

Журнал неорганической химии, 2020, T. 65, № 3, стр. 302-308
Биомиметический синтез карбонатгидроксиапатита в присутствии хондроитинсульфата
О. А. Голованова *
Омский государственный университет им. Ф.М. Достоевского
644077 Омск, пр-т Мира, 55а, Россия
* E-mail: golovanoa2000@mail.ru
Поступила в редакцию 25.07.2019
После доработки 15.10.2019
Принята к публикации 27.10.2019
Аннотация
Проведен синтез карбонатгидроксиапатита (КГА) из прототипа синовии в присутствии хондроитинсульфата. Установлено, что наличие полисахарида в составе модельного раствора не влияет на состав твердой фазы, но способствует уменьшению площади удельной поверхности образцов. Для выявления закономерностей взаимодействия хондроитинсульфата с поверхностью карбонатгидроксиапатита проведен адсорбционный эксперимент и показано, что адсорбция хондроитинсульфата на КГА описывается моделью Ленгмюра. Установлено, что растворение синтезированных образцов в ацетатном буферном растворе (pH 5.5) и 0.9%-ном NaCl является двухстадийным процессом. Образцы, содержащие хондроитинсульфат, имеют более низкие скорости растворения по сравнению с беспримесной твердой фазой.
ВВЕДЕНИЕ
В настоящее время большое внимание уделяется созданию биокомпозитных материалов на основе фосфатов кальция, предназначенных для реконструкции костных дефектов, образующихся в результате патологических изменений (коксартроз, остеопороз, деформирующий артроз и др.), травм, хирургических вмешательств [1–4]. Такие материалы должны обладать биосовместимостью с костной тканью человека. Традиционно в ортопедии используется керамика на основе гидроксиапатита (ГА), который в химическом плане наиболее близок к минеральной составляющей костной ткани. Материалы на его основе не вызывают отрицательных реакций организма и биологически активны в отношении интеграции с костной тканью [5–7].
На сегодняшний день определяющим стал подход регенеративной медицины, основанный на применении биоматериалов, способных инициировать регенерацию костной ткани и замещаться вновь образованной [5, 6, 8]. Перспективными в этом плане являются композиты на основе карбонатгидроксиапатита (КГА) с выполняющими роль матрицы органическими полимерами, которые сочетают в себе структуру и свойства натуральной кости человека [9–11]. В качестве органической компоненты композитов особый интерес представляют полисахариды. Они являются высокоэластичными, биосовместимыми и обладают адгезией к хрящевой ткани [12].
Согласно литературным данным, гетерополисахарид хондроитинсульфат (ХС) является самым распространенным гликозаминогликаном в организме человека [13]. Он обладает уникальными физико-химическими свойствами, улучшает кровоток в синовиальной ткани и субхондральной зоне кости, что приводит к стабилизации показателей обмена хряща, замедляет ее резорбцию, уменьшает потерю кальция, улучшает фосфорно-кальциевый обмен в хрящевой ткани, ускоряет процессы ее восстановления, а также тормозит процессы разрушения хрящевой и соединительной тканей [14, 15].
Известно, что хондроитинсульфат принимает участие в формировании костной ткани, однако его роль в данном процессе изучена недостаточно [15, 16]. При этом представляется возможным получение в ходе “мокрого” синтеза композитов, включающих КГА и ХС.
Целью настоящей работы является синтез твердых фаз, формирующихся из прототипа синовиальной жидкости (синовии) человека в присутствии ХС и изучение их физико-химических свойств.
ОБЪЕКТЫ И МЕТОДЫ
Синтез КГА осуществляли из модельной среды, приближенной по ионно-электролитному составу, pH и ионной силе к синовиальной жидкости человека, по методике [17]. Эксперименты проводили из раствора при пятидесятикратном пересыщении по осадкообразующим ионам Са2+ и ${\text{НРО}}_{{\text{4}}}^{{{\text{3}}--}}$ по сравнению с физиологическим значением данного параметра для синовии. Для приготовления модельных растворов использовали соли CaCl2 ⋅ 2H2O, Na2HPO4 ⋅ 12H2O, MgCl2 ⋅ 6H2O, NaHCO3, KCl, Na2SO4 и NaCl марки “ч. д. а.” Содержание ХС в модельном растворе варьировали от 0.5 до 2.5 г/л с шагом 0.5 г/л. Раствор, содержащий катионы, приливали к раствору с анионами и полисахаридом со скоростью 5 мл/мин. Кислотность модельной системы поддерживали 10%-ным раствором HCl (pH 7.40 ± 0.05). Синтез проводили при комнатной температуре (t = 22–25°С). Образовавшийся осадок выдерживали под маточным раствором в течение 7 сут (время было установлено в ходе предыдущих экспериментов), затем отфильтровывали, промывали водой и сушили при 80°C до полного удаления химически несвязанной воды, после чего взвешивали и исследовали физико-химическими методами анализа.
Количественное определение ХС в твердой фазе осуществляли по разности начальных и конечных концентраций полисахарида в модельном растворе. Для определения концентрации ХС использовали анализ, основанный на цветной реакции D-глюкуроновой кислоты до 5-карбоксифурфурола и цветной реакции последнего с карбазолом с получением интенсивно окрашенного фиолетового комплекса и их последующем спектрофотометрическом определении. Для измерений использовали спектрофотометр ПЭ-5400УФ (λ = 530 ± 2 нм) [18].
Фазовый состав полученных порошков исследовали методом рентгенофазового анализа (дифрактометр D8 Advance Bruker). Для идентификации фаз использовали базу данных ICDD PDF для порошковой дифракции. Расчет размеров кристаллитов (областей когерентного рассеяния) проводили при помощи программы TOPAS 3.0 (Bruker). Погрешность не превышала 5%.
ИК-спектры осадков регистрировали на спектрофотометре ФСМ-2202. Пробы готовили прессованием таблеток с KBr. Морфологию и особенности строения поверхности твердых фаз исследовали методом растровой электронной микроскопии (РЭМ) на электронном микроскопе JSM-6610LV Jeol. Термический анализ проводили на синхронном термическом анализаторе STA-449C Netzsch. Пробы прокаливали в платиновых тиглях на воздухе от 25 до 1000°C со скоростью 10 град/мин. Количественные данные массовых потерь при отжиге получены с помощью программного обеспечения Proteus 7.10. Удельную поверхность образцов исследовали по методике одноточечной адсорбции азота при 77.4 K на адсорбционном приборе Сорбтометр производства ООО “Катакон” (Россия). Расчет полученных величин Syд выполнен по методу БЭТ.
Для проведения адсорбционного эксперимента навески синтезированного без хондроитинсульфата КГА массой 0.3 г заливали растворами ХС разной концентрации (0.025–0.25 мас. %), устанавливали pH 7.40 ± 0.05, встряхивали в течение 30 мин и оставляли на 48 ч. Затем содержимое колб фильтровали и определяли содержание полисахарида в фильтрате. Для установления уравнения, описывающего адсорбцию, проводили обработку экспериментальных данных с позиции теории Ленгмюра и теории Фрейндлиха [19].
Для моделирования активной и пассивной фаз резорбции образцы растворяли при постоянном перемешивании в ацетатном буфере (pH 5.5) и в 0.9%-ном растворе хлорида натрия (рН 7). По методу прямой потенциометрии фиксировали значения pCa в растворе с использованием иономера И-160М. Полученные зависимости обрабатывали с помощью регрессионного анализа (программный пакет SigmaPlot 12.5) [17].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Исследование образцов, полученных из прототипа синовиальной жидкости, методом РФА показало, что присутствие ХС в модельном растворе не влияет на фазовый состав твердой фазы (рис. 1). Все образцы представлены гидроксиапатитом.
Рис. 1.
Дифрактограммы образцов при содержании ХС 0 (1), 0.5 (2), 1.0 (3), 1.5 (4), 2.0 (5) и 2.5 г/л (6).
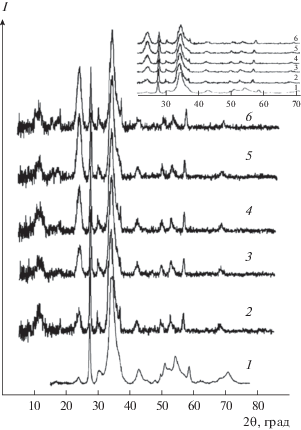
Кристаллографические параметры образцов, синтезированных в присутствии ХС, близки к таковым для стехиометрического апатита (табл. 1). С ростом содержания добавки полисахарида наблюдается уменьшение параметра а и увеличение параметра с по сравнению с беспримесным образцом. Размеры кристаллитов полученных образцов вдоль направления [001] составляют 20–22 нм. Данные табл. 1 обработаны статистически с вероятностью Р = 0.95 и выявлено, что размеры кристаллитов принадлежат одной генеральной совокупности. Характеристики кристаллической решетки образцов, полученных в присутствии ХС, близки к таковым для нестехиометрических карбонатсодержащих кальцийдефицитных гидроксиапатитов (например, JCPDS № 46-0905), составляющих минеральную основу костной ткани человека [4, 6, 17].
Таблица 1.
Кристаллографические параметры решетки, размеры кристаллитов и удельная поверхность КГА
| CХС, г/л | a, Å | с, Å | с/а | D, нм | Syд, м2/г |
|---|---|---|---|---|---|
| 0 | 9.459 ± 0.002 | 6.874 ± 0.002 | 0.727 | 21.6 | 130 ± 7 |
| 0.5 | 9.416 ± 0.005 | 6.881 ± 0.005 | 0.731 | 21.3 | 128 ± 6 |
| 1.0 | 9.418 ± 0.005 | 6.884 ± 0.005 | 0.731 | 20.8 | 103 ± 5 |
| 1.5 | 9.412 ± 0.005 | 6.884 ± 0.005 | 0.731 | 20.8 | 100 ± 5 |
| 2.0 | 9.412 ± 0.005 | 6.884 ± 0.005 | 0.731 | 20.8 | 97 ± 5 |
| 2.5 | 9.416 ± 0.005 | 6.882 ± 0.005 | 0.731 | 20.7 | 88 ± 4 |
| Костная ткань | 9.410 | 6.891 | 0.732 | 5–10 | – |
Предположение о включении групп ${\text{СО}}_{{\text{3}}}^{{{\text{2}}--}}$ в состав осажденного ГА подтверждается данными ИК-фурье-спектроскопии (рис. 2). Так, в спектрах всех полученных образцов наблюдаются валентные (ν(Н2О) при 3400–3440 см–1) и деформационные (δ(Н–О–Н) при 1610–1650 см–1) колебания Н2О; асимметричные валентные и деформационные колебания иона ${\text{PО}}_{{\text{4}}}^{{{\text{3}}--}}$ (ν3(Р–О) при 1030–1090 см–1, ν4(О–Р–О) при 605–564 см–1 и ν2(О–Р–О) при 470–475 см–1); дублет асимметричных валентных колебаний связи ν3(С–О) при 1422, 1460 см–1 и деформационное колебание ν2(О–С–О) при 875 см–1 в ${\text{СО}}_{{\text{3}}}^{{{\text{2}}--}},$ указывающие на механизм замещения фосфатных тетраэдров карбонат-ионами в структуре полученных ГА по Б-типу [20–24].
Рис. 2.
ИК-спектры образцов при содержании ХС 0 (1), 0.5 (2), 1.0 (3), 1.5 (4), 2.0 (5) и 2.5 г/л (6).
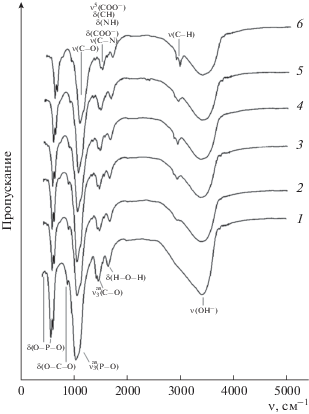
На присутствие в составе синтезированных образцов ХС указывают моды колебаний органических группировок, см–1: 1032–1037 – валентные ν(С–О) пиранового цикла; 1422–1440 – симметричные валентные νs(СОО–); 1460 – деформационные метильных δ(СН) и метиленовых групп δ(СН2); 1507–1543 – деформационные δ(NH) и δ(СОО–), валентные ν(C–N) амида II; 1715–1798 – асимметричные валентные νas(С=О) сложноэфирных групп; 2800–3600 – валентные ν(С–H); 3420–3600 – ν(ОН–), ассоциированные внутримолекулярными водородными связями, а также асимметричные и симметричные колебания OSO3-группы (1230 и 1067 см–1 ) [11, 25].
С увеличением содержания ХС можно отметить уменьшение интенсивности дублета полосы поглощения связей δ(О–Р–О) в ионе ${\text{РО}}_{{\text{4}}}^{{{\text{3}}--}}$ (564–610 см–1), косвенно характеризующего содержание фосфат-ионов в составе твердой фазы. Кроме того, можно отметить увеличение количества полос поглощения органических групп в интервале 2700–4000 см–1, что указывает на большее содержание полисахарида в составе осадка.
Исследование морфологии и особенностей строения поверхности твердых фаз выполнено методом растровой электронной микроскопии (РЭМ) (рис. 3). Видно, что беспримесный образец представляет собой конгломераты частиц неправильной формы размером 0.2–200 мкм и имеет пористую поверхность (рис. 3а). На снимках образца, полученного в присутствии 2.0 г/л ХС, наблюдается уменьшение размера частиц до 0.2–100 мкм, а поверхность становится менее пористой (рис. 3б).

Методом БЭТ установлено, что увеличение добавки ХС в составе исходного раствора способствует уменьшению площади удельной поверхности образцов (табл. 1). Данные табл. 1 обработаны статистически с вероятностью Р = 0.95 и выявлено, что полученные значения не принадлежат одной генеральной совокупности. Таким образом, с увеличением содержания полисахарида в исходном растворе наблюдается уменьшение площади удельной поверхности образцов. Такое влияние на поверхностные свойства твердой фазы может быть связано с менее пористой поверхностью образцов, полученных в присутствии ХС [25–28]. Это подтверждает сделанное ранее предположение по данным РЭМ.
Для установления механизма взаимодействия ХС с поверхностью КГА проведен адсорбционный эксперимент. В результате обработки полученных изотерм сорбции были рассчитаны значения молекулярной (Г) и предельной адсорбции (Г∞), а также степень заполнения поверхности адсорбента. После обработки данных адсорбционных экспериментов получены уравнения, описывающие адсорбцию ХС на поверхности КГА с позиции теории Лэнгмюра и теории Фрейндлиха (рис. S1, табл. 2). Установлено, что адсорбция ХС при рН 7.4 имеет хорошую корреляцию в линейных координатах обеих моделей, но лучшее значение R2 характерно для изотермы в соответствии с моделью Ленгмюра, что указывает на образование мономолекулярного слоя на поверхности ГА [19, 28].
Таблица 2.
Уравнения Фрейндлиха и Ленгмюра для адсорбции хондроитинсульфата на КГА
| Модель Фрейндлиха | |||||
|---|---|---|---|---|---|
| Уравнение | 1/n | n | lnK | K | R2 |
| у = 0.257х + 1.897 | 0.257 | 3.891 | 1.897 | 6.67 | 0.910 |
| Модель Ленгмюра | |||||
| Уравнение | 1/Г∞, кг/моль | Г∞, моль/кг | 1/b Г∞, кг/моль | b | R2 |
| у = 0.214х + 0.004 | 0.214 | 4.684 | 0.004 | 53.37 | 0.980 |
Термический анализ образцов в температурном интервале 25–1000°C позволил выделить четыре этапа, соответствующих превращениям разных по природе составляющих (рис. 4). Первый этап (25–280°С, эндотермический эффект) характеризуется разложением легколетучих компонентов и удалением адсорбированной воды:
(1)
$\begin{gathered} \text{[}{\text{КГА}} \cdot n{{{\text{H}}}_{{\text{2}}}}{\text{O}}] \cdot m{{{\text{Н}}}_{{\text{2}}}}{{{\text{O}}}_{{\left( {{\text{тв}}} \right)}}} \to \\ \to {\text{КГА}} \cdot n{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\left( {{\text{тв}}} \right)}}} + m{{{\text{Н}}}_{{\text{2}}}}{{{\text{O}}}_{{\left( {\text{г}} \right)}}} + {\text{Q}}. \\ \end{gathered} $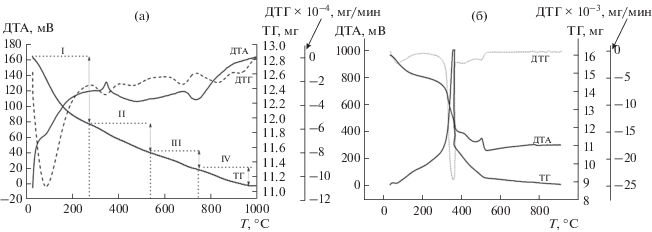
Второй этап (280–470°С, экзотермический эффект) сопровождается удалением хондроитинсульфата (tпл ≈ 225°С) и структурно связанной кристаллизационной воды по схеме [27]:
(2)
${\text{КГА}} \cdot n{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\left( {{\text{тв}}} \right)}}} \to {\text{ КГ}}{{{\text{А}}}_{{{\text{(тв)}}}}} + m{{{\text{Н}}}_{{\text{2}}}}{{{\text{O}}}_{{\left( {\text{г}} \right)}}} + {\text{Q}}{\text{.}}$На третьем этапе (470–750°С, экзотермический эффект) происходит десорбция кристаллизационной воды, на четвертом этапе (750–1000°С) – преобразование КГА в стехиометричную фазу ГА (3) или β-Са3(РО4)2 (4) [7, 20]:
(3)
$\begin{gathered} {\text{Сa}}{{(}_{{10--}}}{{_{x}}_{{/2}}})({\text{P}}{{{\text{O}}}_{4}}){{(}_{{6--}}}_{x}){{({\text{C}}{{{\text{O}}}_{3}})}_{x}}{{({\text{OH}})}_{2}} \to \\ \to {\text{С}}{{{\text{а}}}_{{10}}}{{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{6}}{{\left( {{\text{ОН}}} \right)}_{2}} + {\text{ }}6{\text{С}}{{{\text{О}}}_{2}} \uparrow --\,{\text{Q}}, \\ \end{gathered} $(4)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{\text{9}}}}{\kern 1pt} {{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{{{\text{6}}--x--y}}}{\kern 1pt} {{\left( {{\text{НР}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{y}}{\kern 1pt} {{\left( {{\text{С}}{{{\text{О}}}_{{\text{3}}}}} \right)}_{x}}{\kern 1pt} {{\left( {{\text{ОН}}} \right)}_{{{\text{2}}--{\kern 1pt} y({\text{тв}})}}}{\kern 1pt} \xrightarrow{{800^\circ {\text{C}}}} \\ \to {\text{3}}\beta {\text{ - С}}{{{\text{а}}}_{{\text{3}}}}{{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{{{\text{2}}({\text{тв}})}}} + х{\text{С}}{{{\text{О}}}_{{{\text{2}}\left( {\text{г}} \right)}}} + {\text{2}}{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\left( {\text{г}} \right)}}}--{\text{Q}}. \\ \end{gathered} $Установлено, что термическое преобразование образцов, полученных в присутствии полисахарида, сопровождается наибольшими термическими эффектами на втором и третьем этапах. Из рис. 4 и табл. S3 видно увеличение общей потери массы с ростом содержания ХС. Поэтапное удаление химически связанной воды (этап 3) у образцов, содержащих ХС, проявляется в виде двух пиков на кривых ДТГ при температурах 510 и 710°С.
Важным свойством веществ, рассматриваемых в качестве биоматериалов для инженерии костной ткани, является их способность к растворению при контакте с биологическими жидкостями. Для оценки данной характеристики проведено их растворение в растворителях, подобранных с учетом условий резорбции in vivo. Исследование растворения образцов в ацетатном буферном растворе (рН 5.5) проведено для моделирования активной фазы резорбции остеокластами (рис. 5).
Рис. 5.
Кривые растворения образцов в ацетатном буферном растворе при содержании ХС 0 (1), 0.5 (2), 1 (3), 1.5 (4), 2 (5) и 2.5 г/л (6).
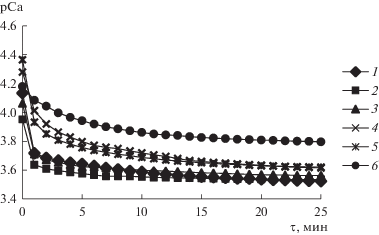
Для определения скорости растворения осадков все кинетические кривые проанализированы с помощью регрессионного анализа [6, 17]. Установлено, что процесс растворения образцов является двухстадийным. На начальном участке кривых зависимость концентрации ионов кальция в растворе от времени C(τ) = $ - \lg {{C}_{{{\text{C}}{{{\text{a}}}^{{2 + }}}}}}$ описывается линейной функцией. Истинная скорость для линейной зависимости определяется как тангенс угла наклона прямой.
Следующая стадия процесса растворения образцов в ацетатном буфере характеризуется экспоненциальной зависимостью:
где С0 – условная начальная концентрация, Сm – концентрация насыщения, b – коэффициент, τ – время.Данная зависимость соответствует кинетике первого порядка [29]. В качестве количественной меры скорости растворения на данном этапе можно рассматривать условную начальную концентрацию.
На линейном и экспоненциальном участках полученной зависимости (табл. S4) наблюдается уменьшение скорости растворения образцов с увеличением содержания ХС в исходном модельном растворе. Полученные результаты можно объяснить тем, что данный полисахарид обладает высокой связывающей способностью по отношению к ионам Са2+, при этом катионы взаимодействуют с отрицательно заряженным ХС, находящимся в ионизированном состоянии, с образованием устойчивого соединения, что препятствует их выделению в раствор [11, 14, 30]. Таким образом, увеличение добавки ХС способствует уменьшению скорости растворения синтезированных образцов.
Растворение образцов в 0.9%-ном растворе NaCl изучали для моделирования пассивной фазы резорбции (рис. 6).
Рис. 6.
Кривые растворения образцов в 0.9%-ном растворе NaCl при содержании ХС 0 (1), 0.5 (2), 1 (3), 1.5 (4), 2 (5) и 2.5 г/л (6).
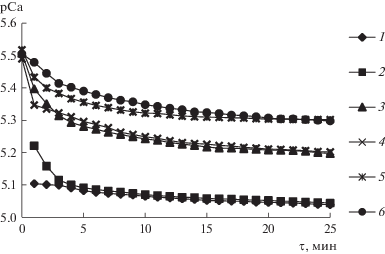
Обработка кинетических кривых с помощью регрессионного анализа показала, что в 0.9%-ном растворе NaCl скорость растворения образцов уменьшается с увеличением содержания ХС (табл. S5). Полученные данные, как и в случае растворения образцов в ацетатном буферном растворе, можно объяснить взаимодействием катионов Са2+ с полисахаридом [11, 14, 30].
ЗАКЛЮЧЕНИЕ
Получен КГА из прототипа синовии в присутствии ХС. Установлено, что наличие полисахарида в модельном растворе не влияет на размеры формирующихся кристаллитов, но приводит к уменьшению площади удельной поверхности образцов за счет уменьшения их пористости. Выявлено, что адсорбция хондроитинсульфата на КГА подчиняется уравнению Ленгмюра. При термической обработке образцов зафиксированы максимальные термические эффекты в диапазоне температур 340–350°C. Данные динамического растворения в ацетатном буфере и 0.9%-ном растворе NaCl показали, что скорость резорбции образцов уменьшается с увеличением содержания ХС в исходном модельном растворе.
Список литературы
Jabr S. Al-Sanabani, Ahmed A. Madfa, Fadhel A. Al-Sanabani // Int. J. Biomater. 2013. V. 2013. P. 12.
Dorozhkin S.V. // Mater. Sci. Eng. 2015. V. 55. P. 272.
Ebrahim M., Bortelho M. // J. Data Brief. 2017. V. 10. P. 93.
Epple M., Ganesan K., Heumann R. et al. // J. Mater. Chem. 2010. V. 20. № 1. P. 18.
Горшенев В.Н., Ершов Ю.А., Телешев А.Т. и др. // Медицинская техника. 2014. № 1. С. 30.
Баринов С.М. // Успехи химии. 2010. № 1. С. 15.
Гудимов Н.В., Беляков А.В. // Высокотемпературные материалы и технологии 2016. Т. 30. № 7. С. 32.
Сафронова Т.В., Путляев В.И. // Наносистемы: физика, химия, математика. 2013. № 4. С. 24.
Bourgeat-Lami E. // J. Nanosci. Nanotechnol. 2002. V. 2. № 1. P. 1.
Rasskazova L.A., Lytkina D.N., Shapovalova Y.G. et al. // Adv. Mater. Res. 2015. V. 1085. P. 394.
Пономарева Н.И., Попрыгина Т.Д., Карпов С.И. и др. // Вестник ВГУ. Сер. Химия. Биология. Фармация. 2012. № 2. С. 82.
Захаров Н.А., Сенцов М.Ю. // Краткие сообщения. Сорбционные и хроматографические процессы. 2011. Т. 11. № 2. С. 178.
Watanabe H., Ikoma T., Chen G. et al. // Key Eng. Mater. 2006. V. 309. P. 533.
Шкарина Т.Н., Исаева И.В., Ковалева С.В. и др. // Фармация. 2007. № 3. С. 42.
Панасюк А.Ф., Ларионов Е.В. // Научно-практическая ревматология. 2000. № 2. С. 12.
Moldovan L., Craciunescu O., Oprita E. et al. // Roumanian Biotechnol. Lett. 2009. V. 14. № 3. P. 4459.
Gerk S.A., Golovanova O.A., Odazhiu V.N. // Inorg. Mater. 2018. V. 54. P. 305. https://doi.org/10.1134/S0020168518030044
Шкарина Т.Н., Исаева И.В., Ковалева С.В. и др. // Фармация. 2007. № 3. С. 42.
Щукин Е.Д., Перцов А.В., Амелина Е.А. М.: Юрайт, 2017. 444 с.
Frank-Kamenetskaya O., Kol’tsov A., Kuz’mina M., Zorina et al. // J. Molecular Structure. 2011. T. 992. № 1–3. P. 9.
Rodicheva G.V., Orlovsky V.P., Privalov V.I. et al. // Russ. J. Inorg. Chem. 2001. V. 46. № 11. P. 1631.
Gabuda S.P., Kozlova S.G., Pletnev R.N. // Dokl. Chemistry. 2007. V. 413. № 2. P. 86.
Shi J., Klocke A., Zhang M. et al. // Eur. J. Mineral 2005. V. 17. P. 769. https://doi.org/10.1127/09351221/2005/0017-0769
Solonenko A.P., Golovanova O.A. // Russ. J. Inorg. Chem. 2014. V. 59. № 11. P. 1228.
Fadeeva T.V., Golovanova O.A. // Russ. J. Inorg. Chem. 2019. V. 64. № 7. P. 847.
Cao G. Nanostructures and Nanomaterials: Synthesis, properties and application. London: Imperial College Press, 2010. 581 p.
Malikova T.V., Golovanova O.A., Chikanova E.S. // Inorg. Mater. 2018. V. 54. № 9. P. 957.
Голованова О.А., Головченко К.К. // Журн. физ. химии. 2019. Т. 93. № 11. С. 1714.
Романовский Б.В. Основы химической кинетики. М.: Экзамен, 2006. 416 с.
Uchisawa H., Okuzaki B., Ichita J. et al. // ICS. 2001. V. 1223. P. 205.
Дополнительные материалы
- скачать Golovanova RJIC.docx
- Дополнительные материалы
Инструменты
Журнал неорганической химии