Реакция K28-P8W48 с 6 экв {UO2}. Навеску K28-P8W48 (0.500 г, 0.034 ммоль) растворяли в 10 мл 1 M раствора LiCl (pH раствора 6.2), затем добавляли навески UO2(NO3)2 · 6H2O (0.100 г, 0.199 ммоль) и хлорида калия (0.125 г, 1.678 ммоль); раствор становился мутно-желтым. Значение pH реакционной смеси устанавливали равным 1.5 добавлением нескольких капель концентрированной соляной кислоты. Полученный мутный раствор нагревали до кипения (при этом осадок растворялся), затем кипятили в течение 24 ч с обратным холодильником. Далее желтый раствор медленно охлаждали до комнатной температуры, в процессе охлаждения образовывался желтый кристаллический продукт. Выход 0.368 г (66% в расчете на исходный K28-P8W48). Для K26[(UO2)7P8W48O184] · ⋅ 50H2O вычислено, %: K 6.40. Найдено, %: K 6.35 (по ААС). ТГА: потеря ~50 молекул воды при нагревании от комнатной температуры до 250°C соответствует удалению кристаллизационных молекул воды.
Реакция K28-P8W48 с 18 экв {UO2}. Процесс проводили аналогично предыдущему, отличие заключается в утроенном количестве нитрата уранила (0.300 г, 0.598 ммоль). Выход 0.452 г (80% в расчете на исходный K28-P8W48). Для K24[(UO2)8P8W48O184] · · 50H2O · 3LiCl вычислено, %: Li 0.13, K 5.79. Найдено, %: Li 0.13, K 5.8 (по ААС). ТГА: потеря ~50 молекул воды при нагревании от комнатной температуры до 250°C соответствует удалению кристаллизационных молекул воды.
Реакция (NH4)21-P8W48 с 6 экв {UO2}. Навеску (NH4)21-P8W48 (0.500 г, 0.036 ммоль) растворяли в 25 мл 1 M раствора LiCl (pH раствора 6.6), затем добавляли навески UO2(NO3)2 · 6H2O (0.108 г, 0.216 ммоль) и хлорида аммония (0.350 г, 6.542 ммоль). Раствор становился мутно-желтым. Величину pH реакционной смеси установили равной 1.5 добавлением нескольких капель концентрированной соляной кислоты. Полученный мутный раствор нагревали до кипения (при этом осадок растворялся), а затем кипятили в течение 24 ч с обратным холодильником. Далее желтый раствор охлаждали до комнатной температуры и оставляли медленно упариваться в открытой колбе Эрленмейера. Желтый кристаллический продукт был получен через 3 сут. Выход 0.182 г. Для (NH4)18H8[(UO2)7P8W48O184] · 50H2O вычислено, %: H 1.20, N 1.66. Найдено, %: H 1.15, N 1.60 (по H,N-анализу). ТГА: потеря ~50 молекул воды при нагревании от комнатной температуры до 250°C соответствует удалению кристаллизационных молекул воды.
Реакция (NH4)21-P8W48 с 18 экв {UO2}. Процесс проводили аналогично предыдущему, отличие заключается в утроенном количестве нитрата уранила (0.325 г, 0.647 ммоль). Выход 0.252 г. Для (NH4)18H8[(UO2)7P8W48O184] · 50H2O + + (NH4)16H8[(UO2)8P8W48O184] · 50H2O вычислено, %: H 1.16, N 1.54. Найдено, %: H 1.15, N 1.55 (по H,N-анализу). ТГА: потеря ~50 молекул воды при нагревании от комнатной температуры до 250°C соответствует удалению кристаллизационных молекул воды.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Цель настоящей работы заключалась в том, чтобы включить катионы уранила в полость циклического “суперлакунарого” фосфовольфрамата [P8W48O184]40–. В качестве источника [P8W48O184]40– использовали литий-калиевую соль K28Li5H7[P8W48O184] · 92H2O (K28-P8W48), полученную в [37], и литий-аммонийную соль Li17(NH4)21H2[P8W48O184] · 85H2O ((NH4)21-P8W48), описанную в [46]. Основное отличие между этими двумя соединениями связано с занятостью внутренней полости полианиона и ее доступностью для конкурентного включения других катионов. В структуре K28-P8W48 во внутренней полости находятся восемь катионов калия, каждый из которых занимает одну из двух возможных позиций – имеется восемь аксиальных позиций с половинной занятостью и четыре экваториальные, полностью занятые. Эти катионы калия играют важную роль в стабилизации полилакунарного [P8W48O184]40–. Они образуют прочные связи с основными участками внутренней полости, и их довольно трудно удалить оттуда [51]. При этом [P8W48O184]40– отдает предпочтение вхождению в полость катионов калия, а не аммония [46]. Поэтому для повышения потенциальной доступности внутренней полости для катионов уранила мы использовали (NH4)21-P8W48. Исходя из полилакунарной природы [P8W48O184]40–, позволяющей включать большое количество катионных фрагментов, мы выбрали два соотношения реагентов {UO2}/{P8W48} для экспериментов – 6 и 18. Из литературы известно, что для включения катионных “гостей”, за исключением синтеза [Mn8(H2O)26(P8W48O184)]24– [44], необходимо нагревание от 30 до 80°C для различных переходных металлов [38–44] или даже гидротермальные условия (140°C) в случае лантанидов [45]. Предварительные эксперименты показали, что нагревания до 100°C достаточно для растворения всех реагентов в наших системах, и мы использовали обычное продолжительное кипячение в колбе Эрленмейера с обратным холодильником.
При охлаждении реакционной смеси из реакции K28-P8W48 с нитратом уранила (как в 6-кратном, так и в 18-кратном соотношении) до комнатной температуры кристаллизация начинается немедленно и желтые кристаллические продукты, обозначенные A и B, быстро отделяются от ярко-желтого маточного раствора. Реакции с (NH4)21-P8W48 проводили при тех же условиях. Ярко-желтые растворы оставляли стоять в открытой колбе Эрленмейера, а желтые кристаллические продукты, обозначенные C и D, формировались постепенно, в течение 2–3 сут для получения приемлемых выходов.
Все продукты были охарактеризованы методами ИК- и КР-спектроскопии. Согласно литературным данным, катион уранила имеет три основные частоты колебаний: валентное симметричное (856–865 см–1), валентное асимметричное (931–943 см–1) и деформационное (~210 см–1) [52, 53]. Однако, как было показано ранее, при его координации с полиоксометаллатами наблюдается значительное ослабление связей в уранильном фрагменте, что приводит к изменению частоты колебаний [29, 30]. В ИК-спектрах наблюдаются практически все сигналы исходного {P8W48} без изменения, что подтверждает стабильность полианиона в процессе реакции. Дополнительные же полосы около 860 см–1 в обоих случаях можно объяснить асимметричными колебаниями U–O, смещенными из-за ослабления связи (рис. S1 ).
В спектрах КР также хорошо видны сигналы исходных полиоксометаллатов. Наблюдается дополнительный широкий пик при 786 см–1 для калиевых солей и при 790 см–1 для аммонийных, связанный со смещенными симметричными колебаниями катиона уранила (рис. S2 ). Аналогичное смещение было описано ранее в работах [29, 30].
Масс-спектрометрия ИЭР. Калиевые соли {P8W48} обычно имеют низкую растворимость в воде. Кристаллические продукты A и B были предварительно растерты в порошок и помещены в воду, где их перемешивали при 60°C до полного растворения. В масс-спектре продукта A наблюдается два основных сигнала, отвечающих зарядовым состояниям 9– и 8–, а также два малоинтенсивных пика, соответствующих зарядам 10– и 7– (рис. S3a ). Значения m/z для этих сигналов хорошо согласуются с формулой {nK+ + + [(UO2)7P8W48O184]26–}(26–n)–. В масс-спектре продукта B наблюдается три сигнала с зарядовыми состояниями от 9– до 7– (рис. 1). Значения m/z хорошо согласуются с формулой {nK+ + + [(UO2)8P8W48O184]24–}(24–n)–.
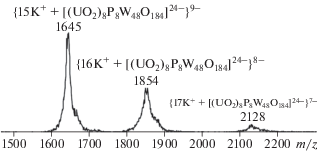
Растворимость аммонийных солей C и D выше калиевых. Масс-спектры обоих продуктов в области m/z 1000–3000 содержат по четыре группы сигналов, отвечающих зарядовым состояниям от 9– до 6–. В масс-спектре C основной сигнал каждой группы был отнесен к аниону [(UO2)7P8W48O184]26– в сочетании с различным количеством катионов. Малоинтенсивные сигналы относятся к [(UO2)6P8W48O184]28– и [(UO2)8P8W48O184]24– (рис. S3b ). В масс-спектре D каждая группа состоит из сигналов двух анионов – [(UO2)8P8W48O184]24– и [(UO2)7P8W48O184]26– в сочетании с различным количеством катионов (рис. S3c ). В табл. 2 приведены наиболее вероятные составы ионов для всех наблюдаемых сигналов в масс-спектрах соединений A, C и D.
Таблица 2.
Соотнесение значений m/z наблюдаемых сигналов в масс-спектрах соединений A, C и D составам ионов
| Заряд иона | m/z | Состав иона |
|---|---|---|
| A | ||
| 10– | 1445.7 | {16K+ + [(UO2)7P8W48O184]26–}10– |
| 9– | 1624.3 | {17K+ + [(UO2)7P8W48O184]26–}9– |
| 8– | 1826.5 | {18K+ + [(UO2)7P8W48O184]26–}8– |
| 7– | 2096.9 | {19K+ + [(UO2)7P8W48O184]26–}7– |
| C | ||
| 9– | 1566.0 | {11${\text{NH}}_{{\text{4}}}^{ + }$ + 6H+ + [(UO2)7P8W48O184]26–}9– |
| 8– | 1765.7 | {12${\text{NH}}_{{\text{4}}}^{ + }$ + 6H++ [(UO2)7P8W48O184]26–}8– |
| 7– | 2020.7 | {13${\text{NH}}_{{\text{4}}}^{ + }$ + 6H++ [(UO2)7P8W48O184]26–}7– |
| 6– | 2359.9 | {14${\text{NH}}_{{\text{4}}}^{ + }$ + 6H++ [(UO2)7P8W48O184]26–}6– |
| D | ||
| 9– | 1567.8 | {12${\text{NH}}_{{\text{4}}}^{ + }$ + 5H+ + [(UO2)7P8W48O184]26–}9– |
| 1601.1 | {12${\text{NH}}_{{\text{4}}}^{ + }$ + 3H+ + [(UO2)8P8W48O184]24–}9– | |
| 8– | 1767.9 | {13${\text{NH}}_{{\text{4}}}^{ + }$ + 5H+ + [(UO2)7P8W48O184]26–}8– |
| 1801.4 | {13${\text{NH}}_{{\text{4}}}^{ + }$ + 3H+ + [(UO2)8P8W48O184]24–}8– | |
| 7– | 2023.3 | {14${\text{NH}}_{{\text{4}}}^{ + }$ + 5H+ + [(UO2)7P8W48O184]26–}7– |
| 2062.8 | {14${\text{NH}}_{{\text{4}}}^{ + }$ + 3H+ + [(UO2)8P8W48O184]24–}7– | |
| 6– | 2365.3 | {15${\text{NH}}_{{\text{4}}}^{ + }$ + 5H+ + [(UO2)7P8W48O184]26–}6– |
| 2411.6 | {15${\text{NH}}_{{\text{4}}}^{ + }$ + 3H+ + [(UO2)8P8W48O184]24–}6– | |
Таким образом, данные масс-спектрометрии указывают на образование двух анионов – [(UO2)7P8W48O184]26– (1) и [(UO2)8P8W48O184]24– (2). Согласно данным атомно-абсорбционной спектроскопии, элементного и термогравиметрического анализа, полученные продукты были идентифицированы следующим образом:
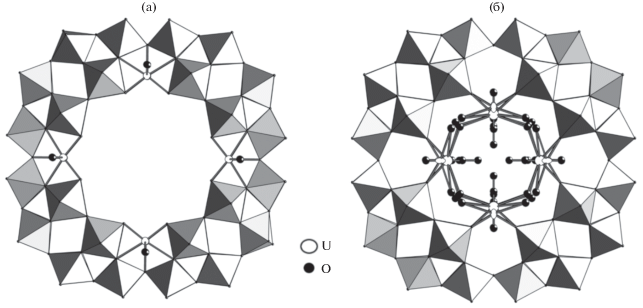
Рентгеноструктурное исследование K24[(UO2)8P8W48O184] · 50H2O · 3LiCl и (NH4)16H8[(UO2)8P8W48O184] · 50H2O. Кристаллическая структура K24[(UO2)8P8W48O184] · 50H2O · · 3LiCl построена из анионов [(UO2)8P8W48O184]24–, которые образуют двухслойную кристаллическую упаковку (ОЦК-топология анионной подрешетки) с ориентацией слоев вдоль кристаллографического направления [110 ] (рис. S4 ). Слои связываются через сольватированные катионы калия, тогда как кристаллическая структура имеет бесконечные каналы, расположенные в кристаллографическом направлении [010] и заполненные молекулами кристаллизационной воды и, возможно, сильно разупорядоченными катионами. Восемь уранильных групп разупорядочены по 32 позициям над двумя ободами аниона {P8W48} (четыре позиции в независимой части). Есть два типа координированных групп {UO2}2+. Первый тип занимает внутренние пентагональные позиции (рис. 2a), находящиеся между двумя строительными блоками {P2W12}, второй – внешнее пространство также между двумя блоками {P2W12} (рис. 2б). Уточнение указывает на 30%-ную занятость для первого типа и 70%-ную занятость для второго. Причиной такого распределения, возможно, является конкуренция между катионами {UO2}2+ и K+ (рис. 3).
Рис. 2.
Позиции разупорядоченных {UO2}2+-групп (а – внутренние, б – внешние) в калиевой соли аниона 2.
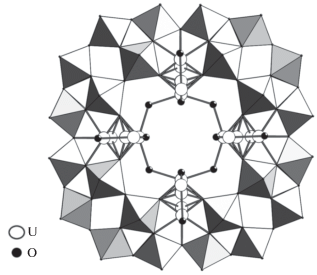
В случае аммонийной соли (NH4)16H8[(UO2)8P8W48O184] · 50H2O группы {UO2}2+ также занимают позиции обоих сортов. Часть ионов уранила находится в пентагональных позициях между строительными блоками {P2W12}, второй тип также связывает два фрагмента {P2W12}, но располагается в центральной полости аниона {P8W48} (рис. 4). Занятость U в позициях первого типа не превышает 10%, тогда как сумма занятостей позиций второго типа ∼90%. Фактически только четыре {UO2}2+-группы располагаются на каждом ободе {P8W48}, занимая различные позиции, и образуют большое число позиционных изомеров, что хорошо согласуется с большим количеством сигналов в спектре ЯМР 31P. По сравнению с K24[(UO2)8P8W48O184] · 50H2O · 3LiCl занятость пентагональных позиций значительно ниже. Это означает, что катионный эффект можно использовать для изменения способа координации {UO2}2+-групп. В кристаллической структуре анион [(UO2)8P8W48O184]24– формирует двухслойную кристаллическую упаковку (ОЦК-топология анионной подрешетки) с ориентацией слоев вдоль кристаллографического направления [110 ] (рис. S5 ). Анионы не связаны между собой и участвуют только в образовании водородных связей с ${\text{NH}}_{{\text{4}}}^{ + }$ и молекулами кристаллизационной воды. Следовательно, кристаллическая ОЦК-упаковка анионов [(UO2)8P8W48O184]24– подавляет любые изменения катионного состава.
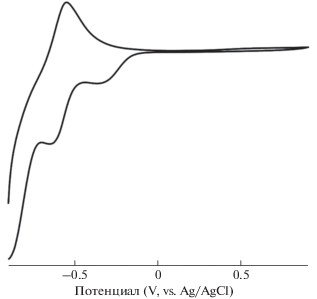
Электрохимическое исследование K24[(UO2)8P8W48O184] · 50H2O · 3LiCl. На циклической вольтамперограмме K24[(UO2)8P8W48O184] · ⋅ 50H2O · 3LiCl в H2O с 1 M LiCl в качестве электролита при pH 2 наблюдается необратимое восстановление при Ec = –0.31 В (отн. Ag/AgCl) с последующим квазиобратимым восстановлением при E1/2 = –0.58 В (отн. Ag/AgCl) при скорости сканирования 10 мВ/с (рис. 5). Разность между потенциалами анодной (Ea) и катодной (Ec) волн (∆E) для второго окислительно-восстановительного процесса составляет 65 мВ, что близко к величине 59 мВ, характерной для обратимого одноэлектронного процесса. Эта величина значительно возрастает с увеличением скорости сканирования, что указывает на потерю электрохимической обратимости (уже при 50 мВ/с значение ∆E составляет 107 мВ). Происхождение этого окислительно-восстановительного процесса, скорее всего, связано с восстановлением вольфрама. Это предположение подтверждается протеканием аналогичного процесса восстановления при E1/2 = –0.48 В (∆E = 120 мВ) и 50 мВ/с для свободного {P8W48}, изученного в тех же условиях, а также литературными данными [54]. Необратимое восстановление при –0.31 В не наблюдается для свободного {P8W48}. Таким образом, можно предположить, что оно относится к восстановлению урана, по-видимому, до U(IV).
Рис. 5.
ЦВА K24 · 2 · 50H2O · 3LiCl в H2O с 1 M LiCl в качестве электролита при pH 2 и скорости сканирования 10 мВ/с.
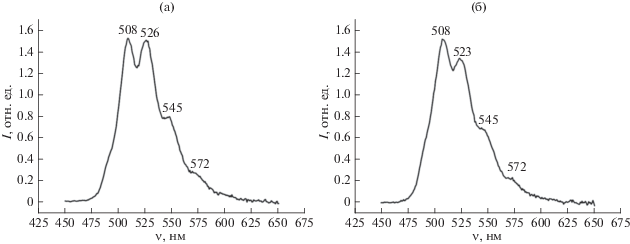
Рентгенолюминесценция UO2(NO3)2, K26[(UO2)7P8W48O184] · 50H2O и K24[(UO2)8P8W48O184] · 50H2O · 3LiCl. На рис. S6 показан спектр рентгенолюминесценции для UO2(NO3)2. Спектр практически идентичен хорошо известным спектрам фотоиндуцированной фосфоресценции нитрата уранила [55]. Он имеет характерную форму с группой из пяти эквидистантных (в энергетическом масштабе) максимумов в зеленоватой области из-за перехода от нижнего возбужденного триплетного состояния к синглетному основному состоянию линейной уранильной части с колебательной субструктурой, связанной с симметричным O=U=O колебанием основного состояния. Еще две более слабые линии хорошо видны на длинноволновом и коротковолновом краях спектра (619 и 473 нм). Пять основных линий, отмеченных на рисунке, и ширина линий ∼9 нм довольно типичны для фотоиндуцированных спектров излучения твердого нитрата уранила. Это свидетельствует о том, что высокоэнергетическое рентгеновское возбуждение создает те же локальные электронные возбужденные состояния, которые ответственны за излучение при фотовозбуждении. На рис. 6 показаны спектры рентгенолюминесценции для K26 · 1 · ⋅ 50H2O (a) и K24 · 2 · 50H2O · 3LiCl (б), записанные в одинаковых экспериментальных условиях. Эти два спектра очень похожи и аналогичны спектру нитрата уранила. Однако хорошо видно, что колебательная структура проявляется менее явно, спектры стали более компактными по длине волны, и большинство характерных максимумов наблюдается при длинах волн, попадающих между максимумами на рис. 6.
ЗАКЛЮЧЕНИЕ
Лакунарный фосфовольфрамат со структурой неорганического кавитанда [P8W48O184]24– способен координировать до 8 ионов уранила путем включения их во внутреннюю полость. При этом они случайным образом занимают доступные для координации позиции двух типов; в присутствии ионов калия наблюдается конкуренция между калием и уранилом за координационные места. Продукты являются превосходными рентгеновскими фотоэмиттерами с примерно равной интенсивностью излучения на единицу U, что также указывает на то, что наблюдаемая эмиссия из-за локализованного электронного возбуждения на отдельных ионах уранила одинаково проявляется как в низкомолекулярном нитрате уранила, так и в полиядерных полиоксосоединениях.
Список литературы
Katz J.J., Morss L.R., Seaborg G.T. The Chemistry of the Actinide Elements. London: Chapman and Hall, 1986. P. 1137. https://doi.org/10.1007/978-94-009-3155-8
Bagnall K.W. Comprehensive Coordination Chemistry. Oxford: Pergamon, 1987. V. 3. P. 1187.
Novikov S.A., Serezhkina L.B., Grigor’ev M.S. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 1. P. 47. [Новиков С.А., Сережкина Л.Б., Григорьев М.С. и др. // Журн. неорган. химии. 2017. Т. 62. № 1. С. 53. https://doi.org/10.7868/S0044457X17010123]https://doi.org/10.1134/S0036023617010120
Krivovichev S.V., Kahlenberg V., Kaindl R. et al. // Angew. Chem. Int. Ed. 2005. V. 44. № 7. P. 1134. https://doi.org/10.1002/anie.200462356
Krivovichev S.V., Kahlenberg V., Tananaev I.G. et al. // J. Am. Chem. Soc. 2005. V. 127. № 4. P. 1072. https://doi.org/10.1021/ja0436289
Burns P.C., Ewing R.C., Miller M.L. // J. Nucl. Mater. 1997. V. 245. № 1. P. 1. https://doi.org/10.1016/S0022-3115(97)00006-8
Burns P.C., Kubatko K.A., Sigmon G. et al. // Angew. Chem. Int. Ed. 2005. V. 44. № 14. P. 2135. https://doi.org/10.1002/anie.200462445
Sigmon G.E., Weaver B., Kubatko K.A. et al. // Inorg. Chem. 2009. V. 48. № 23. P. 10907. https://doi.org/10.1021/ic9020355
Unruh D.K., Burtner A., Pressprich L. // Dalton Trans. 2010. V. 39. P. 5807. https://doi.org/10.1039/C0DT00074D
Ling J., Wallace C.M., Szymanowski J.E.S. et al. // Angew. Chem. Int. Ed. 2010. V. 49. № 40. P. 7271. https://doi.org/10.1002/anie.201003197
Unruh D.K., Baranay M., Baranay M. et al. // Inorg. Chem. 2010. V. 49. № 15. P. 6793. https://doi.org/10.1021/ic100871z
Ling J., Qiu J., Sigmon G.E. et al. // J. Am. Chem. Soc. 2010. V. 132. № 38. P. 13395. https://doi.org/10.1021/ja1048219
Ling J., Qiu J., Szymanowski J.E.S. et al. // Chem. Eur. J. 2011. V. 17. № 9. P. 2571. https://doi.org/10.1002/chem.201003481
Unruh D.K., Ling J., Qiu J. et al. // Inorg. Chem. 2011. V. 50. № 12. P. 5509. https://doi.org/10.1021/ic200065y
Sigmon G.E., Burns P.C. // J. Am. Chem. Soc. 2011. V. 133. № 24. P. 9137. https://doi.org/10.1021/ja2013449
Ling J., Qiu J., Burns P.C. // Inorg. Chem. 2012. V. 51. № 4. P. 2403. https://doi.org/10.1021/ic202380g
Miro P., Pierrefixe S., Gicquel M. et al. // J. Am. Chem. Soc. 2010. V. 132. № 50. P. 17787. https://doi.org/10.1021/ja1053175
Nyman M., Rodriguez M.A., Alam T.M. // Eur. J. Inorg. Chem. 2011. V. 2011. № 14. P. 2197. https://doi.org/10.1002/ejic.201001355
Miro P., Bo C. // Inorg. Chem. 2012. V. 51. № 6. P. 3840. https://doi.org/10.1021/ic300029d
Gil A., Karhanek D., Miro P. et al. // Chem. Eur. J. 2012. V. 18. № 27. P. 8340. https://doi.org/10.1002/chem.201200801
Nyman M., Alam T.M. // J. Am. Chem. Soc. 2012. V. 134. № 49. P. 20131. https://doi.org/10.1021/ja308673f
Armstrong C.R., Nyman M., Shvareva T. et al. // Proc. Natl. Acad. Sci. U.S.A. 2012. V. 109. № 6. P. 1874. https://doi.org/10.1073/pnas.1119758109
Mal S.S., Dickman M.H., Kortz U. // Chem. Eur. J. 2008. V. 14. № 32. P. 9851. https://doi.org/10.1002/chem.200801583
Miró P., Ling J., Qiu J. et al. // Inorg. Chem. 2012. V. 51. № 16. P. 8784. https://doi.org/10.1021/ic3005536
Senchyk G.A., Wylie E.M., Prizio S. et al. // Chem. Commun. 2015. V. 51. P. 10134. https://doi.org/10.1039/C5CC01524C
Kim K.C., Pope M.T. // J. Am. Chem. Soc. 1999. V. 121. № 37. P. 8512. https://doi.org/10.1021/ja9909125
Kim K.C., Pope M.T. // J. Chem. Soc., Dalton Trans. 2001. P. 986. https://doi.org/10.1039/B008162K
Kim K.C., Gaunt A., Pope M.T. // J. Cluster Sci. 2002. V. 13. № 3. P. 423. https://doi.org/10.1023/A:1020507201056
Gaunt A.J., May I., Copping R. et al. // Dalton Trans. 2003. P. 3009. https://doi.org/10.1039/B302955G
Gaunt A.J., May I., Collison D. et al. // J. Mol. Struct. 2003. V. 656. № 1–3. P. 101. https://doi.org/10.1016/S0022-2860(03)00272-2
Khoshnavazi R., Eshtiagh-Hossieni H., Alizadeh M.H. et al. // Polyhedron. 2006. V. 25. № 9. P. 1921. https://doi.org/10.1016/j.poly.2005.12.019
Tan R., Wang X., Chai F. et al. // Inorg. Chem. Commun. 2006. V. 9. № 12. P. 1331. https://doi.org/10.1016/j.inoche.2006.07.044
Khoshnavazi R., Eshtiagh-Hossieni H., Alizadeh M.H. et al. // Inorg. Chim. Acta. 2007. V. 360. № 2. P. 686. https://doi.org/10.1016/j.ica.2006.07.095
Alizadeh M.H., Mohadeszadeh M. // J. Cluster Sci. 2008. V. 19. № 2. P. 435. https://doi.org/10.1007/s10876-008-0184-7
Creaser I., Heckel M.C., Neitz R.J. et al. // Inorg. Chem. 1993. V. 32. № 9. P. 1573. https://doi.org/10.1021/ic00061a010
Dickman M.H., Gama G.J., Kim K.C. et al. // J. Cluster Sci. 1996. V. 7. № 4. P. 567. https://doi.org/10.1007/BF01165802
Contant R., Tézé A. // Inorg. Chem. 1985. V. 24. № 26. P. 4610. https://doi.org/10.1021/ic00220a036
Mal S.S., Kortz U. // Angew. Chem. Int. Ed. 2005. V. 44. № 24. P. 3777. https://doi.org/10.1002/anie.200500682
Mal S.S., Nsouli N.H., Dickman M.H. et al. // Dalton Trans. 2007. P. 2627. https://doi.org/10.1039/B706762N
Müller A., Pope M.T., Todea A.M. et al. // Angew. Chem. Int. Ed. 2007. V. 46. № 24. P. 4477. https://doi.org/10.1002/anie.200700441
Mal S.S., Dickman M.H., Kortz U. et al. // Chem. Eur. J. 2008. V. 14. № 4. P. 1186. https://doi.org/10.1002/chem.200701424
Mitchell S.G., Gabb D., Ritchie C. et al. // Cryst. Eng. Comm. 2009. V. 11. P. 36. https://doi.org/10.1039/B813066C
Bassil B.S., Ibrahim M., Mal S.S. et al. // Inorg. Chem. 2010. V. 49. № 11. P. 4949. https://doi.org/10.1021/ic100050r
Chen S.-W., Boubekeur K., Gouzerh P. et al. // J. Mol. Struct. 2011. V. 994. № 1–3. P. 104. https://doi.org/10.1016/j.molstruc.2011.03.003
Zimmermann M., Belai N., Butcher R.J. et al. // Inorg. Chem. 2007. V. 46. № 5. P. 1737. https://doi.org/10.1021/ic0624423
Boyd T., Mitchell S.G., Gabb D. et al. // Chem. Eur. J. 2011. V. 17. № 43. P. 12010. https://doi.org/10.1002/chem.201101666
Kalneus E.V., Melnikov A.R., Korolev V.V. et al. // Appl. Magn. Reson. 2013. V. 44. № 1–2. P. 81. https://doi.org/10.1007/s00723-012-0397-7
Evtushok D.V., Melnikov A.R., Vorotnikova N.A. et al. // Dalton Trans. 2017. V. 46. P. 11738. https://doi.org/10.1039/C7DT01919J
Sheldrick G.M. // Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053229614024218
Hübschle C.B., Sheldrick G.M., Dittrich B. // J. Appl. Crystallogr. 2011. V. 44. № 6. P. 1281. https://doi.org/10.1107/S0021889811043202
Korenev V.S., Floquet S., Marrot J. et al. // Inorg. Chem. 2012. V. 51. № 4. P. 2349. https://doi.org/10.1021/ic202346r
Jones L.H. // J. Chem. Phys. 1955. V. 23. № 11. P. 2105. https://doi.org/10.1063/1.1740675
Gatehouse B.M., Comyns A.E. // J. Chem. Soc. 1958. P. 3965. https://doi.org/10.1039/JR9580003965
Keita B., Lu Y.W., Nadjo L. et al. // Electrochem. Commun. 2000. V. 2. № 10.P. 720. https://doi.org/10.1016/S1388-2481(00)00104-1
Wang G., Su Y., Monts D.L. // J. Phys. Chem. A. 2008. V. 112. № 42. P. 10502. https://doi.org/10.1021/jp802327f
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии