

Журнал неорганической химии, 2020, T. 65, № 12, стр. 1710-1720
Оптимизация катализаторов окисления CO для термокаталитических и полупроводниковых газовых сенсоров
А. А. Васильев a, *, А. С. Лагутин a, Ш. Ш. Набиев a
a Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Курчатова, 1, Россия
* E-mail: a-a-vasiliev@yandex.ru
Поступила в редакцию 26.06.2020
После доработки 10.07.2020
Принята к публикации 20.07.2020
Аннотация
Изучена кинетика процессов каталитического окисления CO на поверхности допированных благородными металлами металлооксидных катализаторов, используемых в качестве газочувствительных материалов сенсоров. Большинство оксидных материалов (SnO2, Al2O3, цеолит и др.), декорированных палладием и родием, имеют кинетическую особенность, приводящую к неоднозначному отклику сенсора. Исключением является CeO2/ZrO2, допированный палладием; скорость окисления на нем однозначно зависит от концентрации CO. Процессы окисления CO на инертных и химически активных носителях описываются в рамках единой кинетической модели. Существуют два режима работы катализатора: ниже и выше некоторого концентрационного предела, в каждом из режимов реакция имеет первый кинетический порядок по CO, а суперпозиция этих процессов может приводить к эффективному “минус первому” порядку реакции окисления. Переход катализатора из одного состояния в другое носит характер “фазового перехода”, протекающего при изменении концентрации CO, а не температуры катализатора. Кинетика окисления в каждом состоянии описывается небольшим набором параметров, характерных для каждого из катализаторов; энергия активации для каждого катализатора одинакова при всех значениях концентрации CO и кислорода.
ВВЕДЕНИЕ
Реакция каталитического окисления СО очень важна в силу необходимости нейтрализации вредных компонентов газовых выхлопов двигателей внутреннего сгорания, очистки воздуха, разработки химических сенсоров. Не меньше ее значимость для фундаментальных исследований, поскольку с помощью этой реакции можно изучать процессы окисления поверхности и активные центры катализаторов. Начиная с классической работы Ленгмюра [1], она широко применяется для изучения фундаментальных аспектов функционирования гетерогенных катализаторов, процессов окисления поверхности и исследования активных центров катализаторов.
Реакцию окисления СО на металлах платиновой группы обычно рассматривают в рамках классических механизмов Ленгмюра–Хиншельвуда [1, 2] и Или–Ридила [3], согласно которым данная реакция протекает при взаимодействии атомарного кислорода, который активируется на поверхности металла в диссоциативной форме (состояние I), и молекулы СО из газовой фазы (механизм Или–Ридила) либо хемосорбированной на поверхности (механизм Ленгмюра–Хиншельвуда). При этом реализация механизма Или–Ридила считается менее вероятной и подвергается сомнению частью исследователей [4, 5]. Известно, что молекулы СО склонны к сильной адсорбции на поверхности металлов платиновой группы, образуя карбонильную связь по донорно-акцепторному механизму. Высокая теплота адсорбции СО препятствует его окислению при низких температурах. Этот процесс является основой так называемого отравления катализатора хемосорбированным оксидом углерода (состояние II). Как будет показано далее, механизм реакции окисления при высокой и низкой концентрации CO существенно отличается, и переход катализатора из состояния I в состояние II и обратно происходит при изменении соотношения концентрации кислорода и CO в газовой фазе, причем этот переход происходит скачкообразно и носит характер фазового перехода.
В работах Тернера, Сейлса и Мэпла [6, 7] был предложен модифицированный механизм Ленгмюра–Хиншельвуда, в котором учитывалось воздействие реакционной среды на поверхность металла, приводящее к переходу поверхности в оксидное состояние.
Для описания каталитического окисления СО на поверхности оксидов металлов помимо механизма Ленгмюра–Хиншельвуда применяют механизм Марса и ван Кревелена [8]. Этот механизм учитывает участие решеточного кислорода оксида в окислении адсорбированных молекул СО, в результате которого образуются вакансии, восполняющиеся за счет диссоциативно сорбирующегося кислорода газовой фазы. Использование оксидов, в которых легко образуются дефекты типа вакансий за счет переменной степени окисления и на поверхности которых возможно образование слабосвязанного кислорода, позволяет снизить энергию активации реакции окисления. Таким образом, использование решеточного кислорода открывает новый реакционный путь окисления СО. Этот механизм реакции наблюдается в нашем случае при использовании таких носителей, как диоксид олова и особенно CeO2/ZrO2, в которых возможно изменение степени окисления олова и церия соответственно.
Кинетика гетерогенной реакции окисления СО характеризуется сложным динамическим поведением, связанным с возникновением критических явлений, эффекта гистерезиса и хаотического либо автоколебательного протекания каталитической реакции [9–12].
Явление гистерезиса, которое заключается в зависимости свойств системы не только от воздействующих на нее внешних параметров, но и от предыстории данного состояния системы, распространено в гетерогенном катализе [13–17]. Это явление может быть связано с локальным перегревом активных центров за счет экзотермичности протекающих на них реакций с множеством стационарных состояний катализатора и/или нелинейностью механизма каталитической реакции [18–21]. В работе [21] детально исследован концентрационный гистерезис в реакции низкотемпературного окисления метана на дисперсном катализаторе Pt/γ-Al2O3, а в [22] – каталитические свойства катализаторов Pd/TiO2 в реакции окисления CO. Авторы работ [21, 22] предложили два возможных механизма такого гистерезиса, не исключая также и их комбинацию. Первый – это изменение электронного состояния активного компонента катализатора при изменении окислительного потенциала реакционной среды, например, за счет его перехода в металлическое состояние в восстановительных условиях и обратного окисления при увеличении доли кислорода в системе. Второй – изменение покрытия поверхности активного компонента тем или иным реагентом в зависимости от соотношения их парциальных давлений и/или равновесия адсорбции–десорбции [23, 24]. Применительно к окислению метана это может означать отравление платины прочно связанным кислородом при избытке последнего для малоактивного состояния и активированную кислородом адсорбцию метана для активного состояния платинового катализатора подобно модели, предложенной для окисления CO [25]. Скачкообразное повышение активности платиновых катализаторов (зажигание) в обогащенных метаном смесях связано, по мнению авторов [26], с переходом от слабо сорбирующей метан поверхности платины при ее покрытии атомно-адсорбированным кислородом к поверхности, покрытой промежуточными продуктами парциального окисления метана – CO, H и легче хемосорбирующей метан.
В работе [22] подробно исследован температурный гистерезис в реакции окисления СО на катализаторах Pt/γ-Al2O3 и PdO/γ-Al2O3. Это явление может быть результатом того, что экспериментальные точки, полученные при снижении температуры, определяются при существенном перегреве частиц активного компонента по сравнению с температурой газовой смеси вследствие недостаточно высокой теплопроводности носителя [22]. В этом случае образцы катализаторов с различным размером нанесенного активного компонента, характеризующиеся различной площадью контакта с носителем, должны различаться и по величине наблюдаемого гистерезиса. Однако корреляций между размером нанесенных частиц платины и величиной гистерезиса в данной работе экспериментально не обнаружено. К существенному снижению величины наблюдаемого гистерезиса (вплоть до исчезновения) приводило добавление к активному компоненту различных промоторов (Се, Мn).
В работе [27] проведен поиск кинетических параметров гетерогенной каталитической реакции окисления СО на платиновом катализаторе с использованием двух механизмов протекания реакции: взаимодействием с атомарным кислородом и взаимодействием с молекулярным кислородом. Установлено, что механизм взаимодействия СО с молекулярным кислородом лучше согласуется с данными эксперимента.
Возможной причиной возникновения гистерезиса в гетерогенных каталитических реакциях при окислении углеводородов на палладиевых катализаторах является закоксовывание поверхности при низких температурах [28]. Однако, как подчеркнуто в обзоре [29], объяснения происхождения гистерезиса в гетерогенных каталитических реакциях очень часто противоречат друг другу. Разброс результатов экспериментов велик, а спектр изученных катализаторов и реакций, используемых для изучения эффекта гистерезиса, минимален.
Цель настоящей работы – исследование кинетики окисления CO на поверхности допированных (декорированных) благородными металлами катализаторов для оптимального подбора катализаторов газовых сенсоров полупроводникового и термокаталитического типов. Предложенная в работе модель позволяет описать всю совокупность кинетических данных по гетерогенно-каталитическому окислению оксида углерода всех исследованных катализаторов в рамках единого подхода независимо от их химического состава и структуры.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Нанодисперсный порошок диоксида олова как основу для катализатора Pd-SnO2 получали модифицированным золь-гель методом, описанным в работе [30]. В качестве исходного реагента использовали сульфат олова(II), который в водной среде окисляли с помощью перекиси водорода. Преимуществом этого метода синтеза, который проводится при близких к нейтральным значениях pH раствора, является то, что оловянная кислота образуется сразу в виде суспензии изолированных частиц, не требующих последующего размола. Образовавшийся порошок оловянной кислоты отделяли центрифугированием и многократно промывали деионизованной водой до исчезновения реакции с хлоридом бария. Порошок оловянной кислоты высушивали при 150°C и отжигали в течение 1 ч при 600°C на воздухе для удаления воды. В результате такой обработки получали порошок оксида олова. Средний размер частиц SnO2 составлял ~20 нм, а определенная газосорбционным методом (изотерма BET (Брунауэра–Эммета–Теллера)) удельная поверхность была равна 56 м2/г. Нанесение палладия на поверхность носителя катализатора уменьшало удельную поверхность порошка до 48 м2/г. Палладий наносили следующим образом. Порошок диоксида олова диспергировали в воде, затем в воду добавляли расчетное количество хлористого палладия. Хлористый палладий восстанавливался формиатом натрия, при этом частицы порошка диоксида олова являлись центрами кристаллизации палладия и покрывались кластерами палладия размером 2–3 нм. Полученный порошок отмывали в воде, фильтровали, просушивали и отжигали при температуре 600°C.
В работе также исследовали другие носители катализатора: оксид алюминия, стабилизированный оксидом лантана, оксид церия-циркония, синтетический цеолит ZSM-5. При этом использовали коммерческие носители, применяемые в автомобильных катализаторах дожигания оксида углерода.
Состав и основные характеристики исследованных катализаторов приведены в табл. 1. Все катализаторы, за исключением палладиевого катализатора на диоксиде олова, были получены пропиткой соответствующего порошка водным раствором Pd(NH3)4(NO3)2 с последующей сушкой и отжигом при температуре 600°С.
Таблица 1.
Состав изученных катализаторов
| Катализатор | Состав | Удельная поверхность, м2/г |
Дисперсность, или характерный размер частиц |
|---|---|---|---|
| Pd-SnO2 | Палладий (3 вес. %), нанесенный на порошок диоксида олова | 52 | Pd – 2 нм SnO2 – 20 нм |
| Pd-Al2O3 | Палладий (4 вес. %), нанесенный на порошок оксида алюминия с оксидом лантана (2 вес. %, W.R.Grace Co.) | 176 | Pd – 2 нм Al2O3/La2O3 – 7 нм |
| Pd-Al2O3-mix | Палладий (4 вес. %), нанесенный на порошок Al2O3/La2O3 (2 вес. % La2O3). Катализатор разбавлен на 99% чистым порошком Al2O3/La2O3 (2 вес. % La2O3) | 176 | Pd – 2 нм Al2O3/La2O3 – 7 нм |
| Напыленный Pd | Пленка Pd, напыленная на палочку из алюмооксидной керамики | – | Толщина пленки 50 нм |
| Pd-ZSM | Палладий (0.25 вес. %), нанесенный на цеолит ZSM-5 | ~ 550 | – |
| Pd-CeO2/ZrO2 | Палладий (4 вес. %), нанесенный на носитель CeO2/ZrO2 (70/30 мол. %) производства Degussa GmbH | 87 м2/г | Размер частиц носителя ~10 нм, кластеров Pd ~ 2 нм |
| Rh-ZSM | Родий (0.25 вес. %) в цеолите ZSM-5 | ~ 550 | – |
Для сравнения исследовали также каталитические характеристики палладия, нанесенного магнетронным распылением металла на держатель из керамического оксида алюминия. Толщина покрытия составляла 50 нм.
После приготовления все катализаторы помещали в агатовую ступку, в ней полученный порошок смешивали с водой для получения однородного пастообразного материала. Эту пасту наносили на держатель (99.6% α-Al2O3) размером 2 × × 22 мм, просушивали при температуре 150°С и прокаливали при 730 ± 10°С в течение 15 мин. Толщина слоя катализатора на держателе составляла ~10 мкм.
Исследования кинетики реакции окисления СО проводили в проточном химическом реакторе, который состоял из кварцевой трубки длиной l = 22 мм с внутренним радиусом r = 5 мм. Держатель с нанесенным катализатором был расположен по оси реактора, а сам реактор – внутри печки, позволявшей контролировать и стабилизировать его температуру и нагревать газ до поступления в реактор.
Геометрия реактора позволяла, по возможности, избежать влияния осевой и радиальной диффузии в газе на точность определения при комнатной температуре констант скоростей гетерогенных химических реакций при максимальной скорости газового потока 20 см3/мин (v ≈ 0.4 см/с).
Очень важным условием является степень изотермичности измерений, т.е. выяснение того, в какой мере измеряемая температура стенки реактора соответствует температуре катализатора, не перегревается ли он значительно за счет тепла, выделяющегося в ходе экзотермической реакции окисления CO. Это необходимо для того, чтобы исключить влияние неконтролируемого разогрева на так называемое “зажигание” катализатора. В проведенных экспериментах использовали весьма малые концентрации CO (≤0.8 об. %) и низкие скорости газового потока (не более 20 см3/мин). В этих условиях максимальная мощность, выделяющаяся за счет химической реакции окисления CO, составляет
где Qtotal = 20 см3/мин – суммарный расход газа в реакторе, C(CO) = 0.8% – концентрация оксида углерода на входе в реактор, ΔH ∼ 400 кДж/моль – тепловой эффект реакции (изменение энтальпии).Эта мощность может нагреть каталитический элемент до температуры, которая была оценена исходя их следующих соображений. Экспериментально было установлено [31], что газовый сенсор, имеющий площадь поверхности 2 мм2, можно нагреть до температуры 400°С электрической мощностью 500 мВт. Потери тепла при атмосферном давлении практически линейно зависят от разности температур нагретого элемента и стенки реактора и от площади поверхности в интервале 0–400°С. Поэтому держатель с катализатором, имеющий площадь поверхности ~40 мм2, не может быть нагрет более чем на 2–4°С выше температуры окружающего газа и стенки реактора. Эта оценка была подтверждена экспериментально прямым измерением температуры газа на выходе реактора с помощью термопары.
Для регулирования расхода газов использовали контроллеры компании MKS. Поскольку заводская точность этих регуляторов была недостаточной, они были дополнительно откалиброваны и соответствующие поправки учтены при измерениях.
Для измерения концентрации CO на входе и выходе реактора использовали прибор компании Rosemount, измерение концентрации в котором проводили методом недисперсионного поглощения ИК-излучения. Точность измерения составляла ±10 м.д. в интервале концентраций 0–5000 м.д. и ±20 м.д. в интервале концентраций 0–10 000 м.д.
МОДЕЛИРОВАНИЕ КРИВЫХ “ЗАЖИГАНИЯ” КАТАЛИЗАТОРОВ
Эксперименты, проведенные в настоящей работе, были выполнены, в отличие от большинства предыдущих исследований, во-первых, в изотермических условиях, которые исключали самопроизвольный разогрев катализатора в результате протекания на его поверхности экзотермической реакции окисления оксида углерода, а во-вторых, в условиях, которые исключали влияние диффузии на кинетику исследуемой реакции. Последнее обстоятельство исключало проскок реагентов, и степень превращения достигала 100% при достаточно мягких условиях проведения эксперимента. Это позволяло тщательно определить форму кривой, особенно вблизи точки так называемого “зажигания” катализатора.
В связи с изотермическим характером эксперимента нас не устраивали кинетические модели, в которых “зажигание” катализатора объяснялось кризисом теплообмена и его самопроизвольным разогревом при повышении температуры. В этих условиях такая положительная обратная связь (ускорение реакции при повышении температуры, которое, в свою очередь, разогревает катализатор) не возникала из-за того, что были использованы низкие концентрации и низкий расход газа.
Для применения катализаторов окисления CO в качестве чувствительного материала термокаталитических газовых сенсоров, в которых концентрация газа определяется по теплоте его каталитического окисления, очень важной является однозначность зависимости скорости окисления аналита от концентрации. При применении катализаторов с точкой “зажигания” такое требование однозначности не выполняется: одна и та же скорость реакции наблюдается при двух и даже трех разных значениях концентрации.
Прямые кинетические измерения показывают, что при понижении концентрации ниже некоторого значения происходит скачкообразное увеличение скорости реакции, при этом выше и ниже этого фазового перехода реакция сохраняет первый кинетический порядок по концентрации CO, но имеет место точка разрыва кривой (фазовый переход).
При моделировании кривых “зажигания” мы исходили из этого наблюдения, считали, что существует два разных режима работы катализатора – состояние I и состояние II, которые соответственно ниже и выше некоторого концентрационного предела Cinv. Когда концентрация на поверхности катализатора достигает этого значения, скорость реакции изменяется скачком. При этом в обоих состояниях реакция имеет первый порядок по CO, но константа скорости отличается примерно в 4–5 раз.
Концентрация CO из-за его окисления уменьшается при движении газа вдоль катализатора и в некоторой точке достигает порогового значения, при котором константа скорости реакции скачкообразно возрастает. Этот эффект был учтен при построении модели.
Положение этой границы на поверхности катализатора движется при изменении температуры катализатора. Реакция окисления CO в обоих состояниях катализатора подчиняется кинетическому закону первого порядка (рис. 1). Время, необходимое для инверсии катализатора в активное состояние, много меньше времени, необходимого для обратного превращения (несколько секунд по сравнению с ~2 ч, если концентрация CO близка к точке зажигания). Это различие времени инверсии катализатора в прямом и обратном состоянии приводит к гистерезису кривых зависимости скорости превращения от температуры при ее увеличении и уменьшении.
Рис. 1.
Окисление CO на катализаторе Pd-SnO2 при начальной концентрации CO 2310 м.д. и концентрации кислорода 16% для трех температур реактора.
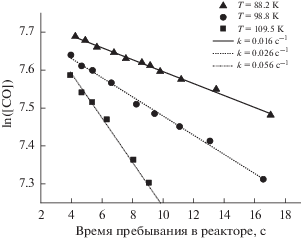
Изменение концентрации газа со временем описывается обычным уравнением первого порядка:
где С0 – начальная концентрация газа, k – константа скорости реакции окисления, С – концентрация газа в момент времени τ. В условиях газового потока τ = l/v, где l – длина катализатора, v – скорость потока газа.Константа скорости реакции подчиняется закону Аррениуса:
где k0 – предэкспоненциальный множитель, Eact – энергия активации реакции, T – температура, при которой происходит химическая реакция, R – универсальная газовая постоянная.Время пребывания газа τ при температуре реактора (r – радиус реактора) связано с линейной скоростью газового потока v и комнатной температурой следующим соотношением:
(4)
$\tau = \left( {\frac{l}{{{{{v}}_{0}}}}} \right)\left( {\frac{{{{T}_{0}}}}{T}} \right) = \left( {\frac{{l\pi {{r}^{2}}}}{Q}} \right)\left( {\frac{{{{T}_{0}}}}{T}} \right).$В условиях газового потока τ = l/v, где l – длина катализатора, v – скорость потока газа, Т0 = 300 K, Q – расход газовой смеси, 20 см3/мин.
Окончательно уравнение, описывающее процесс окисления CO на поверхности катализатора, имеет вид:
(5)
$\frac{C}{{{{C}_{0}}}} = \exp \left( { - {{k}_{0}}\left( {\frac{{l\pi {{r}^{2}}}}{Q}} \right)\left( {\frac{{{{T}_{0}}}}{T}} \right)\exp \left( { - \frac{{{{E}_{{{\text{act}}}}}}}{{RT}}} \right)} \right).$В расчетной модели использовали следующие параметры: Eact – энергия активации в состоянии I, предэкспонент k10, константа скорости реакции k2 в фазе 2 и Cinv – критическая концентрация для перехода из фазы 1 в фазу 2. Все эти параметры, указанные в табл. 2 для всех исследованных катализаторов, определяли численным моделированием с использованием выражения (5), количество элементов разбиения по длине катализаторов составляло от 100 до 150.
Таблица 2.
Оптимальные расчетные параметры модели
| Катализатор | Eact, кал/моль | k10, с–1 | k2, с–1 | Cinv, м.д. |
|---|---|---|---|---|
| Pd-SnO2 | 21 | 5.2 × 1010 | 2.9 | 1450 |
| Pd-Al2O3 | 30 | 5.0 × 1015 | 2.6 | 1250 |
| Pd-Al2O3-mix | 30 | 5.6 × 1013 | 3.0 | 2020 |
| Напыленный Pd | 30 | 0.16 × 1014 | 4.0 | 5400 |
| Pd-ZSM | 30 | 1.25 × 1012 | 4.0 | 6670 |
| Pd-CeO2/ZrO2 | 19 | 0.37 × 1011 | – | – |
| Rh-ZSM | 45 | 5.5 × 1018 | 0.88 | 7300 |
Нами проведено три типа экспериментов. (1) Измерены кривые “зажигания” катализаторов разных типов, т.е. зависимости формы и параметров, описывающих кривые, от концентрации реагентов и активной фазы катализатора. В этих экспериментах определены энергии активации реакции для всех катализаторов. (2) Измерена зависимость концентрации от времени пребывания и построены соответствующие кривые, демонстрирующие, что реакция подчиняется кинетическому закону первого порядка при всех значениях концентраций и температур. Кроме того, при измерении констант скорости реакции при разных температурах было показано, что энергия активации, полученная из кривых “зажигания”, соответствует значению, определенному из прямого измерения констант скоростей реакций. (3) Измерены зависимости концентрации на выходе из реактора от концентрации CO на входе. Цель этих экспериментов – показать, что резкое увеличение константы скорости реакции, наблюдаемое на кривых “зажигания” катализатора, связано не с кризисом теплообмена, как считается обычно, а является результатом резкого увеличения константы скорости реакции при уменьшении (!) концентрации CO в газе.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Катализатор Pd-SnO2. Результаты измерений степени превращения в реакции окисления CO представлены на рис. 2 для двух концентраций кислорода в газе. Анализ этих кривых показывает, что каждая из них не может быть описана одним кинетическим уравнением, так как резкое изменение скорости химической реакции (так называемое “зажигание” катализатора) происходит очень быстро и практически при постоянной температуре без разогрева катализатора. В наших условиях разогрев катализатора за счет теплоты экзотермической реакции окисления CO отсутствует. Мы предположили, что этот эффект связан с известным из литературы отравлением палладиевого катализатора хемосорбированным CO [32] при более низкой температуре и прекращением этого отравления при более высокой температуре. Катализатор, таким образом, может находиться в одном из двух состояний: он может быть покрыт хемосорбированным оксидом углерода (отравление) или хемосорбированным кислородом (отсутствие отравления). В обоих случаях реакция первого порядка протекает по механизму Или–Ридила.
Рис. 2.
Зависимость степени превращения в реакции окисления CO от температуры для катализатора Pd-SnO2. Треугольники – начальная концентрация 2300 м.д., концентрация кислорода – 16%; кружки – начальная концентрация 2140 м.д., концентрация кислорода – 2%. На вставке – окисление CO при начальной концентрации 2310 м.д. и концентрации кислорода 16%. Температура реактора – 382.5 K.
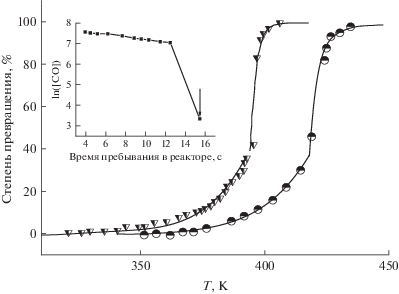
Вставка на рис. 2 иллюстрирует этот эффект. Скорость потока газа в реакторе постепенно уменьшается, поэтому увеличивается время пребывания газа в нем. До некоторой пороговой скорости реакция имеет первый кинетический порядок по концентрации CO (линейная часть графика). Как только концентрация CO в зоне катализатора понижается ниже некоторого порогового значения, процесс каталитического окисления CO становится более эффективным, т.е. при увеличении времени пребывания в реакторе больше некоторого порогового значения концентрация CO на выходе из реактора скачкообразно уменьшается более чем на порядок. Соответствующая точка на графике отмечена стрелкой. При этом, очевидно, что данный эффект нельзя объяснить изменением температуры катализатора, т.е. “зажигание” (light-off) – это чисто кинетический эффект, не связанный с кризисом теплообмена, а связанный с изменением механизма каталитической реакции.
Кинетические кривые, построенные в результате эксперимента по обычной для кинетики схеме (измерение концентрации на выходе из реактора как функции времени пребывания в реакторе), показывают, что кинетика реакции подчиняется уравнению первого порядка по оксиду углерода (рис. 1). Эти измерения проведены при температуре ниже “температуры зажигания”, показанной на рис. 2, а кинетические параметры (энергия активации реакции, предэкспоненциальный множитель и константа скорости реакции в активном состоянии катализатора), которые были получены при моделировании кривой, представлены в табл. 2. Мы сравнили величины энергии активации реакции окисления CO, полученные в результате моделирования кривой “зажигания” катализатора и из графиков, представленных на рис. 1. Эти два независимых метода дали близкие значения энергии активации: 22 ± 2 и 17 ± 3 ккал/моль соответственно, что является подтверждением принятой нами кинетической модели. Как и во всех других случаях, которые будут рассмотрены ниже, мы наблюдали явление гистерезиса. Кривая (рис. 2) при понижении температуры лежит левее кривой при повышении температуры, это связано с тем, что окисленное (активное) состояние катализатора сохраняется в течение длительного времени.
Необходимо отметить, что энергия активации реакции окисления CO при температуре ниже температуры “зажигания” не зависит от концентрации кислорода. При этом предэкспоненциальный фактор пропорционален концентрации кислорода, что подтверждает справедливость механизма Или–Ридила в этой реакции.
Предложенный механизм объясняет все особенности поведения катализаторов, содержащих палладий, и, по крайней мере, катализаторов, содержащих родий.
Рассмотрим поведение других катализаторов окисления оксида углерода.
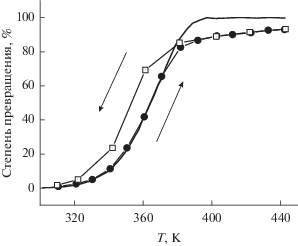
Катализатор Pd-Al2O3. В целом поведение катализатора Pd-Al2O3 аналогично поведению катализатора Pd-SnO2. Мы наблюдали “зажигание” катализатора (рис. 3), которое также не было связано с кризисом теплообмена на каталитической поверхности и соответствовало резкому переходу катализатора в более активное состояние.
Рис. 3.
Зависимости степени превращения в реакции окисления CO от температуры при начальной концентрации CO 2250 м.д. и концентрации кислорода – 15%. Кружки – катализатор Pd-Al2O3. Треугольники – катализатор Pd-Al2O3-mix. На вставке – концентрация CO на выходе из реактора как функция концентрации на входе для катализатора Pd-Al2O3. Температура реактора – 389.3 K, концентрация кислорода – 15%.
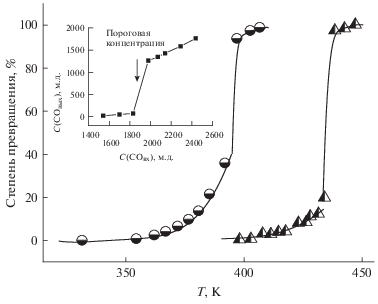
При всех исследованных концентрациях оксида углерода энергия активации процесса ниже точки зажигания равна 30 ± 2 ккал/моль. Это значение заметно и воспроизводимо выше значения энергии активации окисления CO, найденного для палладия, нанесенного на диоксид олова.
Предэкспоненциальный фактор в выражении для константы скорости реакции окисления CO при постоянной концентрации оксида углерода пропорционален концентрации кислорода в газовой фазе, тогда как константа скорости реакции в “активном” состоянии катализатора практически не зависит от концентрации кислорода. Точка зажигания катализатора лежит на ~10–20 град выше, чем температура зажигания катализатора на основе диоксида олова.
Разбавление этого катализатора в 100 раз инертным материалом (чистым носителем Al2O3/La2O3) не меняет энергию активации, а только уменьшает предэкспоненциальный фактор также примерно в 100 раз.
Чтобы еще раз подтвердить нашу кинетическую модель окисления CO на палладийсодержащем катализаторе, мы измерили зависимость концентрации оксида углерода на выходе из реактора от его концентрации на входе при температуре ниже температуры “зажигания”. При уменьшении концентрации ниже порогового значения константа скорости реакции в катализаторе Pd-Al2O3 резко возрастает, что приводит к резкому уменьшению концентрации CO на выходе из реактора (вставка на рис. 3). Этот скачок константы скорости носит концентрационный, а не тепловой характер, так как он происходит при уменьшении концентрации, а не при ее увеличении. Таким образом, и здесь имеет место резкий переход катализатора из состояния с низкой активностью в состояние с высокой активностью, причем это изменение никак не связано с изменением температуры катализатора или газа.
Пленка палладия, напыленная на поверхности оксида алюминия, была исследована с целью исключить влияние материала носителя на свойства палладиевого катализатора.
Поведение катализатора очень сильно зависит от его предварительной обработки. Свежеприготовленный образец имеет очень низкую каталитическую активность. Для увеличения активности необходима обработка катализатора при температуре 300°С в среде, содержащей 0.8% CO и 2% кислорода.
Кривые зависимости концентрации оксида углерода на выходе из реактора от температуры имеют достаточно хорошо выраженную точку зажигания и могут быть описаны тем же кинетическим уравнением первого порядка, что и предыдущие графики. Это описание справедливо при всех исследованных концентрациях кислорода в реакционной смеси. Энергия активации в низкоактивном состоянии и здесь составляет 30 ± 2 ккал/моль, константа скорости в высокоактивном состоянии равна 2 ± 1 с–1.
Синтетический цеолит ZSM-5 – очень популярный объект кинетических исследований, поскольку существует предположение, что этот цеолит с внедренными ионами меди может служить заменой катализаторов с благородными металлами в качестве автомобильных катализаторов дожигания CO [33]. Катализатор Pd-ZSM представляет интерес, так как при низкой концентрации палладия ионы расположены в матрице цеолита изолированно, не образуя кластеров.
Кинетика окисления оксида углерода на этом катализаторе описывается тем же кинетическим уравнением первого порядка, что и во всех предыдущих случаях. Энергия активации окисления CO в области низкой активности катализатора составляет 30 ± 2 ккал/моль. Соответствующие графики представлены на рис. 4. Константа скорости в активном состоянии равна 2.5 ± 1.5 с–1.
Рис. 4.
Температурная зависимость степени превращения в реакции окисления CO на катализаторе Pd-ZSM. Начальная концентрация CO – 8500 м.д., концентрация кислорода – 2%. На вставке – зависимость степени превращения CO в реакции окисления от концентрации CO на его входе в реактор. Начальная концентрация CO – 2400 м.д., концентрация кислорода – 15%, температура реактора – 430 K.
На вставке к рис. 4 показано изменение степени превращения CO в CO2 для катализатора Pd-ZSM как функция концентрации CO на входе. Эта зависимость имеет ступеньку при некоторой концентрации оксида углерода, которая соответствует переходу катализатора из низкоактивного состояния (при высокой концентрации CO) в высокоактивное состояние (при низкой концентрации CO).
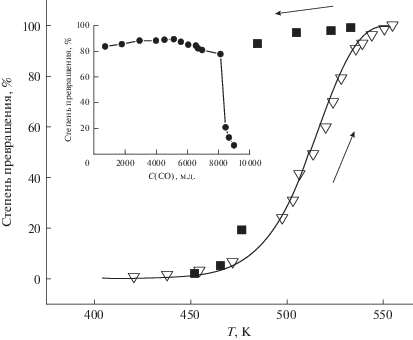
Катализатор Pd-CeO2/ZrO2. В качестве носителя катализатора использовали порошок, содержащий 70 мол. % CeO2 и 30 мол. % ZrO2, произведенный фирмой Degussa GmbH. Как и в предыдущих экспериментах, 4 вес. % палладия были нанесены пропиткой порошка водным раствором Pd(NH3)4(NO3)2. Удельная поверхность порошка катализатора после прокаливания в течение 12 ч при температуре 600°С составляла 87 м2/г. Для синтеза нанодисперсного носителя катализатора использовали метод, аналогичный примененному в работах [34, 35], где характерный размер частиц носителя составлял величину <10 нм.
Поведение катализатора совершенно отличается от поведения трех предыдущих каталитических материалов: палладия на оксиде алюминия, палладия, напыленного на керамику, и палладия на цеолите. Для катализатора на основе палладия, нанесенного на церий-циркониевый материал, вообще не наблюдалось зажигания катализатора. Вся кинетическая кривая может быть описана простым экспоненциальным уравнением, представленным выражением (5).
Результаты моделирования экспериментальной кривой представлены на рис. 5. Во всех расчетах использовали одно и то же значение энергии активации процесса окисления оксида углерода, равное 19 ккал/моль.
Рис. 5.
Температурная зависимость степени превращения в реакции окисления CO на катализаторе CeO2/ZrO2 + 4 вес. % Pd. Начальная концентрация CO – 8250 м.д., концентрация кислорода – 2%.
Такой характер зависимости подтверждает предположение о том, что процесс окисления CO происходит на ионах Ce4+ и палладий является только промотором процесса последующего окисления ионов Ce3+ до состояния Ce4+.
Катализатор Rh-ZSM. Были исследованы катализаторы на основе цеолита ZSM-5 и порошка оксида алюминия, стабилизированного оксидом лантана, допированных 0.25 вес. % родия. Катализаторы готовили таким же способом, как и катализаторы, содержащие палладий. Катализатор Rh-ZSM содержал изолированные ионы родия, что было показано в результате исследования спектров ЭПР образца.
При начальной концентрации CO, равной 8600 м.д., и концентрации кислорода, равной 2%, для катализатора Rh-ZSM также наблюдается “зажигание”, как и для катализаторов, содержащих палладий. При более низких концентрациях оксида углерода в газе катализатор не отравляется хемосорбированным CO (рис. 6), и, таким образом, “зажигание” катализатора при уменьшении концентрации CO не наблюдается.
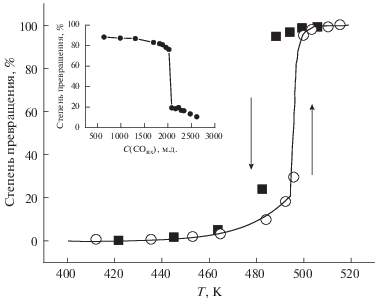
Рис. 6.
Температурная зависимость степени превращения в реакции окисления CO на катализаторе Rh-ZSM. Начальная концентрация CO – 2300 м.д., концентрация кислорода – 15%. На вставке – зависимость степени превращения CO в реакции окисления от концентрации CO на его входе в реактор. Начальная концентрация CO – 8800 м.д., концентрация кислорода – 2%, температура реактора – 480 K.
Только для этого катализатора мы смогли измерить энергию активации реакции окисления CO до и после точки “зажигания”. Это связано с более низкой скоростью окисления CO, в результате чего скорость реакции при низкой концентрации CO достаточно мала для того, чтобы быть измеренной.
Концентрационный предел перехода катализатора лежит при более высокой концентрации, чем для палладия. Катализатор Rh-ZSM переходит в активное состояние при концентрации <7300 м.д. Как и для катализаторов, содержащих палладий, реакция подчиняется закону первого порядка по оксиду углерода как до, так и после точки “зажигания”. Кривая, иллюстрирующая скачок скорости реакции окисления CO при уменьшении концентрации ниже 8000 м.д., представлена на вставке к рис. 6. В высокоактивном состоянии энергия активации реакции равна 31 ± ± 2 ккал/моль. В низкоактивном состоянии энергия активации реакции окисления CO составляет ~45 ккал/моль.
Для катализатора, содержащего родий, возник интересный эффект. При концентрации оксида углерода вблизи перехода катализатора из высокоактивного в низкоактивное состояние наблюдался колебательный режим процесса, детектируемый по колебаниям концентрации оксида углерода на выходе из реактора. Важно, что этот процесс проявлялся при постоянной температуре катализатора. Период этих не очень устойчивых колебаний составляет ~2 с. Подобные колебательные явления наблюдаются при определении концентрации CO с помощью полупроводникового сенсора CO методом шумовой спектроскопии [36].
Применение катализаторов в термокаталитических сенсорах CO. Принцип действия термокаталитических сенсоров основан на измерении теплоты каталитического окисления газа-аналита на катализаторе, нанесенном на терморезистор. Терморезистор, с одной стороны, нагревает катализатор до рабочей температуры, а с другой – измеряет изменение температуры катализатора, связанное с протеканием целевой химической реакции.
Основными требованиями к катализатору являются его активность и однозначность зависимости скорости химической реакции на его поверхности от концентрации газа.
Бóльшая часть катализаторов, исследованных в настоящей работе, второму критерию не удовлетворяет. Это связано с тем, что на кривых зависимости скорости реакции от концентрации оксида углерода (например, рис. 3, вставка) имеется особая точка, в которой скорость химической реакции изменяется скачком приблизительно в 4–5 раз. Поэтому традиционные катализаторы дожигания CO на основе инертных носителей (напыленный палладий, палладий на оксиде алюминия, палладий на цеолите и т.д.) не могут быть использованы для изготовления термокаталитических сенсоров CO.
Единственным исключением из числа исследованных каталитических материалов является палладий на церий-циркониевом катализаторе. Он обеспечивает монотонность зависимости скорости каталитической реакции от концентрации оксида углерода и, следовательно, однозначность отклика термокаталитического сенсора от концентрации.
ЗАКЛЮЧЕНИЕ
Показана возможность описания кинетики окисления CO на всех исследованных катализаторах с использованием одной и той же кинетической схемы. Выявлены две области концентраций CO, в которых поведение катализатора сильно различается. Это состояние I (ниже) и состояние II (выше) концентрационного предела Cinv, при котором скорость окисления CO на поверхности катализатора изменяется скачком. Обратный переход из состояния II в состояние I сопровождается гистерезисом по концентрации СО.
Установлено, что энергия активации процесса окисления CO на всех катализаторах, нанесенных на инертный носитель (палладий на Al2O3/La2O3, палладий, напыленный на керамику, цеолит ZSM-5), практически одинакова и равна 30 ккал/моль. Энергия активации процесса окисления на катализаторах, нанесенных на химически активный носитель, в котором возможно изменение окислительного состояния иона металла (SnO2, CeO2/ZrO2, а также система Ce/Zr/Pr), существенно ниже этой величины и зависит от химических свойств носителя. Это связано с тем, что в таких системах палладий является промотором, способствующим переходу иона металла из восстановленного в окисленное состояние (например, переходу Ce+3 → Ce+4).
Примерно такая же картина характерна и для катализаторов на основе родия. Только для этого катализатора наблюдается колебательный режим протекания реакции окисления оксида углерода.
Полученные результаты позволяют обоснованно выбирать катализаторы для термокаталитических сенсоров CO. Показано, что из числа исследованных только катализатор на основе оксидов церия и циркония, допированный палладием, обеспечивает однозначность зависимости скорости реакции окисления CO от концентрации оксида углерода.
Кроме того, осмысленный выбор условий протекания реакции окисления CO в каталитическом дожигателе выхлопных газов позволит сэкономить до 80% палладия или существенно улучшить степень очистки газов.
Список литературы
Langmuir I. // Trans. Faraday Soc. The Royal Soc. Chem. 1922. V. 17. P. 621.
Hinshelwood C., Grant G. // Nature. 1933. V. 131. P. 361.
Rideal E.K. // Math. Proc. CambridgePhilos. Soc. 1939. V. 35. № 1. P. 130.
Малахов В.Ф., Шмачков В.А., Васильев В.Ю., Колчин A.M. // Кинетика и катализ. 1977. Т. 18. С. 572.
Engel T., Ertl G. // J. Chem. Phys. 1978. V. 69. № 3. P. 1267.
Turner J.E., Sales B.C., Maple M.B. // Surf. Sci. 1981. V. 109. № 3. P. 591.
Turner J.E., Sales B.C., Maple M.B. // Surf. Sci. 1981. V. 103. № l. P. 54.
Mars P., van Krevelen D.W. // Chem. Eng. Sci. 1954. V. 3. P. 41.
Imbihl R., Ertl G. // Chem. Rev. 1995. V. 95. № 3. P. 697.
Беляев В.Д., Слинько M.M., Тимошенко В.И., Слинько М.Г. // Кинетика и катализ. 1973. Т. 14. № 3. С. 810.
Slinko M.M., Jaeger N.I. Oscillatory heterogeneous catalytic systems, studies in surface science and catalysis. Amsterdam: Elsevier, 1994. V. 86. 393 p.
Ertl G. // Adv. Catal. 1990. V. 37. P. 213.
Datye A.K., Bravo J., Nelson T.R. et al. // Appl. Catal. A: General. 2000. V. 198. № 1-2. P. 179.
Colussi S., Trovarelli A., Vesselli E. et al. // Appl. Catal. A: General. 2010. V. 390. № 1-2. P. 1.
Raj R., Harold M.P., Balakotaiah V. // Chem. Eng. J. 2016. V. 281. P. 322.
Kinnunena N.M., Hirvia J.T., Kallin K. // Appl. Catal. B: Environ. 2017. V. 207. P. 114.
Яшник С.А., Винокуров З.С., Сальников А.В. и др. // Кинетика и катализ. 2018. Т. 59. № 1. С. 92.
Chakrabarty T., Silveston P.L., Hudgins R.R. // Can. Soc. Chem. Eng. 1984. V. 62. P. 651.
Lang S.M., Fleischer I., Bernhardt T.M. et al. // ACS Catal. 2015. V. 5. № 3. P. 2275.
Calderón J.C., Ráfales M.R., Nieto-Monge M.N. et al. // Nanomaterials (Basel). 2016. V. 6. № 10. P. 187.
Пахаруков И.Ю., Бекк И.Э., Матросова М.М. и др. // Докл. АН. 2011. Т. 439. № 2. С. 211.
Шутилов А.А., Зенковец Г.А., Пахаруков И.Ю., Просвирин И.П. // Кинетика и катализ. 2014. Т. 55. № 1. С. 11.
Berdau M., Yelenin G.G., Karpowicz A. et al. // J. Chem. Phys. 1999. V. 110. № 23. P. 11551.
Allian A.D., Takanabe K., Fujdala K.L. et al. // J. Am. Chem. Soc. 2011. V. 133. № 12. P. 4498.
Buendia G.M., Machado E., Rikvold P.A. // J. Chem. Phys. 2009. V. 131. № 18. P. 184704.
Lyubovsky M., Smith L.L., Castaldi M. et al. // Catalysis Today. 2003. V. 83. № 1-4. P. 71.
Чернова Л.Е., Митричев И.И., Женса А.В., Кольцова Э.М. // Успехи в химии и химической технологии. 2017. Т. 31. № 8. С. 51.
Bugaev A.L., Usoltsev O.A., Guda A.A. et al. // J. Phys. Chem. C. 2018. V. 122. P. 12029. https://doi.org/10.1021/acs.jpcc.7b11473
Al Soubaihi R.M., Saoud K.M., Dutta J. // Catalysts. 2018. V. 8. P. 660. https://doi.org/10.3390/catal8120660
Васильев А.А. Физико-химические принципы конструирования газовых сенсоров на основе оксидов металлов и структур металл/твердый электролит/полупроводник. Дис. … докт. техн. наук. М., 2004.
Kim J.H., Sung J.S., Son Yu.M. et al. // Sensors and Actuators B. 1997. V. 44. P. 452.
Atsushi Satsuma, Kaoru Osaki, Masatoshi Yanagihara et al. // Catalysis Today. 2015. V. 258. P. 83.
Feng Bin, Xaolin Wei, Kwan San Hui. // Appl. Catal. B: Environ. 2015. V. 162. P. 282.
Simonenko T.L., Simonenko N.P., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1475.
Bugaenko V.V., Pshenichnyi R.N. // Russ. J. Inorg. Chem. 2019. V. 63. P. 169.
Угрюмов Р.Б., Шапошник А.В., Воищев В.С. и др. // Шумовые и деградационные процессы в полупроводниковых приборах. Мат-лы докл. междунар. научно-техн. семинара. (Москва, 3–6 декабря 2002 г.) М.: МНТОРЭС им. А.С. Попова, МЭИ, 2003. С. 97.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии