

Журнал неорганической химии, 2019, T. 64, № 9, стр. 916-922
Синтез монетита из гидроксиапатита кальция и монокальцийфосфата моногидрата в условиях механической активации
Т. В. Сафронова 1, *, И. С. Садилов 1, К. В. Чайкун 1, Т. Б. Шаталова 1, Я. Ю. Филиппов 1
1 Московский государственный университет имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: t3470641@yandex.ru
Поступила в редакцию 11.01.2019
После доработки 17.02.2019
Принята к публикации 15.03.2019
Аннотация
Порошок монетита СаНРО4 с размером частиц 100–300 нм синтезирован из монокальцийфосфата моногидрата Ca(H2PO4)2. H2O и гидроксиапатита кальция Ca10(PO4)6(OH)2 в среде ацетона при использовании механической активации в планетарной мельнице. По данным РФА, после термообработки в интервале 900–1100оС фазовый состав образцов представлен β-пирофосфатом кальция β‑Ca2P2O7. Синтезированный порошок может быть использован для получения кальцийфосфатных резорбируемых керамических материалов.
ВВЕДЕНИЕ
Фосфаты кальция используют как пищевые добавки [1], в качестве катализаторов при проведении некоторых химических реакций [2, 3], в сельском хозяйстве в качестве удобрений [4], а также для создания материалов с уникальными свойствами. Известны материалы с люминесцентными свойствами на основе пирофосфата кальция Са2Р2О7 [5, 6]. Ведутся исследования по созданию и применению для лечения дефектов костной ткани биосовместимых материалов на основе различных фосфатов кальция, к которым относятся гидроксиапатит кальция Ca10(PO4)6(OH)2, трикальцийфосфат Са3(РО4)2, пирофосфат кальция Са2Р2О7, октакальциевый фосфат Са8(НРО4)2(РО4)4 · 5Н2О, брушит СаНРО4 · ⋅ 2Н2О, монетит СаНРО4, гидратированный пирофосфат кальция Са2Р2О7.хН2О [7–9].
Для синтеза порошков фосфатов кальция высокого качества используют химическое осаждение, термическую конверсию, твердофазный синтез [10]. При синтезе применяют дополнительные воздействия, такие как ультразвуковое, гидротермальное воздействие [11] или механическую активацию [12]. Для синтеза фосфатов кальция используют различные среды, в том числе моделирующие ионный состав плазмы крови [13]. Для получения биосовместимых композитов используют порошки фосфатов кальция, синтезированные из смешанно-анионных растворов [14–17].
Для реализации регенеративных методов лечения требуются резорбируемые материалы. Растворимость биосовместимых фосфатов кальция возрастает с уменьшением мольного отношения Са/Р. Для гидроксиапатита кальция Ca10(PO4)6(OH)2 мольное отношение Са/Р = 1.67, и это нерезорбируемый фосфат кальция, известный как неорганический компонент костной ткани [18, 19]. Для лечения дефектов костной ткани широко используют материалы на основе трикальцийфосфата Са3(РО4)2, мольное отношение Са/Р = 1.5 [20]. Исследователи обращают внимание и на фосфаты кальция с отношением Са/Р = 1 из-за присущей им биосовместимости и более высокой резорбируемости. Отношение Са/Р = 1 присуще следующим фосфатам кальция: Са2Р2О7, Са2Р2О7. ⋅ хН2О, СаНРО4, СаНРО4. 2Н2О. При этом высокотемпературный пирофосфат кальция Са2Р2О7 в форме γ- и β-модификаций может быть получен в результате термической конверсии гидратированных и/или кислых фосфатов кальция, а также в результате гетерофазных реакций в порошковых смесях, включающих соединения с отношением Са/Р, большим и меньшим 1, например, по реакциям (1) [17], (2) [21] или (3) [22]:
(1)
${\text{2СаНР}}{{{\text{О}}}_{{\text{4}}}}{\cdot 2}{{{\text{Н}}}_{{\text{2}}}}{\text{О = С}}{{{\text{а}}}_{{\text{2}}}}{{{\text{Р}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{7}}}} + {\text{5}}{{{\text{Н}}}_{{\text{2}}}}{\text{О,}}$(2)
${\text{С}}{{{\text{а}}}_{{\text{2}}}}{{{\text{Р}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{7}}}} \cdot x{{{\text{Н}}}_{{\text{2}}}}{\text{О = С}}{{{\text{а}}}_{{\text{2}}}}{{{\text{Р}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{7}}}} + x{{{\text{Н}}}_{{\text{2}}}}{\text{О,}}$(3)
$\begin{gathered} {\text{2СаС}}{{{\text{О}}}_{{\text{3}}}} + {\text{2(N}}{{{\text{H}}}_{{\text{4}}}}{{{\text{)}}}_{{\text{2}}}}{\text{HP}}{{{\text{O}}}_{{\text{4}}}}{\text{ = }} \\ {\text{ = }}\,\,{\text{С}}{{{\text{а}}}_{{\text{2}}}}{{{\text{Р}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{7}}}} + {\text{4N}}{{{\text{H}}}_{{\text{3}}}} + {\text{2}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}} + {\text{2C}}{{{\text{O}}}_{{\text{2}}}}{\text{.}} \\ \end{gathered} $Синтез гидратированных кислых фосфатов кальция лежит в основе получения брушитного/монетитного цементного камня [23]. Для синтеза брушита или монетита необходимо присутствие в зоне реакции одновременно Са2+ (из соединения основной природы) и ${{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{{\text{4}}}^{--}$ (из соединения кислой природы):
(4)
${\text{C}}{{{\text{a}}}^{{{\text{2}} + }}} + {{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{{\text{4}}}^{--} + {\text{2}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{CaHP}}{{{\text{O}}}_{{\text{4}}}} \cdot {\text{2}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {{{\text{H}}}^{ + }}.$В обзоре [23] перечислены щелочные компоненты: трикальцийфосфат Са3(РО4)2, гидроксиапатит кальция Ca10(PO4)6(OH)2, тетракальциевый фосфат Са4(РО4)2О, оксид кальция СаО, гидроксид кальция Са(ОН)2, а также компоненты кислого характера: фосфорная кислота Н3РО4, монокальцийфосфат моногидрат Ca(H2PO4)2. H2O, серная кислота H2SO4, пирофосфорная кислота H4P2O7, лимонная кислота C3H5O(COOH)3. Выбор компонентов для получения кальцийфосфатного цемента, использование которого часто предполагается in situ в неинвазивных методах компенсации дефектов костной ткани, учитывает природу образующегося сопутствующего продукта реакции. При использовании большинства перечисленных компонентов в любом сочетании сопутствующим продуктом реакции является вода – нетоксичное вещество для тканей организма.
Таким образом, синтез брушита CaHPO4. ⋅ 2H2O или монетита СаНРО4 может быть осуществлен при взаимодействии растворов соответствующих солей или в пастах при формировании цементного камня. При этом синтез кислого ортофосфата кальция с мольным отношением Са/Р = 1 (монетит СаНРО4) в условиях механической активации до настоящего времени в литературе не описан, хотя данный метод позволяет получать активные порошки в качестве исходного материала для получения керамики или композитов с полимерной матрицей.
Цель настоящей работы – синтез фосфата кальция с мольным отношением Са/Р = 1 (монетит СаНРО4) из плохо растворимых соединений – кислой соли кальция (монокальцийфосфата моногидрата Ca(H2PO4)2. H2O) и основной соли кальция (гидроксиапатита кальция Ca10(PO4)6(OH)2) в условиях механической активации. Использование данной пары прекурсоров позволит получить порошок, не содержащий токсичных сопутствующих продуктов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Количество исходных солей для проведения синтеза определили по реакции (5), обеспечивающей синтез фосфата кальция с мольным отношением Са/Р = 1:
(5)
$\begin{gathered} {\text{4Ca(}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{{\text{O}}}_{{\text{4}}}}{{{\text{)}}}_{{\text{2}}}} \cdot {{{\text{H}}}_{{\text{2}}}}{\text{O}} + \\ + \,\,{\text{C}}{{{\text{a}}}_{{{\text{10}}}}}{{{\text{(P}}{{{\text{O}}}_{{\text{4}}}}{\text{)}}}_{{\text{6}}}}{{{\text{(OH)}}}_{{\text{2}}}} = {\text{14СаНР}}{{{\text{О}}}_{{\text{4}}}}{\text{ + 6}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}}{\text{.}} \\ \end{gathered} $Рассчитанные количества гидроксиапатита Ca10(PO4)6(OH)2 (CAS № 1306-06-5, puriss. p.a. ≥90%, Riedel-deHaen, Sigma-Aldrich Laborchemikalien, 04238, lot 70080, Германия) и монокальцийфосфата моногидрата Ca(H2PO4)2. H2O (CAS № 10031-30-8 puriss. p.a. ≥85%, Sigma-Aldrich) помещали в емкости из диоксида циркония. К порошковой смеси добавляли мелющие тела из диоксида циркония при соотношении масса порошка : масса мелющих тел = 1 : 5. После добавления ацетона (ГОСТ 2603-79) емкости с порошками и ацетоном закрывали и закрепляли в планетарной мельнице. Продолжительность механической активации при скорости вращения 600 об./мин составляла 20 мин. После завершения обработки в планетарной мельнице порошок сушили на воздухе при комнатной температуре в течение 2 ч. После сушки порошки пропускали через сито с размером ячеек 200 мкм. Из полученных порошков на ручном прессе Carver Laboratory Press model c (USA) изготавливали компактные порошковые заготовки в форме таблеток диаметром 12 мм и высотой 2–3 мм при давлении прессования 100 МПа без использования временного технологического связующего. Сформованные порошковые заготовки обжигали в печи при различных температурах в интервале 900–1100°С (скорость нагрева 5 град/мин, выдержка при заданной температуре 2 ч, охлаждение вместе с печью).
Линейную усадку и геометрическую плотность образцов керамики определяли, измерив их массу и размеры (с точностью ±0.05 мм) до и после обжига.
Рентгенофазовый анализ (РФА) синтезированного порошка, порошковых смесей после проведения синтеза в условиях механической активации и образцов после обжига проводили на дифрактометре Rigaku D/Max-2500 с вращающимся анодом (Япония) с использованием CuKα-излучения. Для проведения качественного фазового анализа использовали базу данных ICDD PDF2 [24].
Синхронный термический анализ (ТА) выполняли на термоанализаторе NETZSCH STA 409 PC Luxx (NETZSCH, Германия) при скорости нагревания 10 град/мин. Масса образца составляла не менее 10 мг. Исследование состава образующейся при разложении образцов газовой фазы проводили при помощи квадрупольного масс-спектрометра QMS 403C Aëolos (NETZSCH, Германия), совмещенного с термоанализатором NETZSCH STA 409 PC Luxx. Масс-спектры (MS) записывали для массовых чисел 18 и 17 (Н2О+ и ОН+), 44 (${\text{CO}}_{2}^{ + }$).
Микроструктуру образцов исследовали методом растровой электронной микроскопии на электронном микроскопе LEO SUPRA 50VP (Carl Zeiss, Германия; автоэмиссионный источник); съемку осуществляли при ускоряющем напряжении 3–20 кВ во вторичных электронах (детектор SE2). На поверхность образцов напыляли слой хрома (до 10 нм).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
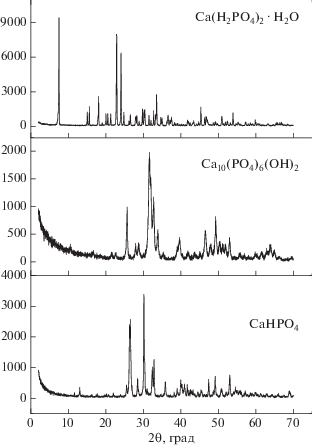
На рис. 1 представлены результаты РФА исходных реагентов и синтезированного монетита СаНРО4, которые позволяют сделать вывод о том, что после механической активации порошковой смеси в ацетоне в течение 20 мин все исходные реагенты исчерпаны, а фазовый состав порошка представлен монетитом СаНРО4 (карточка PDF 9-80). Действительно, реакция (5) свидетельствует о том, что при взаимодействии монокальцийфосфата моногидрата Ca(H2PO4)2. H2O (карточка PDF 9-347) и гидроксиапатита Ca10(PO4)6(OH)2 (карточка PDF 74-566) количества воды достаточно для образования монетита СаНРО4. И хотя оба исходных вещества обладают низкой растворимостью (~17 г/л для Ca(H2PO4)2. H2O и ~0.0003 г/л для Ca10(PO4)6(OH)2) [25], а количества воды в зоне реакции невелико (даже с учетом некоторого количества воды, присутствующего в ацетоне), механическая активация обеспечивает протекание реакции (5). Следует отметить, что растворимость монетита СаНРО4 занимает промежуточное положение между растворимостью гидроксиапатита Ca10(PO4)6(OH)2 и монокальцийфосфата моногидрата Ca(H2PO4)2. H2O и составляет ~0.048 г/л. Однако более высокая растворимость монокальцийфосфата моногидрата Ca(H2PO4)2. H2O по сравнению с растворимостью гидроксиапатита кальция и монетита, по всей видимости, обеспечивает в водных сегментах (каплях) в условиях механической активации уровень рН, благоприятный для образования и сохранения монетита СаНРО4, а не гидроксиапатита.
Рис. 1.
РФА исходных веществ Ca(H2PO4)2. H2O (карточка PDF 9-347), Ca10(PO4)6(OH)2 (карточка PDF 74-566) и синтезированного фосфата кальция СаНРО4 (карточка PDF 9-80).
На рис. 2 представлены микрофотографии исходных порошков и синтезированного монетита СаНРО4. Размер пластинок монокальцийфосфата моногидрата Ca(H2PO4)2. H2O достаточно большой и составляет 50–300 мкм при толщине 15–20 мкм (рис. 2а). Размер частиц гидроксиапатита Ca10(PO4)6(OH)2 составляет 100–200 нм (рис. 2б). Размер частиц синтезированного монетита СаНРО4 может быть оценен как 100–300 нм (рис. 2в). При этом на фото можно заметить частицы как столбчатой морфологии, так и с формой, близкой к изометричной. Известно, что для монетита СаНРО4, полученного из растворов, характерна пластинчатая морфология частиц [26]. В данном случае для монетита СаНРО4, полученного в условиях механической активации из плохо растворимых в воде фосфатов кальция в присутствии ацетона, пластинчатая морфология частиц не наблюдается. Такой результат может быть обусловлен тем, что химическая реакция (5) синтеза монетита СаНРО4 протекает на поверхности гидроксиапатита Ca10(PO4)6(OH)2. Можно считать, что в описанных условиях происходит гетерогенный синтез на поверхности раздела твердое/жидкость, где твердое – гидроксиапатит кальция, а жидкость – водный раствор монокальцийфосфата моногидрата Ca(H2PO4)2. H2O. Ацетон при этом выполняет роль среды дезагрегации, обеспечивающей подвижность мелющих тел, частиц порошка и сегментов (капель) водного раствора. Некоторое увеличение размера частиц монетита СаНРО4 по сравнению с размером частиц гидроксиапатита кальция Ca10(PO4)6(OH)2 может быть обусловлено тем, что из одного моля гидроксиапатита Ca10(PO4)6(OH)2 (М = 1004 г/моль) с плотностью 3.16 г/см3 образуется 14 молей монетита (М = 136 г/моль) с плотностью 2.93 г/см3. При этом объем отдельно взятой частицы может увеличиться примерно в два раза.
Рис. 2.
Микрофотографии исходных веществ Ca-(H2PO4)2. H2O (а), Ca10(PO4)6(OH)2 (б) и синтезированного фосфата кальция СаНРО4 (в).

Данные термического анализа исходных веществ и синтезированного монетита СаНРО4 представлены на рис. 3 и 4. Данные ТА и MS для исходных веществ – Ca(H2PO4)2. H2O и Ca10(PO4)6(OH)2 – даны для сравнения. Сопоставление данных ТА и MS подтверждает отсутствие исходных веществ в синтезированном порошке, что согласуется с результатами РФА (рис. 1). Для монетита СаНРО4 при 400°С характерно протекание реакции (6), отражающей превращение монетита CaHPO4 в пирофосфат Ca2P2O7.
(6)
${\text{2CaHP}}{{{\text{O}}}_{{\text{4}}}}{\text{ = C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{P}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{7}}}}{\text{ + }}{{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Рис. 3.
TA исходных веществ Ca(H2PO4)2. H2O, Ca10(PO4)6(OH)2 и синтезированного фосфата кальция СаНРО4.
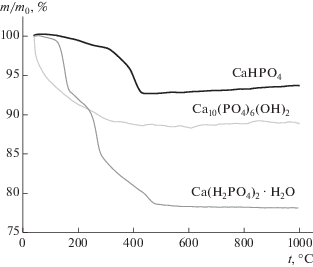
Рис. 4.
MS исходных веществ Ca(H2PO4)2. H2O, Ca10(PO4)6(OH)2 и синтезированного фосфата кальция СаНРО4 для m/z = 18.
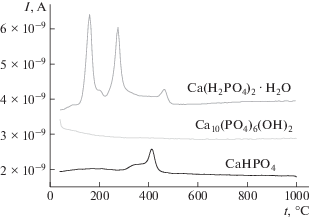
Общая потеря массы порошка монетита СаНРО4 при нагревании составила ∼7% (рис. 3). По данным MS, уменьшение массы монетита СаНРО4 происходит за счет выделения воды в интервале 300–475°С (рис. 4).
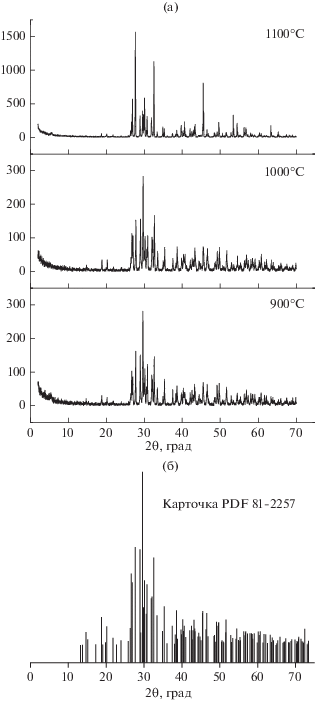
По данным РФА (рис. 5), фазовый состав керамики после тестовых обжигов в интервале температур 900–1100°С представлен β-Са2Р2О7.
Рис. 5.
РФА образцов керамики из порошка СаНРО4, синтезированного из Ca(H2PO4)2. H2O и Ca10(PO4)6(OH)2, после обжига при 900, 1000 и 1100°С (а) и штрих-диаграмма для карточки PDF 81-2257 (β-Ca2P2O7) (б).
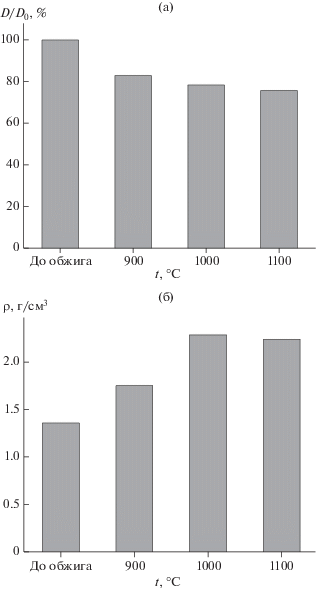
На рис. 6 приведены значения линейной усадки (а) и плотности (б) образцов керамики до и после обжига. Плотность образца, отпрессованного при 100 МПа из синтезированного порошка, по сравнению с теоретической плотностью β-пирофосфата кальция (3.09 г/см3) составила 45%. Линейная усадка образцов после обжига при 900°С составила 17%, а после обжига при 1100°С – 25%. Плотность образцов после обжига при 1000–1100°С составила 74–76% по сравнению с теоретической плотностью β-пирофосфата кальция (3.09 г/см3).
Рис. 6.
Линейные размеры (а) и плотность (б) образцов керамики из порошка СаНРО4, синтезированного из Ca(H2PO4)2 · H2O и Ca10(PO4)6(OH)2, после обжига при 900, 1000 и 1100°С.
На рис. 7 представлена микроструктура керамики на основе β-Са2Р2О7 после обжига при различных температурах в интервале 900–1100°С. Размер зерен после обжига при 900–1000°С составляет 2–4 мкм. При этом микроструктура керамики после обжига при 900°С выглядит более рыхлой и пористой. Размер зерен керамики после обжига при 1100°С составляет 5–10 мкм.

ЗАКЛЮЧЕНИЕ
Порошок монетита СаНРО4 с размером частиц 100–300 нм, синтезированный из монокальцийфосфата моногидрата Ca(H2PO4)2. H2O и гидроксиапатита кальция Ca10(PO4)6(OH)2 в условиях механической активации, может быть использован для получения керамических материалов с фазовым составом, включающим биосовместимую биорезорбируемую фазу – пирофосфат кальция β-Са2Р2О7.
Кроме того, синтезированный порошок может быть использован в качестве наполнителя для создания материалов с полимерной матрицей, а также при допировании соответствующими ионами и в качестве матрицы при создании люминесцентных материалов.
Полученные из синтезированного порошка керамические кальцийфосфатные материалы, содержащие биорезорбируемую и биосовместимую фазу пирофосфата кальция β-Са2Р2О7, могут быть рекомендованы для создания костных имплантатов.
Список литературы
Киселева С.И. Пищевые и биологически активные добавки: учебное пособие. Новосибирск: Изд-во НГТУ, 2013. 48 с. ISBN 978-5-7782-2251-9
Ruthner H., Noller H. // J. Catal. 1975. V. 38. № 1–3. P. 264. https://doi.org/10.1016/0021-9517(75)90087-1
Ghantani V.C., Dongare M.K., Umbarkar S.B. // RSC Advances. 2014. V. 4. № 63. P. 33319. https://doi.org/10.1039/c4ra06429a
Расулов А.А., Бадалова О.А., Алимов У.К. и др. // Universum: технические науки. 2017. Т. 8. № 41. http://7universum.com/ru/tech/archive/item/5081
Kolaya Siddhartha, Basu M., Sudarsana V., Tyagi A.K. // Solid State Sci. 2018. V. 85. P. 26. https://doi.org/10.1016/j.solidstatesciences.2018.09.007
Roman-Lopez J., Lozano I.B., Cruz-Zaragoza E. et al. // Appl. Radiat. Isot. 2017. V. 124. P. 44. https://doi.org/10.1016/j.apradiso.2017.03.004
Dorozhkin S.V. // Ceram. Int. 2015. V. 41. № 10. P. 13913. https://doi.org/10.1016/j.ceramint.2015.08.004
Safronova T.V., Putlyaev V.I. // Inorg. Mater. 2017. V. 53. № 1. C. 17. https://doi.org/10.1134/S0020168516130057
Xu H.H., Wang P., Wang L. et al. // Bone research. 2017. V. 5. P. 17056. https://doi.org/10.1038/boneres.2017.56
Shavandi A., Bekhit A.El-Din A. et al. // J. Biomimetics, Biomaterials and Biomedical Engineering. 2015. V. 25. P. 98. https://doi.org/10.4028/www.scientific.net/JBBBE.25.98
Ouerfelli N., Zid M.F. // J. Struct. Chem. 2016. V. 57. № 3. C. 628. https://doi.org/10.1134/S0022476616030252
Chaikina M.V., Bulina N.V., Ishchenko A.V. et al. // Russ. Phys. J. 2014. V. 56. № 10. Р. 1176. [Чайкина М.В., Булина Н.В. и др. // Изв. вузов. Физика. 2013. Т. 56. № 10. С. 66.]https://doi.org/10.1007/s11182-014-0159-0
Golovanova O.A. // Russ. J. Inorg. Chem. 2018. V. 63. № 12. Р. 1541. [Голованова О.А. // Журн. неорган. химии. 2018. Т. 63. №. 12. С. 1530.]https://doi.org/10.1134/S0036023618120094
Solonenko A.P., Blesman A.I., Polonyankin D.A. et al. // Russ. J. Inorg. Chem. 2018. V. 63. V. 8. P. 993. [Солоненко А.П., Блесман А.И., Полонянкин Д.А., Горбунов В.А. // Журн. неорган. химии. 2018. Т. 63. № 8. С. 953.]https://doi.org/10.1134/S0036023618080211
Safronova T.V., Putlyaev V.I., Filippov Ya.Yu. et al. // Glass Ceram. 2018. V. 75. № 3–4. P. 118. [Сафронова Т.В., Путляев В.И., Филиппов Я.Ю. и др. // Стекло и керамика. 2018. № 3. С. 41.]https://doi.org/10.1007/s10717-018-0040-7
Ezhova Z.A., Zakharov N.A., Koval E.M. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 8. Р. 1001. [Ежова Ж.А., Захаров Н.А., Коваль Е.М. и др. // Журн. неорган. химии. 2018. Т. 63. № 8. С. 961.]https://doi.org/10.1134/S0036023618080065
Safronova T.V., Knot’ko A.V., Shatalova T.B. et al. // Glass Ceram. 2016. V. 73. № 1–2. Р. 25. [Сафронова Т.В., Кнотько А.В., Шаталова Т.Б. и др. // Стекло и керамика. 2016. № 1. С. 27.]https://doi.org/10.1007/s10717-016-9819-6
Данильченко С.Н. // Вісник СумДУ. Серія Фізика, математика, механіка. 2007. № 2. С. 33.
Герк С.А., Голованова О.А. // Вестник Омского ун-та. 2015. № 4 (78). С. 39.
Carrodeguas R.G., De Aza S. // Acta Biomaterialia. 2011. V. 7. № 10. P. 3536. https://doi.org/10.1016/j.actbio.2011.06.019
Safronova T.V., Putlyaev V.I., Kurbatova S.A. et al. // I-norg. Mater. 2015. V. 51. № 11. P. 1177. [Сафронова Т.В., Путляев В.И., Курбатова С.А. и др. // Неорган. материалы. 2015. Т. 51. № 11. С. 1269.]https://doi.org/10.1134/S0020168515110096
Safronova T.V., Putlyaev V.I., Ivanov V.K. et al. // Refract. Ind. Ceram. 2016. V. 56. № 5. P. 502. [Сафронова Т.В., Путляев В.И., Иванов В.К. и др. // Новые огнеупоры. 2015. № 9. С. 45.]https://doi.org/10.1007/s11148-016-9877-x
Tamimi F., Sheikh Z., Barralet J. // Acta Biomaterialia. 2012. V. 8. № 2. P. 474. https://doi.org/10.1016/j.actbio.2011.08.005
ICDD (2010). PDF-4+ 2010 (Database) / Ed. Soorya Kabekkodu. International Centre for Diffraction Data. Newtown Square. PA. USA. http://www.icdd.com/ products/pdf2.htm
Dorozhkin S.V. // Prog. Biomater. 2016. V. 5. № 1. P. 9. https://doi.org/10.1007/s40204-015-0045-z
Mulongo-Masamba R., El Kassri T., Khachani M. et al. // J. Therm. Anal. Calorim. 2015. V. 124. № 1. P. 171. https://doi.org/10.1007/s10973-015-5130-y
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии