

Журнал неорганической химии, 2020, T. 65, № 10, стр. 1308-1316
Расширение инкапсулирующего макробициклического лиганда с использованием палладий-катализируемой реакции Сузуки–Мияура дииодоклатрохелатного трис-глиоксимата железа(II) с реакционноспособными атомами галогена в апикальных заместителях
А. Ф. Асаченко a, b, М. А. Топчий a, Г. Е. Зелинский b, c, И. П. Лимарёв b, c, П. В. Дороватовский d, А. В. Вологжанина b, Я. З. Волошин b, c, *
a Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский пр-т, 29, Россия
b Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
c Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
d Национальный исследовательский центр “Курчатовский институт”
123098 Москва, пл. Академика Курчатова, 1, Россия
* E-mail: voloshin@ineos.ac.ru
Поступила в редакцию 25.04.2020
После доработки 28.05.2020
Принята к публикации 29.05.2020
Аннотация
Изучена возможность расширения апикальных заместителей при макробициклическом трис-глиоксиматном остове при помощи C–C-кросс-сочетания Сузуки–Мияура, катализируемого палладием. Реакция дииодоклатрохелата железа(II) с фенилборной кислотой успешно проведена в толуоле с использованием каталитической системы Pd(CH3COO)2/SPhos и CsF в качестве неорганического основания. Кипячением реакционной смеси с обратным холодильником при 110°С получен макробициклический комплекс железа(II) с двумя апикальными бифенильными заместителями и исследован спектральными методами и с помощью РСА. Асимметричная ячейка его монокристалла содержит две независимые клатрохелатные молекулы А и В. Их координационные полиэдры FeN6 высотой 2.37 Å с расстояниями Fe–N 1.903(3)–1.916(2) Å и хелатными углами (α) 78.4°–78.6° имеют искаженную тригонально-призматическую–тригонально-антипризматическую геометрию с углами искажения (φ) 18.5° и 18.0° соответственно; инкапсулированный ион железа(II) находится практически в их центрах. Кристаллическая упаковка определяется в основном межмолекулярными гидрофобными взаимодействиями H…H, H…C и C…C, а также водородными связями H…O и H…N.
ВВЕДЕНИЕ
Рациональный дизайн и эффективный синтез жестких макробициклических комплексов с инкапсулированным ионом металла – клатрохелатов [1, 2], молекулы которых содержат терминальные полиароматические или донорные группы в апикальных заместителях, а также заместители с пиридильными терминальными группами [3–6], позволяют получать различные типы клатрохелатных интеркаляторов ДНК и других биоэффекторов [1, 2], а также производные клатрохелатных лигандных синтонов: полиядерные комплексы и металломакроциклы [4–7], координационные капсулы, металлo-органические каркасные соединения (МОКСы) и координационные полимеры [4, 8–11]. Так, макробициклический трис-глиоксимат железа(II) с 3-пиридильными терминальными группами, молекула которого содержит два апикальных N-донорных заместителя, был успешно использован в [9] в качестве металлокомплексного лигандного синтона для получения уникальной тетраэдрической Pd4L8-координационной капсулы, содержащей восемь макробициклических синтонов этого типа. Одним из наиболее подходящих синтетических подходов для получения этих клеточных комплексов является использование металл-катализируемых реакций кросс-сочетания, в том числе гомогенного палладий-катализируемого кросс-сочетания Сузуки–Мияура – эффективного и мощного метода современной органической химии, реализованного ранее для синтеза ароматических соединений. В частности, реакции кросс-сочетания Сузуки арилгалогенидов с фенилборными кислотами использовались в [12–14] для образования новой связи CАрил–CАрил. Этот подход реализован на промышленном уровне и позволяет получать широкий круг фармацевтических и высокотехнологичных органических соединений. В то же время в литературе сообщается об очень ограниченном числе металл-катализируемых превращений координированных лигандов, поскольку процессы их гомо- и кросс-сочетания осложнены побочными реакциями как лигандов, так и координирующих ионов металлов металлокомплексов как субстратов с компонентами каталитической системы. Таким образом, в качестве подходящих субстратов могут быть использованы только координационные соединения с высокой кинетической и термодинамической устойчивостью в жестких условиях вышеуказанных гомогенных реакций сочетания. Инкапсулированный ион металла в клеточных комплексах изолирован от влияния внешних факторов, таких как эффекты растворителя, и побочных процессов координации лигандов. Поэтому полиазометиновые клатрохелаты переходных металлов имеют высокую химическую устойчивость и широкий круг реакционноспособных комплексов этого типа (прежде всего галогеноклатрохелатные предшественники) и подходят для их дальнейшей функционализации с использованием наиболее распространенных реакций современной органической химии. Ранее осуществленные превращения их реберного хелатирующего фрагмента, катализируемые палладием, представлены на схеме 1 . Более того, реакции кросс-сочетания Соногашира, катализируемые палладием (схема 2 ), были успешно использованы в [3] для синтеза бис- и трис-клатрохелатов железа(II) с жесткой стержнеобразной геометрией и функционализирующими апикальными заместителями при сшивающих атомах бора, которые содержат терминальные 4-пиридильные группы. В [15] нами был получен показанный на схеме 1 5-бром-2-фурилсодержащий макробициклический предшественник с двумя терминальными атомами галогена в реберных ароматических заместителях и была проведена его реакция кросс-сочетания Сузуки–Мияура с 4-карбоксилфенилборной кислотой, которая привела к образованию реберно-функционализированных клатрохелатов железа(II) с присущими функционализирующими группами. В [16] было показано, что дииодоклатрохелат железа(II) с присущими атомами галогена в одном из трех его реберных хелатирующих фрагментов, также показанный на схеме 1 , претерпевает палладий-катализируемые кросс-сочетания Сузуки–Мияура и Соногашира в “классических” условиях. Их результатом явилось целевое расширение и функционализация инкапсулирующего макробициклического лиганда. Фенилборная кислота, 4-карбоксифенилборная и 6-этокси-2-нафтилборная кислоты и диэтиловый эфир 4-(этоксикарбонил)фенилборной кислоты были изучены в [16] в качестве борсодержащих компонентов реакций Сузуки–Мияура вышеуказанного дииодоклатрохелатного субстрата в ДМФА и ТГФ как растворителях. Целевые клатрохелатные продукты были выделены [16] с наиболее высокими выходами в ДМФА, в то время как наибольшая активация борсодержащих компонентов наблюдалась при использовании водного раствора Na2CO3.

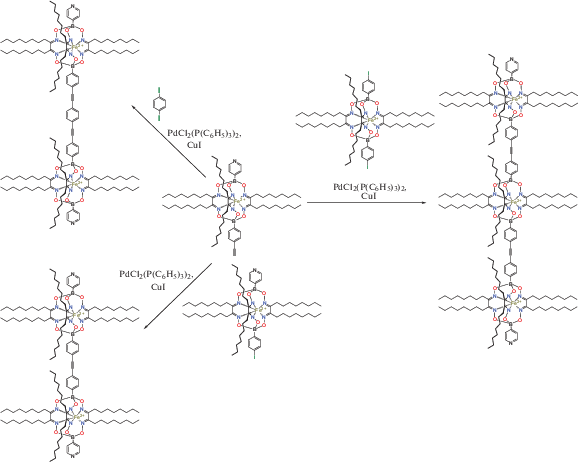
Схема 2.
Попытка провести кросс-сочетание Сузуки–Мияура вышеупомянутого дииодоклатрохелатного предшественника с фенилборной кислотой в указанных условиях реакции привела к полному разрушению клеточного остова, давая также макробициклические продукты гидродегалогенирования и реакции тандемного гидродегалогенирования – замещения (схема 1 ) с низкими выходами. Обнаружено [16] наличие только следовых количеств целевого диарилзамещенного клатрохелатного продукта двойного кросс-сочетания Сузуки–Мияура. Напротив, использование 6-этокси-2-нафтилборной кислоты, молекула которой содержит электронодонорный заместитель, в качестве борсодержащего компонента позволило получить [16] целевые клатрохелатные продукты моно- и бис-кросс-сочетания Сузуки–Мияура с умеренным выходом. Полученные моно- и дифункционализированные клеточные комплексы являются продуктами тандемной реакции гидродеиодинирования – C–C-кросс-сочетания и двойного C–C-кросс-сочетания соответственно. Установлено, что наличие электронодонорного заместителя в молекуле соответствующей борной кислоты существенно влияет на протекание ее реакции (реакций) Сузуки–Мияура, увеличивая реакционную способность соответствующего органопалладиевого интермедиата.
Использование клатрохелатных трис-глиоксиматных лигандных синтонов или соответствующих ДНК-интеркаляторов позволяет избежать стерических затруднений в их полиядерных производных, а также в супрамолекулярных гибридных системах с биомакромолекулами, координационных и супрамолекулярных капсулах, координационных полимерах и МОКСах на их основе. В настоящей работе мы установили возможность расширения апикальных заместителей при макробициклическом трис-глиоксиматном остове с использованием палладий-катализируемой реакции кросс-сочетания Сузуки–Мияуры терминальных атомов иода в соответствующем клатрохелатном предшественнике.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы
Использовали коммерчески доступные реагенты (Sigma–Aldrich ®) FeCl2 · 4H2O, триэтиламин, 4-иодофенилборную кислоту, безводный K3PO4, CsF, Pd(CH3COO)2, комплексы палладия(II) с лигандами dppf и SPhos (их структуры приведены на схеме 2 ), сорбенты и органические растворители. Глиоксим получали конденсацией глиоксаля с гидроксиламином согласно [17], дииодомакробициклический предшественник FeGm3(B4I-Ph)2 – аналогично другим клатрохелатным трис-глиоксиматам железа(II) [18, 19].
Элементный анализ на содержание C, H и N выполняли на приборе Carlo Erba 1106 в Лаборатории микроанализа ИНЭОС РАН.
Масс-спектры MALDI-TOF в положительной и отрицательной областях регистрировали на масс-спектрометре MALDI-TOF-MS Bruker Autoflex II (Bruker Daltonics) в режиме reflecto-mol. Ионизацию проводили УФ-лазером с длиной волны 337 нм. Образец помещали на никелевую пластину, в качестве матрицы использовали 2,5-дигидроксибензойную кислоту. Точность измерений составляла 0.1%.
Спектры ЯМР 1H и 13C{1H} регистрировали для раствора в ДМСО-d6 на спектрометре Bruker Avance 600. Измерения проводили с использованием остаточных сигналов этого дейтерированного растворителя.
ЭСП раствора комплекса в ДМСО записывали в диапазоне 250–800 нм на спектрофотометре Varian Cary 100. Разложение спектра на индивидуальные гауссовы компоненты проводили с использованием программы Fityk [20].
ИК-спектр твердого образца (таблетка KBr) в диапазоне 400–4000 см–1 регистрировали на спектрометре Perkin Elmer FT-IR Spectrum BX II.
Синтез
FeGm3(B4-biPh)2. Комплекс FeGm3(B4I-Ph)2 (0.185 г, 0.25 ммоль), фенилборную кислоту (0.122 г, 1 ммоль), соль палладия(II) Pd(CH3COO)2 (0.023 г, 4 мол. %), лиганд SPhos (0.082 г, 8 мол. %) и безводный CsF (0.152 г, 1 ммоль) растворяли/суспендировали в сухом толуоле (40 мл) в атмосфере аргона. Реакционную смесь кипятили с обратным холодильником в течение 24 ч, а затем охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали, промывали толуолом (несколько порций), ТГФ и экстрагировали дихлорметаном (100 мл). Экстракт упаривали досуха в вакууме, получили желто-оранжевый мелкокристаллический продукт. Выход 0.71 г (44%).
Спектр ЯМР 1H (ДМСО-d6), δ, м.д.: 7.36 (т, J1H–1H = 7.3 Гц, 2H, biPh), 7.47 (т, J1H–1H = 7.6 Гц, 4H, biPh), 7.62 (д, J1H–1H = 7.9 Гц, 4H, biPh), 7.72–7.64 (м, 8H, biPh), 8.27 (с, 6H, Gm). Спектр ЯМР 13C{1H} (ДМСО-d6), δ, м.д.: 125.38, 125.97, 127.06, 129.37 (все уш. синглеты, biPh), 132.57 (с, Gm). ИК-спектр (KВr), ν, см–1: 1555 ν(C=N), 902, 960, 974, 1009 ν(N–O), 1137 м ν(B–O). ЭСП (ДМСО): λmax, нм (ε × 10–3 моль–1 л см–1) 245 (44), 257 (12), 287 (13), 302 (8.4), 322 (2.2), 410 (3.6), 438 (6.8).
Рентгеноструктурный анализ
Интенсивности отражений кристалла FeGm3(B4-biPh)2 были зарегистрированы с помощью пучка К4.4 “Белок” в Курчатовском центре синхротронного излучения (ГНЦ “Курчатовский институт”, Москва, Россия) при длине волны 0.79313 Å с использованием детектора Rayonix CCD 165. Сбор данных осуществляли при температуре 100 K на оборудовании Oxford CryoJet (Oxford Cryosystems). Интегрирование изображения проводили с использованием программы iMosflm [21]. Интегрированные интенсивности были эмпирически скорректированы на поглощение с использованием программы Scala [22]. Параметры кристалла FeGm3(B4-biPh)2 при 100.0(2) K: C30H24B2FeN6O6, M = 642.02, триклинная сингония, пр. гр. $P\overline 1 $, a = 12.160(2), b = 14.420(3), c = = 17.670(4) Å, α = 101.22(3)°, β = 109.03(3)°, γ = = 102.42(3)°, V = 2740.4(11) Å3, Z = 4, ρвыч = = 1.556 г/см3, μ = 0.822 мм–1, 9534 независимых отражения (Rint = 0.0457), 7464 наблюдаемых отражения, окончательные параметры сходимости: R1[I > 2σ(I)] = 0.049, wR(F2) = 0.131, GOF = 1.024. Структура решена прямым методом и уточнена полноматричным методом наименьших квадратов относительно F 2. Неводородные атомы уточнены анизотропно, вычислены положения атомов водорода и все атомы водорода включены в уточнение по модели “наездника” с Uизо(H) = = 1.2Uэкв(X). Все вычисления проводили с использованием программных пакетов SHELXL2014 [23] и OLEX2 [24]. Координаты атомов, величины тепловых параметров и список всех отражений депонированы в Кембриджском банке структурных данных (№ 1982029; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/ data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Модельные палладий-катализируемые реакции Сузуки–Мияура дииодоклатрохелатного предшественника FeGm3(B4I-Ph)2 с фенилборной кислотой проводили в условиях, позволявших использовать борсодержащие арилгалогениды в качестве реакционноспособных арилирующих компонентов такого С–С-кросс-сочетания [25, 26]. Попытки провести реакции этого клатрохелатного предшественника с фенилборной кислотой в ТГФ в качестве растворителя в присутствии 8 мол. % Pd(dppf)Cl2, K3PO4 и воды (10 эквивалентов) при комнатной температуре и при 60°С привели не к получению целевых клатрохелатных продуктов, а лишь к гидродегалогенированию комплекса FeGm3(B4I-Ph)2. Этот процесс является характерным металл-катализируемым превращением галогенокатрохелатных квазиароматических трис-диоксиматов металлов с присущими атомами галогена в реберных хелатирующих фрагментах [2] и основной побочной реакцией в случае их превращений. Стараясь избежать этого нежелательного процесса, мы провели модельное С–С-кросс-сочетание в сухом толуоле как растворителе, используя каталитическую систему Pd(CH3COO)2/SPhos [27] и CsF в качестве неорганического основания. Образование клатрохелатных продуктов арилирования макробициклического предшественника FeGm3(B4I-Ph)2 при комнатной температуре не наблюдалось, в то время как кипячение реакционной смеси с обратным холодильником при 110°C привело к образованию целевого продукта двойного C–C-кросс-сочетания – клатрохелата FeGm3(B4-biPh)2 (схема 3).
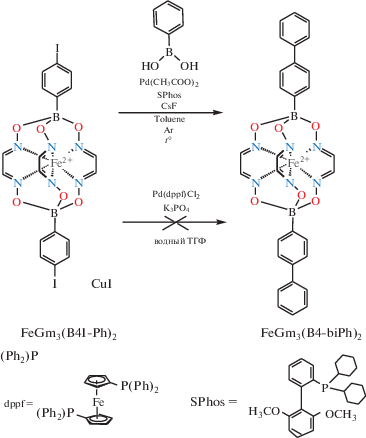
Схема 3.
Комплекс FeGm3(B4-biPh)2 был изучен методами элементного анализа, MALDI-TOF масс-спектрометрии, ЭСП, ЯМР 1H и 13C{1H}, ИК-спектроскопии и РСА.
Наиболее интенсивный пик в положительном диапазоне MALDI-TOF масс-спектра этого клатрохелата железа(II) относится к его молекулярному иону.
Число и положение сигналов в спектрах ЯМР 1H и 13C{1H} его раствора, в частности сигналов протонов апикальных бифенильных заместителей и метиновых протонов хелатирующих глиоксиматных фрагментов, а также соотношение их интегральных интенсивностей в спектре ЯМР 1Н подтвердили состав и C3-симметрию молекулы FeGm3(B4-biPh)2. Число линий в спектре ЯМР 13C{1H} указывает на эквивалентность реберных хелатирующих фрагментов этой молекулы.
ИК-спектр FeGm3(B4-biPh)2 содержит валентные колебания связей C=N, N–O и B–O, характерные для борсодержащих трис-диоксиматных клатрохелатов [1]. В то же время характеристические колебания ν(C=N) глиоксиматных реберных фрагментов наблюдаются около 1550 см–1 и существенно (на ∼30 см–1) смещены в низкочастотную область по сравнению с таковыми в спектрах его алифатических и ароматических макробициклических аналогов.
Разложение ЭСП полученного трис-глиоксиматного клатрохелата железа(II) FeGm3(B4-biPh)2 на гауссовы компоненты позволило выделить в видимой области две интенсивные полосы переноса заряда металл–лиганд Fed → L π* с максимумами в диапазоне 420–440 нм. Этот спектр в УФ-диапазоне содержит также ряд более интенсивных полос, отнесенных к π–π*-переходам в полиазометиновом квазиароматическом остове, а также к таковым в его апикальных бифенильных заместителях.
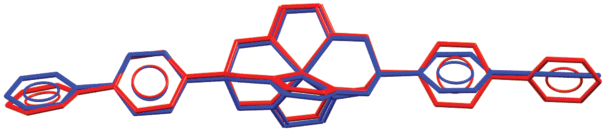
По данным РСА, асимметричная ячейка кристалла FeGm3(B4-biPh)2 содержит две симметрично-независимые клатрохелатные молекулы A и B (рис. 1); основные геометрические параметры их макробициклических остовов приведены в табл. 1. Координационные полиэдры FeN6 этих клеточных молекул имеют искаженную тригонально-призматическую (ТП)–тригонально-антипризматическую (ТАП) геометрию с углами искажения φ = 18.5° и 18.0° соответственно (φ = 0° в случае идеальной ТП и φ = 60° в случае идеальной ТАП). Расстояния Fe–N в молекулах FeGm3(B4-biPh)2 типов A и B изменяются от 1.903(3) до 1.916(2) Å, а инкапсулированный ион железа(II) находится практически в центре их макробициклических остовов. Высота h этих ТП–ТАП полиэдров равна 2.37 Å, а хелатные углы α находятся в диапазоне 78.4°–78.6°. Все основные длины и углы связей в их клеточных остовах аналогичны таковым для других борсодержащих глиоксиматов железа(II) с известными рентгеновскими структурами [9, 18, 19 и 28]. Следует отметить, что хелатные связи С–С в макробициклических трис-глиоксиматных лигандах двух вышеуказанных независимых молекул FeGm3(B4-biPh)2 (1.418(6)–1.427(5) Å, табл. 1) значительно меньше, чем в молекулах их реберно-функционализированных алифатических и ароматических борсодержащих клатрохелатных аналогов с инкапсулированным ионом железа(II) (ср. 1.44 Å) [1, 2]. В то же время отсутствие сильных меж- и внутримолекулярных взаимодействий в изученном методом РСА кристалле FeGm3(B4-biPh)2 и наличие в его молекуле ординарных связей B–C и C–C обусловливают относительно свободное вращение бифенильных апикальных заместителей при клатрохелатном остове. Как видно из рис. 1 (снизу слева), в молекуле FeGm3(B4-biPh)2 типа А оба фениленовых фрагмента при сшивающих атомах бора практически параллельны друг другу, а также одному из трех реберных хелатирующих фрагментов. Напротив, в молекуле FeGm3(B4-biPh)2 типа B средние плоскости первых упомянутых ароматических фрагментов образуют двугранный угол 38.1°, и только один из этих фениленовых остатков практически параллелен соответствующему глиоксиматному фрагменту этой молекулы (рис. 1, снизу справа). Двугранные углы между средними плоскостями фениленовых и фенильных фрагментов их бифенильных апикальных заместителей при сшивающих атомах бора близки к 40° (42.8(1)° и 44.1(1)° в молекуле FeGm3(B4-biPh)2 типа A и 36.4(1)° и 44.8° в молекуле типа B). Двугранные углы между средними плоскостями терминальных фенильных групп одного и того же макробициклического лиганда составляют 117.9° и 29.8° в клатрохелатных молекулах FeGm3(B4-biPh)2 типов A и B соответственно (рис. 2).
Рис. 1.
Общий фронтальный вид двух независимых клатрохелатных молекул FeGm3(B4-biPh)2 (вверху) в представлении атомов как тепловых эллипсоидов (p = 50%) и вид этих молекул вдоль молекулярных C3-псевдоосей симметрии (скелетная модель, внизу).
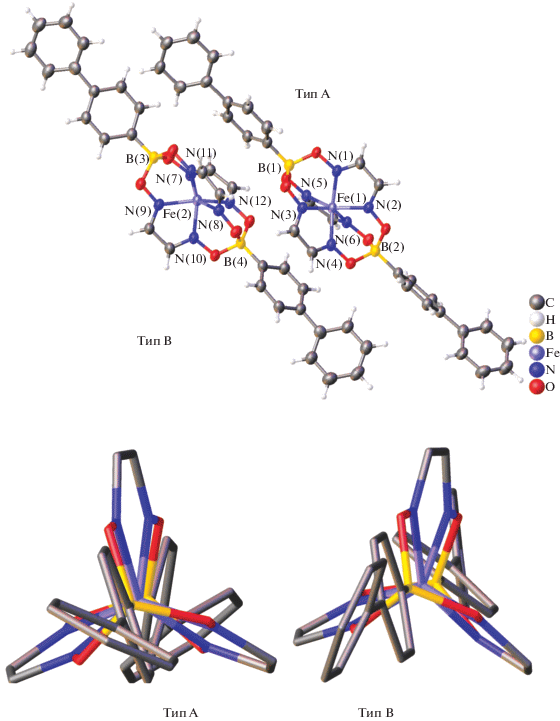
Таблица 1.
Основные геометрические параметры клеточных остовов двух независимых клатрохелатных молекул в кристалле FeGm3(B4-biPh)2
| Параметр | Тип A | Тип B |
|---|---|---|
| Fe–N, Å | 1.907(3)–1.913(3) ср. 1.909 |
1.903(3)–1.916(2) ср. 1.908 |
| B–O, Å | 1.491(4)–1.499(4) ср. 1.496 |
1.487(5)–1.505(3) ср. 1.493 |
| N–O, Å | 1.366(4)–1.377(4) ср. 1.372 |
1.369(4)–1.377(4) ср. 1.374 |
| C=N, Å | 1.296(5)–1.306(5) ср. 1.302 |
1.299(4)–1.302(5) ср. 1.300 |
| C–C, Å | 1.418(6)–1.419(6) ср. 1.418 |
1.418(6)–1.427(5) ср. 1.421 |
| N=C–C=N, град | 5.7(5)–7.3(5) ср. 6.4 |
6.4(5)–7.0(4) ср. 6.7 |
| φ, град | 18.5 | 18.0 |
| α, град | 78.6 | 78.4 |
| h, Å | 2.37 | 2.37 |
Рис. 2.
Различие в конформациях двух независимых клатрохелатных молекул FeGm3(B4-biPh)2 типов A и B, визуализированное путем наложения их неводородных скелетов/атомов (атомы водорода опущены для ясности).
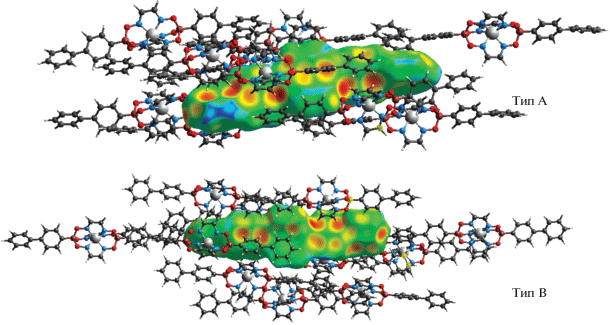
Кристаллическая упаковка этих двух типов клатрохелатных молекул определяется в основном их гидрофобными взаимодействиями, как это было оценено с использованием пакета программ CrystalExplorer17 [29], основываясь на их молекулярных поверхностях Хиршфельда [30], которые показаны на рис. 3. Вклады взаимодействий H…H, H…C и C…C в эти поверхности Хиршфельда составляют соответственно 44.6, 26.8 и 4.8% для молекулы FeGm3(B4-biPh)2 типа A и 42.9, 30.8 и 3.8% для молекулы типа B. Обнаружено, что среди других межмолекулярных взаимодействий важную роль играют водородные связи H…O и H…N. Их вклад в поверхности Хиршфельда составляет 13.7–14.7 и 4.3–4.5% соответственно, в то время как вклад других межмолекулярных контактов не превышает 2.1%. Как видно из рис. 3, водородные связи C–H…O являются наиболее короткими взаимодействиями этого типа. Красные пятна на вышеуказанных молекулярных поверхностях Хиршфельда, которые нанесены при помощи de (точки поверхностей, наиболее близких к внешним атомам), относятся в основном к этим водородным связям, в то время как оранжевые пятна указывают на относительно короткие C–H…π-взаимодействия. Следует отметить, что увеличение числа ароматических гидрофобных фрагментов на молекулу клатрохелата в случае комплекса FeGm3(B4-biPh)2 приводит к более плотной упаковке кристалла. Это свидетельствует о слабых супрамолекулярных взаимодействиях между его клеточным остовом и апикальными полиароматическими заместителями соседних клатрохелатных молекул. В кристаллах других борсодержащих трис-глиоксиматов железа(II), изученных ранее методом РСА, было обнаружено тесное межмолекулярное связывание двух и более макробициклических молекул посредством их реберных глиоксиматных хелатирующих фрагментов [9, 18, 19, 28].
ЗАКЛЮЧЕНИЕ
Оптимизированы условия палладий-катализируемой реакции Сузуки–Мияура дииодоклатрохелатного трис-глиоксимата железа(II) с реакционноспособными атомами галогена в апикальных заместителях путем выбора подходящего палладийсодержащего катализатора, температуры и растворителя. Это позволило получить с умеренным выходом целевой клатрохелатный продукт с расширенными полиароматическими апикальными заместителями. Проведенная модельная реакция C–C-кросс-сочетания может быть распространена на синтез других апикально-функционализированных клеточных комплексов этого типа с терминальными реакционноспособными/полиароматическими группами.
Список литературы
Voloshin Y.Z., Belaya I.G., Krämer R. Cage metal complexes: clathrochelates revisited. Springer, 2017. [Волошин Я.З., Белая И.Г., Кремер Р. Клеточные комплексы металлов: клатрохелаты возвращаются. М.: Граница, 2019].
Voloshin Y.Z., Kostromina N.A., Kraemer R. // Clathrochelates: synthesis structure and properties. Elsevier, 2002.
Lebed E.G., Belov A.S., Dolganov A.V. et al. // Inorg. Chem. Commun. 2013. V. 33. P. 57.
Wise M.D., Ruggi A., Pascu M. et al. // Chem. Sci. 2013. V. 4. P. 1658.
Pascu M., Marmier M., Schouwey C. et al. // Chem. Eur. J. 2014. V. 20. P. 5592.
Zhang Y.Y., Lin Y.J., Jin G.X. // Chem. Commun. 2014. V. 50. P. 2327.
Ardavan A., Bowen A.M., Fernandez A. et al. // Quant Inform. 2015. V. 1. P. 15012.
Wise M.D., Holstein J.J., Pattison P. et al. // Chem. Sci. 2015. V. 6. P. 1004.
Jansze S., Cecot G., Wise M.D. et al. // J. Am. Chem. Soc. 2016. V. 138. P. 2046.
Janze S.M., Wise M.D., Vologzhanina A.V. et al. // Chem. Sci. 2017. V. 8. P. 1901.
Cecot G., Marmier M., Germia S. et al. // J. Am. Chem. Soc. 2017. V. 139. P. 8371.
Barder T.E., Walker S.D., Martinelli J.R., Buchwald S.L. // J. Am. Chem. Soc. 2005. V. 127. P. 4685.
Alonso F., Beletskaya I.P., Yus M. // Tetrahedron. 2008. V. 64. P. 3047.
Schneider F., Stolle A., Ondruschka B., Hopf H. // Org. Process Res. Dev. 2009. V. 13. P. 44.
Varzatskii O.A., Denisenko I.N., Belov A.S. et al. // Inorg. Chem. Commun. 2014. V. 44. P. 134.
Denisenko I.N., Varzatskii O.A., Selin R.A. et al. // RSC Adv. 2018. V. 8. P. 13578.
Park D.J., Stern A.G., Wilier R.L. // Synth. Commun. 1990. V. 20. P. 2901.
Zelinskii G.E., Chuprin A.S., Belov A.S. et al. // Inorg. Chim. Acta. 2016. V. 453. P. 210.
Zelinskii G.E., Belov A.S., Vologzhanina A.V. et al. // Polyhedron 2019. V. 160. P. 108.
Wojdyr M. // J. Appl. Crystallogr. 2010. V. 43. P. 1126.
Battye T.G.G., Kontogiannis L., Johnson O. et al. // Acta Crystallogr., Sect D 2011. V. 67. P. 271.
Evans Ph. // Acta Crystallogr., Sect D. 2006. V. 62. P. 72.
Sheldrick G.M. // Acta Crystallogr. 2015. V. A71. P. 3.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. P. 339.
Lee S.J., Gray K.C., Paek J.S., Burke M.D. // J. Am. Chem. Soc. 2008. V. 130. P. 466.
Fyfe J.W.B., Valverde E., Seath C.P. et al. // Chem. Eur. J. 2015. V. 21. P. 8951.
Barder T.E., Walker S.D., Martinelli J.R., Buchwald S.L. // J. Am. Chem. Soc. 2005. V. 127. P. 4685.
Voloshin Y.Z., Lebedev A.Y., Novikov V.V. et al. // Inorg. Chim. Acta. 2013. V. 399. P. 67.
Wolff S.K., Grimwood J.J., McKinnon J.J. et al. // CrystalExplorer (University of Western Australia, 2012).
Spackman M.A., Jayatilaka D. // Cryst. Eng. Comm. 2009. V. 11. P. 19.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии