

Журнал неорганической химии, 2020, T. 65, № 4, стр. 458-466
Композиты МоО2/С: синтез, свойства, механизм формирования
З. А. Фаттахова a, *, Г. С. Захарова a
a Институт химии твердого тела УрО РАН
620990 Екатеринбург, ул. Первомайская, 91, Россия
* E-mail: fattahova.zilara@yandex.ru
Поступила в редакцию 17.09.2019
После доработки 16.10.2019
Принята к публикации 27.11.2019
Аннотация
Гидротермально-микроволновой обработкой водного раствора пероксомолибденовой кислоты с глюкозой и последующим отжигом прекурсора в инертной атмосфере при 500°С синтезированы композиты на основе диоксида молибдена моноклинной сингонии МоO2/С. Содержание углерода в композитном материале изменяется от 8 до 24 мас. %. Методами рентгенофазового и термогравиметрического анализа, ИК- и КР-спектроскопии, сканирующей и просвечивающей электронной микроскопии, низкотемпературной адсорбции азота определены основные физико-химические характеристики синтезированных соединений. Установлено, что существенным фактором, определяющим состав, текстурные и термические свойства композитов МоО2/С, а также особенности их формирования, является молярное соотношение компонентов реакционной массы Мо : С6Н12О6 = 1 : (0.25–1.0). Предложен механизм образования композита МоО2/С.
ВВЕДЕНИЕ
Благодаря низкому электрическому сопротивлению (8.8 × 10–5 Ом м [1]) и большой теоретической разрядной емкости (838 (мА ч)/г [2]) диоксид молибдена MoO2 считается одним из перспективных анодных материалов литиевых источников тока [3]. Например, MoO2 с морфологией нанопластин обеспечивает удельную разрядную емкость ~300 (мА ч)/г после 20-го цикла заряд/разряд при плотности тока 5 мА/см2 [4]. Однако широкое практическое применение MoO2 в качестве анодного материала сдерживается резким изменением объема при его электрохимическом циклировании [5]. Получение наноразмерного МоО2 с различной морфологией является одним из методов, направленных на решение этой проблемы. Наноразмерный МоО2 получают в виде частиц неправильной формы [6, 7], наностержней [8], нановолокон [9], что обеспечивает большую удельную площадь поверхности и более высокую реакционную активность. Другим методом улучшения характеристик является формирование композита на основе оксида металла с углеродом [10]. Однако наиболее эффективным подходом представляется сочетание перечисленных выше способов. Так, композит MoO2/C с морфологией микросфер при плотности тока 100 мА/г обеспечивает удельную разрядную емкость 768 и 800 (мА ч)/г после первого и 300-го цикла заряд/разряд соответственно [11]. Хорошая циклируемость и стабильные емкостные параметры (327 (мА ч)/г при плотности тока 1000 мА/г с кулоновской эффективностью 92% после 20 разрядных циклов) характеризуют композит MoO2/C с морфологией частиц в виде нанопроволоки [12].
В литературе описаны различные способы получения наноразмерных композитных материалов на основе диоксида молибдена и углерода. Наиболее универсальными методами получения композита МоО2/С являются золь-гель синтез и гидротермальная обработка смеси молибденсодержащих соединений и углеродных компонентов с последующим отжигом в инертной атмосфере. В качестве источника молибдена может быть использован металлический Мо, а также водорастворимые соли молибдена. При этом различные органические соединения (этиленгликоль, глюкоза, этанол, олеиновая кислота, углеродные нанотрубки и т.д.) служат источником углерода. Гидротермальная обработка смеси молибдата аммония (NH4)2MoO4 и углеродных нанотрубок позволяет получать нанакомпозиты МоО2/С без дополнительного отжига [13]. Недостатком данного метода является необходимость предварительной активации поверхности углеродных нанотрубок. Термическим разложением смеси МоО2, поливинилиденфторида –(С2Н2F2)n–, метилпирролидона C4H7NO могут быть получены частицы МоО2/С со сферической морфологией размером 100 нм [14]. Термообработка смеси МоО3 и олеиновой кислоты C17H33COOH приводит к формированию наностержней МоО2/С диаметром 200 нм и длиной 1–8 мкм [15]. Гидротермальная обработка смеси пероксомолибденовой кислоты и этиленгликоля C2H6O2 с последующим отжигом в инертной атмосфере позволяет получать частицы композита МоО2/С, подобные сферам размером 10 нм [16]. Использование гептамолибдата аммония (NH4)6Mo7O24 вместо пероксомолибденовой кислоты приводит к увеличению размера частиц до 30 нм [17]. Сферические частицы МоО2/С размером 200 нм могут быть получены отжигом в атмосфере аргона смеси молибдата аммония (NH4)6Mo7O24 и поливинилпирролидона (C6H9NO)n [18]. Добавление C2H6O2 в реакционную смесь (NH4)6Mo7O24 и поливинилпирролидона приводит к уменьшению размера частиц до 60–80 нм [19]. Благодаря доступности и низкой стоимости в качестве углеродсодержащего соединения широко используется глюкоза. Обработка смеси МоО3 и глюкозы с последующим отжигом в инертной атмосфере позволяет получать композит МоО2/С в виде нанолистов [20, 21]. Добавление этанола в реакционную смесь МоО3 и глюкозы приводит к формированию наноремней шириной 200 нм и длиной 5–10 мкм [22]. Варьирование температуры прокаливания смеси МоО3 · 2H2O и глюкозы позволяет целенаправленно получать МоО2/С с заданной морфологией. Так, при температуре отжига 400°С образуются частицы МоО2/С с морфологией наноремней шириной 10 нм и длиной <25 нм, повышение температуры пиролиза до 600°С приводит к формированию сферических частиц размером ~23 нм, ступенчатый же отжиг при температурах 400 и 700°С позволяет увеличить размер сфер до 39 нм [23]. Гидротермальная обработка с последующим отжигом в атмосфере аргона смеси МоО2 и глюкозы позволяет получать частицы размером 200 нм [24]. Несмотря на универсальность и широкое распространение известных способов получения композитов МоО2/С, имеются определенные технологические сложности, обусловленные многостадийностью и длительностью процесса. Поэтому в последнее время исследователи все чаще используют метод гидротермально-микроволнового синтеза, который позволяет существенно снизить время синтеза, регулировать давление системы, а также гомогенизировать состав реакционной массы за счет перемешивания [25].
В настоящей работе впервые гидротермально-микроволновой обработкой водного раствора пероксомолибденовой кислоты и глюкозы с последующим отжигом в инертной атмосфере синтезирован композит на основе диоксида молибдена и углерода, исследована морфология, текстурные и физико-химические свойства МоО2/С, а также рассмотрен механизм формирования композита.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали порошок металлического молибдена (99.9 мас. % Мо), 30%-ный раствор пероксида водорода марки “ос. ч.” и глюкозу С6Н12О6 марки “ч.”. Получение композита проводили в две стадии. На первой стадии порошок молибдена растворяли при охлаждении (5–10°С) в избытке Н2О2 с образованием желтого раствора пероксомолибденовой кислоты. К полученному раствору добавляли водный раствор глюкозы в молярном соотношении Мо : С6Н12О6 = = 1 : (0.25–1.0). Затем реакционную массу подвергали гидротермально-микроволновой обработке в реакторе Monowave 300 фирмы Anton Parr (США) при температуре 160°С в течение 20 мин. В результате образуется раствор черного цвета, который упаривали досуха. Полученный осадок, использованный в качестве прекурсора, обозначали как МоО2-Х, где Х – мольное содержание глюкозы в реакционной массе. На второй стадии MoO2-Х подвергали прокаливанию в токе азота со скоростью 7 град/мин при температуре 500°С в течение 1 ч. Рентгенофазовый анализ (РФА) образцов выполняли на дифрактометре Shimadzu XRD 7000 (CuKα-излучение, λ = 1.5418 Å). Морфологию порошков изучали с помощью сканирующей электронной микроскопии (СЭМ) (JEOL JSM 6390 LA) и просвечивающей электронной микроскопии (ПЭМ) (JEOL JEM-200CX). Исследования образцов методом ИК-Фурье-спектроскопии проводили на спектрометре Spectrum One (Perkin–Elmer). Спектры комбинационного рассеяния регистрировали на спектрометре U1000 (Renishaw) с использованием твердотельного лазера LCM-S-11 с длиной волны 532 нм и мощностью 40 мВт. Для понижения поглощения лазерного излучения образцом, приводящего к его нагреву и изменению свойств, мощность лазера уменьшили до 1%. Термический анализ выполняли на анализаторе STA 449 F3 Jupiter (Netzsch), совмещенном с масс-спектрометром QMS 403, при скорости нагрева 10 град/мин в атмосфере воздуха (ТГ–ДСК–МС). Для изучения термической стабильности композитов на воздухе был проведен рентгенофазовый анализ порошков после их отжига в муфельной печи в интервале температур 215–315°С с шагом 5°С в течение 15 мин. Текстурные характеристики (площадь удельной поверхности, пористость) композитов определяли методом низкотемпературной адсорбции азота на анализаторе Gemini VII (Micromeritics). Пробоподготовку образцов проводили вакуумированием при 150°С в течение 2 ч. На основе полученных изотерм сорбции азота рассчитывали площадь удельной поверхности по методу Брунауэра–Эммета–Теллера [26]. Анализ пористости материалов выполняли с использованием данных изотерм сорбции по методу Баррета–Джойнера–Халенда [27].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
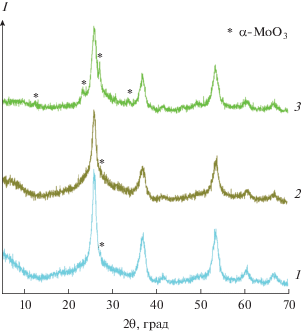
Согласно данным РФА, порошки прекурсоров МоО2-Х являются слабоокристаллизованными (рис. 1а). При этом с увеличением X наблюдается усиление характерных рефлексов МоО2. Термолиз МоО2-Х в атмосфере азота при температуре 500°С в течение 1 ч приводит к формированию композита на основе структуры МоO2 (рис. 1б). С увеличением содержания углеродной компоненты в реакционной массе (X) наблюдается уменьшение интенсивностей дифракционных пиков композитов МоО2/С-Х. Соединения МоО2/С-Х кристаллизуются в моноклинной сингонии с пр. гр. Р21/с, параметры элементарной ячейки которых приведены в табл. 1. Установлено, что с увеличением содержания глюкозы в реакционной массе параметры элементарной ячейки а и с увеличиваются, а параметр b уменьшается. С использованием уравнения Шеррера был рассчитан средний размер кристаллитов МоО2:
где D – размеры кристаллитов, k – безразмерный коэффициент формы частиц, равный 0.9 (постоянная Шеррера), Δ(2θ) – ширина пика на половине высоты, θ – брегговский угол, λ – длина волны рассеянного излучения. Расчет вели по интенсивным и хорошо разрешенным дифракционным пикам (011), (020), (022). Средний размер кристаллитов МоО2 композитов МоО2/С-Х представлен в табл. 1. Для композитов МоО2/С-0.25 и МоО2/С-0.5 средний размер кристаллитов MoO2 практически одинаковый. Значительное уменьшение среднего размера кристаллитов наблюдается для композита МоО2/С-1.Рис. 1.
Дифрактограммы прекурсоров МоО2-Х (а), композитов MoO2/C-Х (б) при X = 0.25 (1), 0.5 (2), 1 (3) и позиции брегговских пиков МоО2 по данным JCPDS 72-4534.
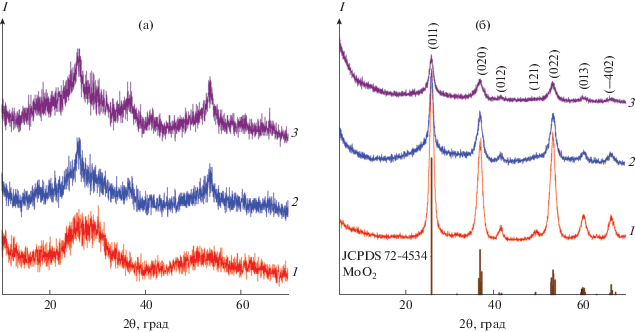
Таблица 1.
Параметры элементарной ячейки, текстурные характеристики композита МоО2/С-Х и МоО2
| Композит | a, Å | b, Å | c, Å | β, град | V, Å3 | Dср, нм | S, м2/г | Dпор, нм |
|---|---|---|---|---|---|---|---|---|
| МоО2/С-0.25 | 5.582(5) | 4.850(7) | 5.501(2) | 118.95(6) | 130.3(4) | 6.2 | 19.7 | 4.4 |
| МоО2/С-0.5 | 5.600(2) | 4.832(1) | 5.636(2) | 120.00(1) | 132.0(9) | 6.1 | 48.2 | 3.6 |
| МоО2/С-1 | 5.657(7) | 4.818(2) | 5.641(6) | 120.60(3) | 132.3(7) | 5.1 | – | – |
| МоО2 (JCPDS 72-4534) | 5.6109 | 4.8562 | 5.6285 | 120.95 | 131.53 | – | – | – |
На рис. 2 показана морфология композитов МоО2/С в зависимости от содержания С6Н12О6 в реакционной массе. По данным СЭМ, порошки композитов MoO2/C-X состоят из сильно агломерированных частиц шарообразной формы. С увеличением содержания глюкозы в реакционной смеси диаметр частиц увеличивается и для композита MoO2/C-1 составляет >500 нм.

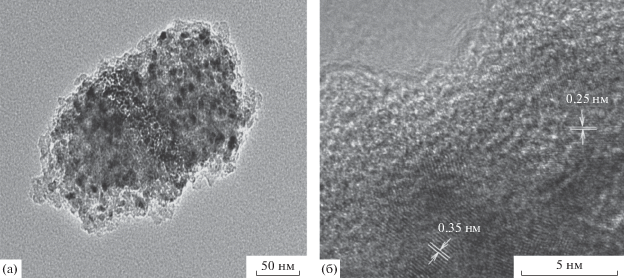
ПЭМ позволила более детально изучить морфологию композитов (рис. 3). ПЭМ-изображение МоО2/С-1 низкого разрешения демонстрирует наличие наночастиц МоО2 и аморфного углеродного слоя толщиной ~10 нм (рис. 3а). Расстояния, обнаруженные в кристаллических областях, составляют около 0.35 и 0.25 нм, что соответствует межплоскостным расстояниям (011) и (020) кристаллической структуры МоО2 (рис. 3б). Исследование с помощью ПЭМ дополнительно подтверждает сосуществование в композитах на основе диоксида молибдена кристаллитов МоО2 и углерода.
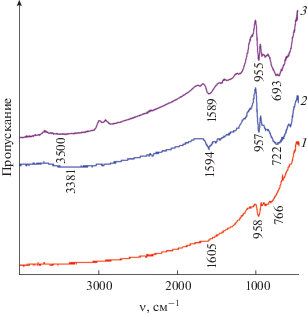
ИК-спектры композитов МоО2/С-Х (рис. 4) соответствуют стандартному спектру диоксида молибдена [28]. В ИК-спектрах МоО2/С-Х наблюдаются интенсивные узкие полосы в интервале 955–958 см–1, отвечающие валентным колебаниям связей ν(Mo=O). Валентные колебания связей ν(Мо–О–Мо) в октаэдре МоО6 проявляются в диапазоне 600–850 см–1. Деформационные колебания адсорбированных молекул воды описываются полосами в области 1589–1605 см–1 [29]. Кроме того, в композитах МоО2/С-0.5, МоО2/С-1 наблюдается широкая малоинтенсивная полоса в интервале 3381–3500 см–1, характерная для валентных колебаний ОН-групп [30].
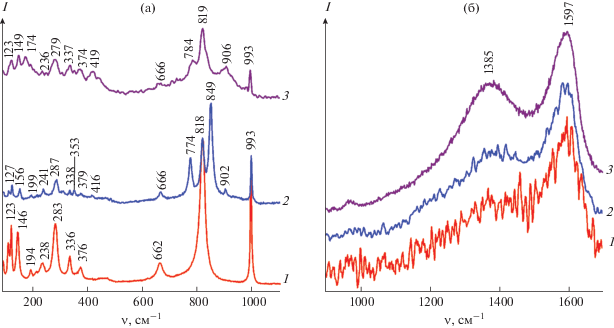
В спектрах КР соединений МоО2/С-Х (рис. 5а) в интервале 90–1100 см–1 наблюдаются вибрационные моды диоксида молибдена [31]. Дополнительно в спектре КР проявляется интенсивная полоса при 993 см–1, которая описывает валентные колебания кратных связей Мо=О и указывает на наличие слоистой структуры, характерной для орторомбической фазы МоО3 [32]. Появление пиков МоО3 объясняется частичным окислением МоО2 при воздействии лазера на композит МоО2/С во время исследования методом спектроскопии КР [33]. Дополнительный анализ спектров КР позволил идентифицировать присутствие углерода в композитах. В спектрах КР МоO2/C-Х (рис. 5б) в интервале 1100–1700 см–1 наблюдаются две характеристические полосы углерода. Пик при 1385 см–1 описывает D-линию, которая соответствует колебаниям атомов углерода с sp3-типом гибридизации и свидетельствует о наличии разупорядочения. Пик при 1597 см–1 описывает G-линию, соответствующую колебаниям атомов углерода в sp2-гибридизации, и указывает на наличие в образце мелкокристаллического графита [34]. Таким образом, исследования выполненные с использованием КР-спектроскопии показали, что при формировании композита МоO2/C происходит образование разупорядоченной мелкокристаллической графитовой составляющей. Для оценки степени упорядоченности углерода использовали показатель, определяемый соотношением интенсивностей D- и G-линий (ID/IG). Для композитов МоO2/C-0.25, МоO2/C-0.5 и МоO2/C-1 величина ID/IG равна 0.53, 0.59 и 0.72 соответственно. Увеличение показателя ID/IG композитов МоO2/C-Х с ростом содержания глюкозы в реакционной массе свидетельствует о понижении степени графитизации конечного продукта. Кроме того, вычисленные значения ID/IG свидетельствуют о более высокой степени упорядочения углеродного каркаса композитов МоO2/C по сравнению с оксидом графена, композитом МоО2/графен, для которых аналогичное значение выше [35]. Подобное явление наблюдалось для композитов состава TiO2/C [36].
Рис. 5.
КР-cпектры композитов MoO2/C-0.25 (1), MoO2/C-0.5 (2) и MoO2/C-1 (3) в диапазоне частот 90–1100 см–1 (а) и 900–1700 см–1 (б).
Для количественного определения содержания углерода в композитах MoO2/C-Х использовали ТГ-ДСК-МС анализ на воздухе (рис. 6). Термическое разложение композитов МоO2/C-Х проходит в несколько стадий. В интервале температур 20–220°С для всех образцов наблюдается убыль массы до ~5.7%, которую можно отнести к удалению адсорбционной воды. Присутствие адсорбционной воды в МоO2/C-Х подтверждено данными ИК-спектроскопии. Относительно сложный характер изменения массы в интервале температур 220–650°С обусловлен одновременным протеканием процессов окисления углеродной составляющей композита до СО2 и МоО2 до МоО3. Выделение диоксида углерода подтверждается интенсивным пиком молекулярного иона ${\text{CO}}_{2}^{ + }$ в масс-спектре с m/z = 44 а.е.м. Различие в температурных режимах выделения CO2 на МС-кривых для композитов МоO2/C-Х в процессе отжига можно объяснить неоднородным состоянием углерода, входящего в состав композита. Эндоэффект, наблюдаемый при температуре 801°С, описывает плавление МоО3, образующегося при окислении диоксида молибдена. Термогравиметрический анализ композитов МоO2/C-X позволил оценить содержание углерода в образцах, которое составляет 8, 18 и ~ 24 мас. % для МоO2/C-0.25, МоO2/C-0.5 и МоO2/C-1 соответственно.
Рис. 6.
Кривые ТГ-ДСК-МС композитов MoO2/C-0.25 (а), MoO2/C-0.5 (б) и MoO2/C-1 (в) в атмосфере воздуха.
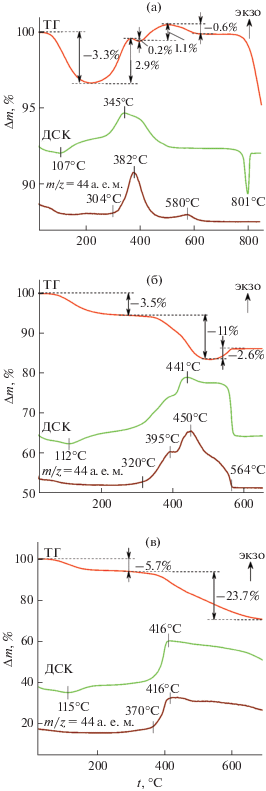
Дополнительно была изучена термическая стабильность композитов МоO2/C-Х на воздухе. Согласно данным РФА, кристаллическая структура МоО2 композитов МоO2/C-0.25, МоO2/C-0.5 и МоO2/C-1 сохраняется до температуры 215, 255 и 285°С соответственно (рис. 7). Повышение температуры выше установленного значения приводит к частичному окислению МоО2 и образованию МоО3 в качестве примесной фазы. Таким образом, увеличение содержания глюкозы в исходной реакционной смеси позволяет повысить термическую стабильность композитов МоO2/C-Х на 70°С.
Рис. 7.
Дифрактограммы композитов MoO2/C-1 (1), MoO2/C-0.5 (2) и MoO2/C-0.25 (3), полученные после отжига на воздухе при температурах 300, 270 и 230°С соответственно.
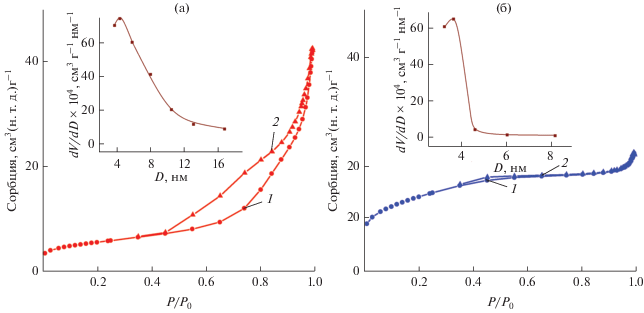
На рис. 8 представлены изотермы сорбции азота и соответствующие кривые распределения пор по размерам композитов MoO2/C-Х. Текстурные параметры образцов MoO2/C-Х зависят от содержания глюкозы в реакционной массе (табл. 1). Определить площадь удельной поверхности и пористость композита MoO2/C-1 не представлялось возможным даже при увеличении времени пробоподготовки до 6 ч. Возможно, это обусловлено высоким содержанием углерода, часть которого слабо связана с матрицей композита, что не позволило достичь состояния равновесия при вакуумировании образца в процессе пробоподготовки. Согласно классификации ИЮПАК [37], изотермы сорбции композита MoO2/C-Х относятся к IV типу с петлей гистерезиса Н3 (Х = = 0.25) и Н4 (Х = 0.5). Для композита МоO2/C-0.5 наблюдается наибольшая величина удельной поверхности, равная 48.2 м2/г, что, по-видимому, обусловлено образованием дефектов в композитном материале при карботермическом восстановлении МоО2 с ростом концентрации углеродной составляющей. Для композитов MoO2/C-Х характерно очень узкое распределение пор по размерам с преобладанием мезопор размером 3–5 нм.
Рис. 8.
Изотермы сорбции (1 – адсорбция, 2 – десорбция) и кривые распределения пор по размерам (вставки) композитов MoO2/C-0.25 (а) и MoO2/C-0.5 (б).
В соответствии с полученными результатами, возможный механизм образования композита МоО2/С может быть описан следующими химическими реакциями:
(2)
$\begin{gathered} {\text{2Mo}} + {\text{1}}0{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}} \to \\ \to {\text{2}}{{{\text{Н}}}^{ + }} + \,\,{\text{ }}{{[{\text{M}}{{{\text{o}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}}{{({{{\text{O}}}_{{\text{2}}}})}_{{\text{4}}}}]}^{{{\text{2}}--}}} + {\text{9}}{{{\text{H}}}_{{\text{2}}}}{\text{O}},{\text{ }} \\ \end{gathered} $(3)
$\begin{gathered} {\text{2}}{{{\text{H}}}^{ + }} + {{\left[ {{\text{M}}{{{\text{o}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}}{{{\left( {{{{\text{O}}}_{{\text{2}}}}} \right)}}_{{\text{4}}}}} \right]}^{{{\text{2}}--}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to {\text{4}}{{{\text{H}}}^{ + }} + {\text{2}}{{\left[ {{\text{Mo}}{{{\text{O}}}_{{\text{2}}}}{{{\left( {{{{\text{O}}}_{{\text{2}}}}} \right)}}_{{\text{2}}}}} \right]}^{{{\text{2}}--}}}, \\ \end{gathered} $(4)
$\begin{gathered} {\text{2}}{{{\text{Н}}}^{ + }} + {{\left[ {{\text{Mo}}{{{\text{O}}}_{{\text{2}}}}{{{\left( {{{{\text{O}}}_{{\text{2}}}}} \right)}}_{{\text{2}}}}} \right]}^{{{\text{2}}--}}} + {\text{3}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{{\text{12}}}}}{{{\text{О}}}_{{\text{6}}}} \to \\ \to {\text{Мо}}{{{\text{О}}}_{{\text{2}}}} + {\text{3}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{{\text{12}}}}}{{{\text{О}}}_{{\text{7}}}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}, \\ \end{gathered} $(5)
$\begin{gathered} {\text{5Mo}}{{{\text{O}}}_{{\text{2}}}} + {{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{{\text{12}}}}}{{{\text{О}}}_{{\text{7}}}} \to \\ \to {\text{5Мо}}{{{\text{О}}}_{{\text{2}}}}{\text{/С}} + {\text{СО}} + {\text{6}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}}. \\ \end{gathered} $Синтез композита начинается с получения раствора пероксомолибденовой кислоты растворением порошка молибдена в избытке водного раствора H2O2 (реакции (2)–(3)) [38]. Процесс сопровождается окислением металлического молибдена до Mo6+. Далее нагревание раствора пероксомолибденовой кислоты и глюкозы в гидротермально-микроволновых условиях приводит, согласно реакции (4), к восстановлению Мо6+ до Мо4+ и окислению глюкозы до глюконовой кислоты С6Н12О7 [39]. Формирование углеродной компоненты композита МоО2/С происходит при прокаливании реакционной массы в инертной атмосфере за счет разложения глюконовой кислоты (реакция (5)). Очевидно, что глюкоза в процессе синтеза выполняет одновременно роль мягкого восстановителя и источника углерода. Стоит отметить, что проведение реакции в условиях жидкофазного взаимодействия исходных реагентов позволяет вводить углерод в состав композита in situ. Такой подход к получению композита МоО2/С обеспечивает простоту и технологичность его выполнения, а также равномерное формирование оксидной и углеродной составляющих композита.
ЗАКЛЮЧЕНИЕ
Установлено, что гидротермально-микроволновой обработкой водного раствора пероксомолибденовой кислоты и глюкозы с последующим отжигом прекурсора в инертной атмосфере могут быть получены композитные материалы на основе диоксида молибдена. Содержание углерода в композитах определяется исходной концентрацией С6Н12О6 в реакционной массе и составляет 8–24 мас. %. Термическая стабильность композита и его текстурные характеристики зависят от содержания углерода, входящего в состав композита.
Список литературы
Лазарев В.Б., Соболев В.В., Шаплыгин И.С. Химические и физические свойства простых оксидов металлов. М.: Наука, 1983.
Sun C. Advanced Battery Materials. Wiley–Scrivener Publishing, 2019.
Hu X., Zhang W., Liu X. et al. // Chem. Soc. Rev. 2015. V. 44. № 8. P. 2376. https://doi.org/10.1039/c4cs00350k
Yang L.C., Gao Q.S., Zhang Y.H. et al. // Electrochem. Commun. 2008. V. 10. № 1. P. 118. https://doi.org/10.1016/j.elecom.2007.11.009
Zhang W.-M., Wu X.-L., Hu J.-S. et al. // Adv. Funct. Mater. 2008. V. 18. № 24. P. 3941. https://doi.org/10.1002/adfm.200801386
Liu Y., Zhang H., Quyang P. et al. // Electrochim. Acta. 2013. V. 102. P. 429. https://doi.org/10.1016/j.electacta.2013.03.195
Chen X., Zhang Z., Li X. et al. // Chem. Phys. Lett. 2006. V. 418. P. 105. https://doi.org/10.1016/j.cplett.2005.09.138
Koziej D., Rossel M.D., Ludi B. et al. // Small. 2011. V. 7. № 3. P. 377. https://doi.org/10.1002/smll.201001606
Yang S., Zhang Y., Wang W. et al. // J. Cryst. Growth. 2006. V. 290. № 1. P. 96. https://doi.org/10.1016/j.jcrysgro.2005.10.149
Li H., Zhou H. // Chem. Commun. 2012. V. 48. № 9. P. 1201. https://doi.org/10.1039/c1cc14764a
Li X., Xiao Q., Gao Y. et al. // J. Alloys. Compd. 2017. V. 723. № 5. P. 1113. https://doi.org/10.1016/j.jallcom.2017.06.274L
Gao Q., Yang L., Lu X. et al. // J. Mater. Chem. 2010. V. 20. № 14. P. 2807. https://doi.org/10.1039/b921001f
Bhaskar A., Deepa M., Rao T.N. // ACS Appl. Mater. Interfaces. 2013. V. 5. P. 2555. https://doi.org/10.1021/am3031536
Tang S., Yang L., Liu J. et al. // Mater. Res. Bull. 2018. V. 102. P. 277. https://doi.org/10.1016/j.materresbull.2018.01.039
Wang Y., Huang Z., Wang Y. // J. Mater. Chem. A. 2015. V. 3. P. 21314. https://doi.org/10.1039/C5TA05345E
Zhou L., Wu H.B., Wang Z. et al. // ACS Appl. Mater. Interfaces. 2011. V. 3. P. 4853. https://doi.org/10.1021/am201351z
Liu Y., Zhang H., Ouyang P. et al. // Mater. Res. Bull. 2014. V. 50. P. 95. https://doi.org/10.1016/j.materresbull.2013.10.024
Liu X., Wu D., Ji W. et al. // J. Mater. Chem. A. 2015. V. 3. № 3. P. 968. https://doi.org/10.1039/C4TA04373A
Wang Z., Chen J.S., Zhu T. et al. // Chem. Commun. 2010. V. 46. P. 6906. https://doi.org/10.1039/c0cc01174f
Jiang J., Yang W., Wang H. et al. // Electrochem. Acta. 2017. V. 240. P. 379. https://doi.org/10.1016/j.electacta.2017.04.103
Ni J.F., Zhao Y., Li L. et al. // Nano Energy. 2015. V. 11. P. 129. https://doi.org/10.1016/j.nanoen.2014.10.027
Yang L., Liu L., Zhu Y. et al. // J. Mater. Chem. 2012. V. 22. № 26. P. 13148. https://doi.org/10.1039/C2JM31364B
Sun X., Shi Y., Fang X. et al. // J. Nanosci. Nanotechnol. 2014. V. 14. P. 4278. https://doi.org/10.1166/jnn.2014.8281
Liu X., Ji W., Liang J. et al. // Phys. Chem. Chem. Phys. 2014. V. 16. № 38. P. 20570. https://doi.org/10.1039/c4cp02960g
Zakharova G.S., Baklanova I.V., Suntsov A.Y. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 12. P. 1584. [Захарова Г.С., Бакланова И.В., Сунцов А.Ю. и др. // Журн. неорган. химии. 2016. Т. 61. № 12. С. 1646.]https://doi.org/10.1134/S0036023616120214
Brunauer S., Emmett P.H., Teller E. // J. Am. Chem. Soc. 1938. V. 60. № 2. P. 309. https://doi.org/10.1021/ja01269a023
Barrett E.P., Joyner L.G., Halenda P.P. // J. Am. Chem. Soc. 1951. V. 73. № 1. P. 373. https://doi.org/10.1021/ja01145a126
Trivedi M.K., Tallapragada R.M., Branton A. et al. // IJMSA. 2015. V. 4. № 5. P. 354. https://doi.org/10.11648/j.ijmsa.20150405.21
Макатун В.Н. Химия неорганических гидратов. Мн.: Наука и техника, 1985.
Накамото К. ИК-спектры и спектры КР неорганических и координационных соединений. Пер. с англ. М.: Мир, 1991.
Dieterle M., Mestl G. // Phys. Chem. Chem. Phys. 2002. V. 4. № 5. P. 822. https://doi.org/10.1039/b107046k
Lee S., Seong M., Tracy C. et al. // Solid State Ionics. 2002. V. 147. № 1–2. P. 129. https://doi.org/10.1016/s0167-2738(01)01035-9
Camacho-Lypez M. A., Escobar-Alarcyn L., Picquart M. et al. // Opt. Mater. 2011. V. 33. P. 480. https://doi.org/10.1016/j.optmat.2010.10.028
Ferrari A.C., Robertson J. // Phys. Rev. B. 2000. V. 61. № 20. P. 14095. https://doi.org/10.1103/physrevb.61.14095
Palanisamy K., Kim Y., Kim H. et al. // J. Power Sources. 2015. V. 275. P. 351. https://doi.org/10.1016/j.jpowsour.2014.11.001
Fattakhova Z.A., Zakharova G.S., Andreikov E.I. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 7. P. 857. [Фаттахова З.А., Захарова Г.С., Андрейков Е.И. и др. // Журн. неорган. химии. 2019. Т. 64. № 7. С. 700. https://doi.org/10.1134/S0044457X19070079]https://doi.org/10.1134/S0036023619070076
Sing K.S.W., Everett D.H., Haul R.A.W. et al. // Pure Appl. Chem. 1985. V. 57. № 4. P. 603. https://doi.org/10.1351/pac198557040603
Li X., Xiao Q., Zhang H. et al. // J. Energy Chem. 2018. V. 27. № 3. P. 940. https://doi.org/10.1016/j.jechem.2017.09.008
Sun X.H., Zheng C.M., Zhang F.H. et al. // J. Phys. Chem. C. 2009. V. 113. № 36. P. 16002. https://doi.org/10.1021/jp9038682
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии